Criptorquidia y patología escrotal
Introducción
En este tema se abordan las patologías testiculares benignas más frecuentes, que comparten como dato común, alteraciones en la exploración escrotal. Para su mejor comprensión y basándose en su origen y curso clínico más habitual, las tratamos según la siguiente clasificación:
1. Criptorquidia o maldescenso testicular: alteraciones del desarrollo y descenso testicular, de origen malformativo.
2. Escroto agudo: alteraciones testiculares adquiridas de curso clínico agudo.
3. Otras patologías escrotales: de origen adquirido o malformativo y curso clínico variable.
Dada la importancia de las posibles repercusiones sobre la viabilidad del testículo en muchas de estas situaciones, el diagnóstico y seguimiento adecuados de los trastornos de descenso testicular o de otras patologías, como el varicocele, así como el reconocimiento de signos de alarma en los casos de escroto agudo, son esenciales y suponen el objetivo principal de este capítulo.
Trastornos del desarrollo y descenso testicular
El descenso testicular al escroto puede continuar en el periodo postnatal, hasta los 6 meses o el año de edad. El testículo no descendido es la malformación genital más frecuente en el varón recién nacido.
Aunque, en muchas ocasiones, se habla de forma generalizada de criptorquidia para referirse a la ausencia de testículo en el escroto o, incluso, de testículo no palpable, es preferible utilizar el término maldescenso testicular (MDT) o testículo no descendido. De este modo, se incluyen de forma más adecuada todas las alteraciones provocadas por un trastorno en el desarrollo y/o descenso testicular, que dan lugar tanto a exploraciones de testículo palpable como no palpable.
El descenso del testículo desde la cavidad abdominal puede completarse de forma espontánea durante los primeros meses de vida, durante los 6 meses en los nacidos a término (constatándose ausencia de descenso testicular en el 1 %, al año) y hasta el primer año en los recién nacidos (RN) prematuros. Así, los testículos no descendidos constituyen la malformación congénita genital más frecuente en el varón recién nacido, con prevalencias variables según la edad gestacional: 1-5 % en RN a término y 30-40 % en RN pretérminos.
Es bilateral en el 30 % de los pacientes. En estos casos de bilateralidad con testículos no palpables o en aquellos casos de MDT en los que se detecta algún otro signo sospechoso de alteración del desarrollo de la diferenciación sexual (hipospadias, escroto bífido, etc.), debe solicitarse valoración endocrinológica y estudio genético.
A pesar del descenso espontáneo, en muchos casos, es aconsejable el seguimiento clínico durante varios años durante la infancia, dada la posibilidad de “reascenso” testicular adquirido (40 %)(1,2).
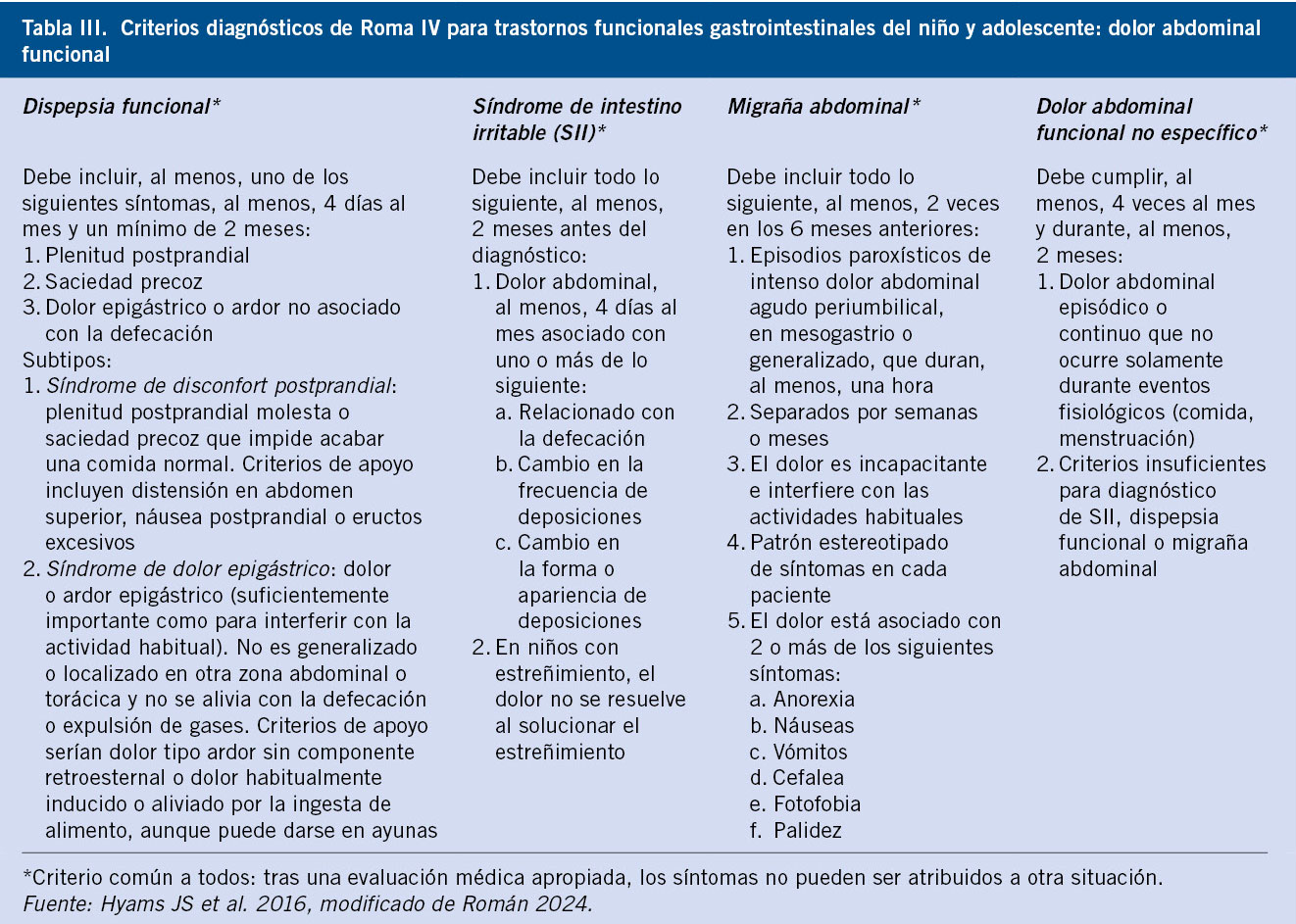
Clasificación
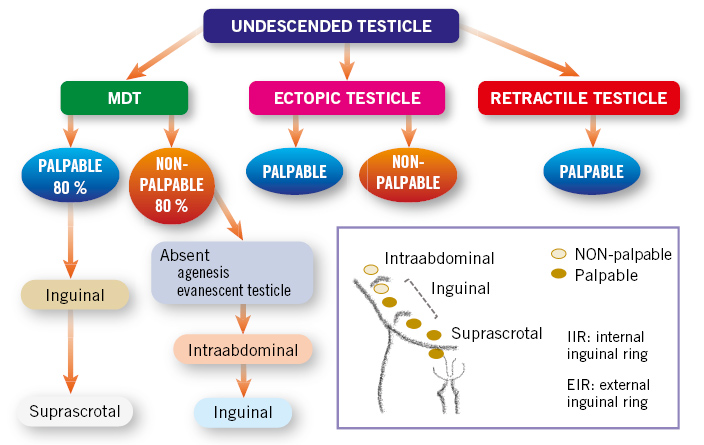
Basada en la etiopatogenia del MDT, y en la exploración física en función de la localización del testículo y de si estamos ante una situación de testículos palpables o no palpables(3-5) (Fig. 1):
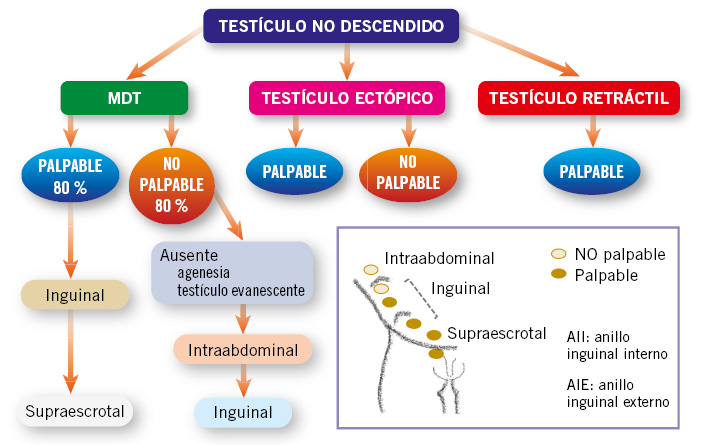
Figura 1. Clasificación del testículo no descendido, en función de su patogenia y localización. Se indica la situación de testículo palpable o no palpable según la posición testicular. MDT: maldescenso testicular.
• Maldescenso testicular congénito: el testículo se encuentra fuera de la bolsa escrotal, en cualquier punto a lo largo del trayecto normal de descenso. Pueden ser: palpables (80 %), cuando se sitúan externos al orificio inguinal interno; o no palpables (20 %), localizados profundos al orificio inguinal interno o a nivel intraabdominal.
En la exploración física, los testículos palpables pueden, en ocasiones, traccionarse hasta el escroto, pero “reascienden” inmediatamente, lo que les diferencia de los testículos retráctiles “en ascensor”.
• Testículo ectópico: el testículo se encuentra fuera del escroto a nivel extra-abdominal, en una posición “aberrante”, diferente al trayecto normal de descenso. Se sitúa más frecuentemente en la región del anillo inguinal superficial o a nivel del pubis. Otras posibilidades, menos frecuentes, son: la región femoral o perineal. Habitualmente, son testículos palpables y no descienden espontáneamente, por lo que requieren tratamiento quirúrgico.
• Testículo ausente, por dos mecanismos patogénicos posibles: 1) la agenesia testicular; o 2) la atrofia testicular secundaria a torsión testicular intrauterina, situación conocida como “testículo evanescente”(5).
• Testículo retráctil, “en ascensor”: se produce por un reflejo cremastérico exagerado. Son siempre palpables en región inguinal o en puntos altos del escroto, y se descienden fácilmente a escroto con la tracción manual, permaneciendo en escroto durante un tiempo. Se considera una situación fisiológica, que no requiere tratamiento, ya que el testículo ha completado su descenso a su posición normal en el escroto. Sin embargo, deben seguirse en consulta con exploraciones periódicas, ya que hasta un tercio de los casos pueden tener un ascenso posterior.
• Testículo no descendido adquirido: el testículo se encuentra en el escroto durante el primer año de vida, pero asciende posteriormente, debido a la falta de crecimiento del cordón espermático, que queda corto y retrae el testículo. Se puede considerar que son los verdaderos testículos retráctiles. Con frecuencia se detecta entre los 5-10 años de edad.
Patogenia y aspectos embriológicos(3,6-9)
En la etiopatogenia de la criptorquidia se describen como principales causas: factores genéticos, entre ellos los que afectan al cromosoma Y o al gen INSL-3, la prematuridad o el bajo peso al nacer.
Formación del testículo
La cresta gonadal indiferenciada se desarrolla hacia la gónada testicular a partir de la 6ª-7ª semana de gestación (SDG), influenciada por la acción del factor de transcripción SRY (sex-determining región Y) y, posteriormente, por otras moléculas (WT1, SF1, SOX9, FGF9), que provocan a su vez el desarrollo de las células de Sertoli y de Leydig del testículo. Las primeras, mediante la secreción de hormona antimulleriana (HAM), provocan la regresión de los conductos de Müller y ejercen una acción trófica para los espermatozoides. Por otra parte, las células de Leydig, mediante la liberación de testosterona, inducen el desarrollo de los conductos de Wolff (que darán lugar al conducto deferente y vesículas seminales) y a través de su metabolito dihidrotestosterona (DHT), el desarrollo de los genitales externos masculinos. Las células de Leydig, además liberan insulin-like factor 3 (INSL3), que produce el crecimiento del gubernaculum testis, que acompañará el descenso del testículo al escroto.
Descenso testicular al escroto
El testículo desciende desde el retroperitoneo hasta el escroto, en dos fases, que pueden finalizar de forma fisiológica hasta los 3-6 meses después del nacimiento:
1. Fase transabdominal: controlada por la hormona insulin-like 3 (INSL3), producida por las células de Leydig, y que estimula el crecimiento del gubernáculo para su posterior migración en dirección caudal. En esta fase, el testículo se desliza por la cavidad abdominal adosado al gubernáculum hasta situarse en el orificio inguinal interno, hacia la 15 SDG.
2. Fase inguinoescrotal, más compleja, e influenciada por la acción de los andrógenos, en la que el gubernaculum debe crecer y migrar 4-5 cm desde la región inguinal hasta el escroto, y guía así el desplazamiento del testículo desde la entrada en el canal inguinal hasta el escroto. Esta es la etapa habitualmente afectada en los casos de MDT, lo que explica que la localización más frecuente de los testículos no descendidos sea la región inguinal. Finaliza en torno a la 35 SDG, pudiendo prolongarse hasta los primeros meses de vida postnatal, ayudado por la elevación transitoria de gonadotropinas en los primeros 6 meses tras el nacimiento, que provoca una estimulación de las células de Leydig y, con ello, un incremento de los niveles de testosterona.
Causas genéticas
• Alteraciones cromosómicas que afecten al cromosoma Y: síndrome de Klinefelter u otras alteraciones estructurales de dicho cromosoma Y.
• Síndromes de base genética con afectación de otros cromosomas: síndrome de Noonan, síndrome de Prader-Willi (alteración en el cromosoma 15) o síndrome de Wiedemann-Beckwith (alteraciones en el cromosoma 11).
• Alteraciones en la secreción de HAM secundarias a disfunciones en la activación del factor transcripcional SRY, y de otros como: WT1, SF1, SOX9, FGF9 y DAX1. Producen anomalías en la diferenciación de los genitales internos, en la secreción de testosterona y en su transformación a dihidrotestosterona, con afectación del desarrollo de los genitales externos y del descenso testicular.
• Mutaciones en el gen INSL-3 (“insulin-like factor 3”) y en su receptor LGR8, involucrados en la masculinización del gubernaculum testis.
Prematuridad
La prematuridad o el bajo peso al nacer, influyen en la aparición de criptorquidia. La prematuridad constituye una situación favorecedora del MDT, debido al descenso del testículo durante el tercer trimestre de gestación, con posibilidad de completarse durante los primeros 3-6 meses tras el nacimiento.
Factores ambientales
Se debe investigar sobre exposición a factores maternos como: hipertensión, tabaco o consumo de analgésicos durante el embarazo. Así mismo, el contacto con sustancias químicas como: pesticidas, filatos y talatos (constituyentes en algunos plásticos), con propiedades estrogénicas, podrían afectar el descenso testicular al actuar como disruptores endocrinos, interfiriendo en la síntesis o acción de los andrógenos.
Atrofia testicular
Por accidente isquémico durante la gestación: torsión intraútero.
Diagnóstico
El diagnóstico del MDT se basa en los datos clínicos y en la exploración física, que se complementa con la ecografía en los casos de testículos no palpables, o en aquellos en los que se sospechen alteraciones de la diferenciación sexual.
Anamnesis
Antecedentes familiares y/o personales de criptorquidia u otras alteraciones del desarrollo genital masculino. Antecedentes obstétricos: edad gestacional, peso al nacimiento, ingesta o contacto con fármacos anti-androgénicos durante la gestación. Preguntar sobre el momento en que se apreció el MDT; así como, si se constató el testículo en el escroto en alguna ocasión o si la alteración se refiere como unilateral o bilateral.
Exploración física
Se debe realizar la exploración en un ambiente cálido y tranquilo. El paciente debe estar en decúbito supino con las piernas flexionadas y en abducción. En caso de duda, se puede explorar en posición de “cuclillas” o con el paciente sentado con las piernas cruzadas (exploración de Taylor). La exploración inguinoescrotal se realiza mediante palpación bimanual en busca de la localización del testículo. Se colocan los dedos a nivel del orificio inguinal interno, y se deslizan a lo largo del canal inguinal hasta el escroto. Debe valorarse: si el testículo es palpable o no palpable y, en el primer caso, su localización, así como el tamaño, consistencia y movilidad. También, si la afectación es unilateral o bilateral. En la inspección del escroto, valorar la morfología del escroto: asimetría o hipoplasia escrotal. En los casos unilaterales, la constatación de un aumento del tamaño escrotal en el lado donde el testículo es palpable, debe hacer sospechar una hipertrofia escrotal compensatoria y el antecedente de atrofia testicular intraútero como causa del testículo no palpable en el lado contralateral.
Se debe descartar la presencia de otras alteraciones del desarrollo genital masculino (hipospadias, alteraciones escrotales, implantación peneana, etc.), que pudieran sugerir la existencia de alteraciones de la diferenciación sexual(3,7).
Estudio hormonal y genético
Ante la presencia de criptorquidia bilateral, alteraciones en genitales externos o testículos no palpables y sospecha de testículo intraabdominal, se debe completar el diagnóstico con la solicitud de cariotipo y pruebas de función hormonal (gonadotropinas, testosterona y sus metabolitos, inhibina y HAM), que serán valoradas durante el seguimiento por los especialistas en Endocrinología(4).
Pruebas de imagen
En el caso de los testículos palpables, el diagnóstico de MDT es clínico y no es necesaria ninguna prueba diagnóstica complementaria. Se reserva la indicación de la ecografía abdominal y de la región inguinal para los casos de testículos no palpables. Esta detecta la existencia de los testículos de localización inguinoescrotal en la mayoría de los casos, con sensibilidad y especificidad cercanas al 100 %. Sin embargo, en los pacientes con testículos intraabdominales, la sensibilidad y especificidad descienden al 50 % y 75 %, respectivamente, por lo que la no detección ecográfica de testículo no descarta la existencia de testículo intraabdominal(4). En cualquier caso, aunque la ecografía puede ayudar en la elección de la vía de abordaje (se puede optar por iniciar la exploración quirúrgica por vía inguinal en los casos en los que se detecte ecográficamente el testículo en el canal inguinal), esta no es imprescindible en los pacientes con testículo no palpable, ya que en estos siempre va a estar indicada la exploración quirúrgica y orquidopexia, en los casos de que se confirme la existencia de testículo viable.
Se recomienda la realización de ecografía abdominal en aquellos pacientes con criptorquidia en los que se sospechen alteraciones del desarrollo sexual, en la búsqueda de testículos intraabdominales, malformaciones de genitales internos o, incluso, la presencia de tumoraciones abdominales de carácter secretor hormonal, causantes de alteraciones del desarrollo sexual. En caso de duda, deberá completarse el estudio radiológico mediante la realización de RM abdominopélvica para confirmar o descartar dichas anomalías.
En los casos en los que la ecografía no pueda confirmar la presencia de testículo y se sospeche la existencia de criptorquidia intraabdominal, está indicada la exploración laparoscópica abdominal en busca de un testículo intraabdominal o, incluso, de atrofia o agenesia testicular(3-5,8,9).
Tratamiento(3-7,9,10)
La edad y la distinción entre testículos palpables y no palpables son los aspectos fundamentales a considerar en el tratamiento del MDT. Ante el diagnóstico de MDT, el tratamiento de elección es la realización de una orquidopexia precoz: descenso y fijación testicular al escroto, preferiblemente antes del año de edad.
El objetivo del tratamiento de los testículos no descendidos es la colocación del testículo en su posición escrotal normal, permitiendo su crecimiento a la temperatura óptima (33ºC) para el correcto desarrollo y diferenciación de las células madre gonadales, minimizando así el riesgo de aparición de secuelas y complicaciones relacionadas con la infertilidad o el cáncer testicular en el futuro. Así mismo, el normal posicionamiento del testículo en el escroto, permite la exploración del mismo y la detección precoz de situaciones de escroto agudo o la presencia de tumoraciones testiculares.
El tratamiento indicado en los pacientes con MDT es quirúrgico. El objetivo es la localización del testículo y su descenso y fijación en el escroto (orquidopexia), o bien, la extirpación de restos testiculares en los casos de atrofia testicular.
Los aspectos fundamentales a tener en cuenta son: la edad indicada para la intervención quirúrgica, la distinción entre testes palpables y no palpables en cuanto a la elección de las técnicas quirúrgicas a realizar, y el seguimiento de los testículos retráctiles (“en ascensor”), hasta poder descartar definitivamente un ascenso testicular adquirido, que requiera tratamiento quirúrgico.
Edad(4,9,11)
El tratamiento quirúrgico debe realizarse en torno al año de edad, a partir de los 6 meses y no antes, por la posibilidad de descenso espontáneo hasta ese momento. Se recomienda la realización de una orquidopexia precoz: hasta los 12 meses o, como muy tarde, los 18 meses de edad, que es el tiempo límite considerado para la recuperación (“catch up”) de una fertilidad potencialmente dañada por los cambios en las células germinales testiculares y la depleción de las células de Leydig, anteriormente descritos. Esta indicación está especialmente establecida en los casos de criptorquidia bilateral, en los testículos no palpables, o en aquellos palpables en los que el testículo se encuentra a un nivel “alto” en la región inguinal proximal, a nivel del orificio inguinal interno. Este consenso, en cuanto a la indicación de la orquidopexia precoz, se basa en los resultados publicados en pacientes con MDT, operados al año de edad, en los que se ha demostrado un mayor crecimiento del volumen testicular y mejores índices de espermiogénesis en estudios histológicos testiculares. También, estudios hormonales han revelado mayores niveles de inhibina B e inferiores de FSH, en pacientes operados antes de los 2 años de edad, en comparación con los que lo hicieron a edades posteriores.
Testículos palpables
La técnica de elección consiste en la orquiolisis y funiculolisis (sección del gubernaculum testis y disección del músculo cremáster y de los elementos del cordón espermático), descenso del testículo al escroto y fijación al mismo: procedimiento conocido como orquidopexia(5). El objetivo de la orquidofuniculolisis es la liberación del testículo de sus fijaciones al pubis, la localización de un conducto peritoneo vaginal persistente (cpvp) para su ligadura y sección, si existe, así como la elongación del cordón espermático para facilitar el descenso testicular. Este primer paso permite el descenso testicular para la posterior orquidopexia.
Este procedimiento quirúrgico puede realizarse, tanto por vía inguinal como por vía escrotal (técnica de Bianchi); reservándose este último abordaje para los testículos localizados caudalmente al orificio inguinal externo, próximos al escroto.
En los pacientes con criptorquidia bilateral, puede considerarse el procedimiento escalonado en dos tiempos separados para cada lado, sobre todo en aquellos en los que los testículos están localizados a un nivel alto en el canal inguinal, y se prevé un procedimiento más complejo, con mayor riesgo de complicaciones perioperatorias en cuanto a sangrado o lesión de los tejidos del cordón espermático o del propio testículo. También, en aquellas situaciones en las que el testículo presenta una localización alta y/o tiene un cordón espermático corto, es necesario realizar la orquidopexia en dos tiempos: dejando en un primer momento el testículo en algún punto del canal inguinal, para su posterior descenso meses después, al escroto, si es posible.
La orquidopexia no requiere tratamiento antibiótico profiláctico y, habitualmente, se lleva a cabo como un procedimiento de cirugía mayor ambulatoria, en ausencia de antecedentes o datos clínicos que obliguen al ingreso hospitalario.
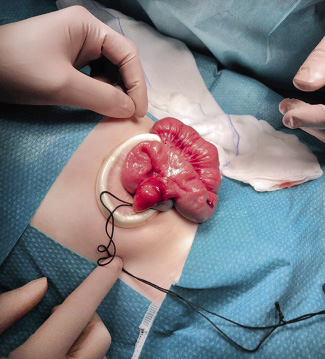
Los puntos “clave” de la técnica de orquidopexia por vía inguinal, se describen a continuación (Fig. 2):

Figura 2. Orquidopexia, abordaje inguinal. A. Gubernaculum testis fijado a la región del pubis (punta de flecha). B, D. Conducto peritoneovaginal persistente-cpvp (asterisco) junto a los elementos del cordón espermático. En la figura D, el cpvp se ha abierto para su disección y ligadura en el momento de la funiculolisis. El epidídimo (e) se encuentra disociado del testículo. C. Orquiolisis y funiculolisis completada. Se muestra el pequeño tamaño de los elementos del cordón espermático (flecha), que se encuentran hipoplásicos. Disociación epidídimo-testicular (e). E. Orquidopexia mediante la fijación con sutura del testículo a la bolsa escrotal. Incisión inguinal realizada para la orquiolisis y funiculolisis en el paso previo (flecha). Fuente: propiedad de la autora.
• Incisión cutánea inguinal y entrada en el canal inguinal mediante su apertura, intentando localizar el nervio ilioinguinal, que discurre próximo al cordón espermático, para su preservación(*).
(*) En el caso de la técnica de Bianchi, los pasos anteriormente descritos se realizan mediante una única incisión escrotal. La elección del abordaje inguinal o escrotal, en los casos de testículos palpables distales al anillo inguinal externo, depende de la experiencia y preferencias del cirujano, al no existir indicaciones expresas de un abordaje u otro en estos casos, ya que los resultados y las complicaciones de la orquidopexia son comparables.
• Orquiolisis: movilización del testículo mediante la sección del gubernaculum, y funiculolisis: disección del músculo cremáster y de los elementos del cordón espermático. Esto permite: 1) la localización de un cpvp si existe, para su sección y ligadura en su base, próximo al anillo inguinal interno; y 2) disminución de tensión en el cordón espermático para el descenso del testículo en dirección escrotal.
Este paso es el más delicado por el riesgo de daño de los vasos espermáticos o del conducto deferente durante la disección de los mismos.
En esta fase se debe realizar la inspección del testículo en relación a su localización exacta, tamaño, aspecto (disociación epidídimo-testicular, estado de los vasos espermáticos y del conducto deferente, o signos de atrofia). Asimismo, se realizará la extirpación de apéndices testiculares o del epidídimo, si se confirman.
• Incisión cutánea en el escroto y creación de bolsa escrotal a nivel sub-dartros, para el alojamiento del testículo.
• Sutura de ambas heridas: inguinal y escrotal.
Testículos no palpables
El objetivo del tratamiento es confirmar o descartar la existencia de testículo y su exploración en el caso de que este esté presente, para realizar la orquidopexia si está en buen estado o extirparlo en el caso de atrofia evidente.
En todos los casos es necesaria siempre, como primera medida, la exploración inguinoescrotal bajo anestesia, ya que esta puede permitir la palpación del testículo y su localización en el canal inguinal en pacientes en los que se diagnosticó de testículos no palpables durante la exploración en la consulta(12). En aquellas situaciones en las que se confirme la presencia de testículo en región inguinal o se palpe el cordón, se continúa la intervención quirúrgica por vía inguinal. Si, por el contrario, no es posible, se opta preferentemente por la exploración por vía laparoscópica para confirmar o descartar la presencia de testículo y/o elementos del cordón espermático en la cavidad abdominal o a la entrada del canal inguinal. Sería posible también, continuar la búsqueda del cordón y testículo no palpables por vía inguinal, mediante la disección hacia el peritoneo a través del anillo inguinal interno, procediendo al descenso testicular por vía inguinal, en el caso de encontrar el testículo, o a la exploración laparoscópica cuando no se localice por esta vía inguinal.
Exploración laparoscópica (Fig. 3)

Figura 3. Laparoscopia exploradora en pacientes con criptorquidia y testículos no palpables. A. Testículo intraabdominal. B. Imagen laparoscópica a nivel del anillo interno. Se observan los vasos espermáticos hipoplásicos con extremo “ciego” (flechas). El conducto deferente también se encuentra amputado a ese nivel (puntas de flecha). Fuente: propiedad de la autora.
Se considera la técnica de elección en el tratamiento de los testículos no palpables. La laparoscopia permite confirmar la presencia o ausencia de testículos intraabdominales y su aspecto (la no visualización ecográfica no descarta su presencia), los anillos inguinales, la persistencia del proceso vaginal, y el examen de los elementos del cordón espermático: conducto deferente y vasos espermáticos.
En la mayoría de los pacientes (80 %), este abordaje permite confirmar la entrada de los elementos del cordón espermático a través del anillo inguinal interno o la existencia de un testículo viable en la cavidad intraabdominal; siendo otras posibilidades menos frecuentes: la localización de testículo atrófico intraabdominal (10 %) o la “amputación” de los vasos espermáticos o del conducto deferente en el retroperitoneo, sin presencia de testículo (10 %).
En los pacientes en los que la exploración laparoscópica demuestre los elementos del cordón espermático entrando en el canal inguinal, se continuará la intervención por vía inguinal, para la búsqueda del testículo y su fijación a la bolsa escrotal si este es viable, o proceder a la extirpación de restos testiculares, si no lo es.
En aquellos casos en los que se objetive la presencia de testículo intraabdominal viable, se puede continuar el procedimiento de descenso y pexia testicular en el escroto, tanto por vía laparoscópica como por vía inguinal, según los hallazgos:
• En aquellas situaciones en las que el cordón espermático sea corto, con imposibilidad para el descenso del testículo al escroto, se indicará un procedimiento secuencial en dos tiempos, según la técnica de Stephens-Fowler. En un primer momento, se realiza la sección de los vasos espermáticos, dejando el aporte vascular del testículo a través de la arteria del conducto deferente; para en una segunda fase (meses después) en la que se prevé el desarrollo de vasos colaterales, proceder al descenso testicular hacia el escroto. El desarrollo de estos vasos colaterales elongados facilita la movilización y el descenso del testículo hasta el escroto, con una tasa de éxito aproximada del 85 %.
• En los casos poco frecuentes, en los que se observe un cordón espermático largo, se podrá realizar el descenso testicular al canal inguinal por vía laparoscópica en un solo tiempo, para completar la orquidopexia por vía inguinal.
• Por último, en los casos en los que se confirme la existencia de un testículo intraabdominal atrófico, se llevará a cabo la orquiectomía laparoscópica, con finalización del procedimiento en este punto.
Al igual que se ha comentado en el tratamiento de los testículos palpables, en el MDT con testículos no palpables bilaterales, y para cualquiera de las técnicas elegidas, se recomienda la orquidopexia en dos tiempos, retrasando el descenso testicular del segundo lado para dar tiempo a evaluar los resultados de la primera orquidopexia. De esta manera, en aquellas situaciones en las que se haya producido, durante la evolución, atrofia testicular del testículo descendido, existe la posibilidad de realizar una intervención quirúrgica menos agresiva en el lado contralateral, con menor disección del cordón espermático y menor riesgo de atrofia testicular. Incluso, es posible plantearse dejar el testículo en una localización palpable, más proximal al escroto, para minimizar los daños testiculares.
Consideraciones generales en el tratamiento quirúrgico del MDT(3-5,7,9)
• Es importante recordar que la biopsia testicular no es un procedimiento rutinario indicado durante las técnicas de orquidopexia, siendo recomendada solo en situaciones especiales, como en los casos de genitales ambiguos, alteraciones cromosómicas o como parte de otros estudios en pacientes con patologías endocrinológicas.
• Durante el periodo postoperatorio se debe evitar el esfuerzo físico y los deportes que provoquen microtraumatismos en la zona escrotal, durante aproximadamente un mes.
El seguimiento postoperatorio se realizará a intervalos variables (comenzando cada 3-6 meses durante el primer año), valorando la posición del testículo y su crecimiento en cada revisión, hasta la pubertad.
• Los cambios inflamatorios locales son frecuentes tras la orquidopexia, pudiendo aparecer en el escroto o incluso en la región inguinal. Otras complicaciones leves menos habituales son: infección, hematoma o dehiscencia de la herida quirúrgica. Otras complicaciones menos frecuentes son: el “reascenso” testicular, que requiere tratamiento quirúrgico, o la lesión del conducto deferente.
• La atrofia testicular es la complicación más severa del tratamiento quirúrgico, con tasas variables en función de la técnica: 2 % en la orquidopexia inguinal, 8 % en la técnica de Stephens-Fowler clásica en dos tiempos, o 28 % en esta última cuando se lleva a cabo en un solo tiempo. Se produce por daño de los vasos espermáticos o por cambios inflamatorios severos postquirúrgicos sobre el cordón espermático o el parénquima testicular.
Tratamiento hormonal
Aunque no está indicado para inducir el descenso testicular(3,7,8,12,13), algunos autores defienden su indicación como tratamiento asociado a la orquidopexia, por la posibilidad de mejorar la fertilidad futura(14). Así, en las guías europeas de urología pediátrica(4), se contempla el tratamiento hormonal con GnRH (hormona liberadora de gonadotropinas) en los pacientes con criptorquidia bilateral, según un nivel de evidencia 4, al no haberse podido demostrar resultados concluyentes al respecto. Por el contrario, se desestima el tratamiento con hCG (gonadotropina coriónica humana), por los efectos secundarios sobre la espermatogénesis al producir apoptosis en las células germinales, cambios inflamatorios y disminución del volumen testicular(3).
Testículos retráctiles y ascenso testicular adquirido
Los testículos retráctiles en ascensor son aquellos que ascienden fuera del escroto como consecuencia de la contracción fisiológica del músculo cremáster y, por lo tanto, no requieren tratamiento. Durante la exploración, se descienden fácilmente a escroto con la tracción manual, permaneciendo en posición escrotal durante la exploración física. Es muy importante diferenciarlos del maldescenso testicular congénito o de aquellos que reascienden con el tiempo por un acortamiento del cordón espermático (también llamados testículos retráctiles verdaderos), situaciones ambas que requieren tratamiento quirúrgico.
El testículo ascendido adquirido puede producirse a partir de los 6 meses de edad, coincidiendo con el descenso de los niveles de andrógenos que han mantenido el músculo cremáster más relajado y el crecimiento del músculo dartos escrotal, durante el periodo denominado “minipubertad”, entre los 2 y 6 meses de edad. A partir de este momento, la disminución de los niveles de testosterona, adelgaza ambas estructuras, favoreciendo el “empuje” del testículo fuera del escroto. También, la retracción del testículo puede deberse a la fibrosis permanente de un cpvp previo que, a su vez, acorta los elementos del cordón espermático(9).
Diferenciar la causa de esta retracción testicular: 1) por contracción muscular en el caso de los testículos en ascensor; o 2) por un acortamiento tisular del cordón espermático, en el caso de los verdaderos testículos retráctiles, es crucial. En el primer caso, nos encontramos ante una situación fisiológica; y en el segundo, ante una situación de maldescenso testicular adquirido, que requerirá la realización de orquidopexia, habitualmente entre los 5 y 10 años de edad. Es por ello, que estos pacientes deben seguirse hasta la pubertad mediante exploración periódica, hasta confirmar o descartar de forma definitiva una situación de MDT(10,12,15).
Fertilidad y cáncer
El desarrollo y crecimiento del testículo a una temperatura corporal anormalmente elevada en el abdomen o región inguinal (37ºC), puede provocar efectos deletéreos sobre el testículo de forma precoz, lo que justifica el tratamiento temprano de estos pacientes, durante el primer año de edad. Las posibles alteraciones acontecen sobre las células germinales testiculares y su transformación a stem cells para la futura espermatogénesis, así como sobre las células de Leydig, que se han visto disminuidas en los estudios histológicos de los testículos criptorquídicos(4,11). De este modo, se ha demostrado que los varones, con criptorquidia no tratada, presentan una fertilidad dañada, con una disminución de las tasas de fertilidad, más probable si la orquidopexia se realiza a partir de los 18 meses de edad. En este sentido, se han descrito tasas de infertilidad del 10-30 % en pacientes con criptorquidia unilateral, asociada a azoospermia en el 13 % de los casos, que se ve incrementada hasta en el 90 % de los pacientes con criptorquidia bilateral no tratada(4). Así mismo, se han publicado alteraciones en relación a hipofunción testicular, disminución del volumen testicular, alteraciones del esperma y disminución funcional de las células de Leydig, en pacientes con antecedentes de criptorquidia(16).
En relación al cáncer testicular, se ha demostrado un incremento del riesgo de degeneración maligna en la edad adulta en los pacientes con antecedentes de MDT, sobre todo en aquellos que no se trataron antes de la pubertad y, más frecuentemente, en los testículos de localización intraabdominal. Varios estudios y metaanálisis publican de hecho una reducción significativa del riesgo de malignización testicular con la orquidopexia prepuberal(4,17).
En la tabla I se resumen las principales recomendaciones relacionadas con el diagnóstico y tratamiento de las alteraciones del descenso testicular, según la última actualización de 2022 de la Asociación Europea de Urología Pediátrica.

Escroto agudo
La torsión de apéndices testiculares, la epididimitis y la torsión de testículo, suponen más del 80 % de las causas de escroto agudo. El escroto agudo requiere siempre una valoración urgente para descartar una situación de torsión testicular, o realizar una exploración quirúrgica urgente en el caso de que esta se sospeche.
Se define por el cuadro clínico de dolor testicular de pocas horas de evolución, asociado habitualmente a signos inflamatorios locales como: tumefacción, eritema y/o aumento de la temperatura escrotal. La torsión del apéndice testicular, la epididimitis u orquiepididimitis o la torsión testicular son las causas más frecuentes (80 %), por este orden.
Se incluyen también, como causas menos frecuentes: el edema escrotal idiopático (que puede cursar sin dolor), la hernia incarcelada, el traumatismo testicular u otros cuadros clínicos, como la púrpura de Schönlein-Henoch o la orquitis viral(18).
Requiere siempre una valoración urgente, que confirme o descarte la existencia de torsión testicular, ya que sin un tratamiento precoz, dará lugar a la necrosis y atrofia del testículo.
En la evaluación es muy importante una correcta exploración física y prestar atención a ciertos signos o síntomas característicos de cada una de estas entidades, que nos orientarán, junto a los datos ecográficos, al diagnóstico en cada caso. De este modo, la evolución y localización del dolor, más agudo, severo y generalizado en el testículo y escroto en los pacientes con torsión testicular, o localizado en el polo superior testicular en los cuadros de torsión de los apéndices testiculares o en la epididimitis, nos orientarán al diagnóstico en cada caso. Por otro lado, los cuadros de torsión testicular neonatal evolucionada, pueden cursar sin dolor asociado.
La fiebre y la detección de infección urinaria deben orientar hacia un episodio de orquitis-orquiepidimitis, que se presenta aproximadamente en el 20 % de los pacientes.
Torsión testicular
La torsión testicular es la tercera causa de escroto agudo. Es más frecuente a partir de la pubertad, producida por una mala fijación y posición del testículo en el escroto. En la época perinatal se produce otro pico de incidencia, pudiendo haberse producido intraútero o tras el nacimiento.
La torsión de testículo (TT) se produce por la rotación del cordón testicular sobre su eje longitudinal, provocando una disminución o ausencia de la vascularización del testículo y, en fases avanzadas, la necrosis y atrofia testicular. El tiempo de evolución y el grado de torsión del cordón espermático son los dos factores principales de riesgo del daño testicular, habiéndose demostrado que el riesgo de necrosis testicular se produce a partir de las 8-10 horas de evolución(4,19).
Con una incidencia aproximada de 1/4.000 varones (< 25 años), es la tercera causa más frecuente de escroto agudo en la población pediátrica (15-20 %)(4,18,20), y debe diferenciarse en el diagnóstico, de la torsión de apéndices testiculares (primera causa de escroto agudo), ya que pueden presentarse con sintomatología inicial muy similar, pero con una evolución, tratamiento y pronóstico muy diferentes.
La congestión y tumefacción testicular de las primeras fases, producida por la compresión de los vasos venosos torsionados, evoluciona hacia la isquemia y necrosis del parénquima testicular por obstrucción arterial, a medida que evoluciona. Esto explica que, en los momentos iniciales, la ecografía pueda mostrar una “falsa hiper-vascularización”, por la congestión de las cubiertas testiculares, con visualización aún de flujo arterial, aunque disminuido, si la torsión no es completa. Este hecho debe tenerse siempre en cuenta en los casos de sospecha de torsión testicular, para evitar fallos o retrasos en su diagnóstico.
La TT presenta dos picos de incidencia: uno menos prevalente en el periodo neonatal (15 %), y otro más frecuente a partir de la pubertad entre los 12 y 18 años de edad(4).
En función de su origen y la disposición del testículo y cordón espermático con respecto a la túnica vaginal, se clasifican los distintos tipos y entidades clínicas(4,18,21):
• Torsión intravaginal (Fig. 4): la más frecuente y de aparición más habitual en torno a la pubertad (65 %), aunque puede aparecer a cualquier edad. Se produce por una mala fijación, polar y estrecha, del testículo a la túnica vaginal, lo que predispone a una mayor movilidad del testículo y a su torsión sobre el eje del cordón espermático en el interior de la túnica vaginal en el escroto. Una deformidad típica es el denominado “testículo en badajo”, en la que el testículo se dispone horizontalizado, suspendido por el cordón testicular dentro de la vaginal, muy móvil; y, por lo tanto, con una mayor predisposición a la torsión. Su prevalencia se estima en el 12 % de los varones, siendo bilateral en el 40 % de los casos.
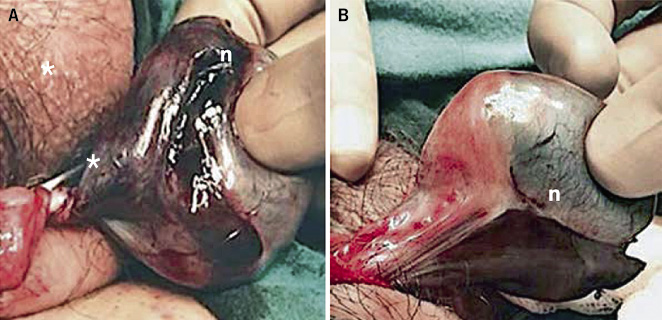
Figura 4. Torsión testicular intravaginal. Adolescente de 17 años de edad con torsión completa de 360º sobre cordón espermático (asterisco), de 12 horas de evolución. A. Exploración quirúrgica por vía escrotal: se observa necrosis (n) del parénquima testicular. B. Detorsión escrotal sin signos de viabilidad testicular, por lo que se indicó la realización de orquiectomía. Fuente: propiedad de la autora.
• Torsión extravaginal: se produce por una rotación del testículo y la túnica vaginal de forma conjunta sobre el eje del cordón espermático en la región inguinal. Es la que se presenta en la etapa prepuberal.
Cuando se diagnostica en el periodo neonatal, la TT se ha producido durante la gestación o en algún momento tras el nacimiento, durante el primer mes de vida. Se manifiesta como torsión testicular hasta en el 20 % de los casos, y supone el 15-20 % de los casos de TT. Hay que tener en cuenta que esta puede estar infraestimada, por el hecho de que muchos casos se diagnostican más tarde como criptorquidia o atrofia testicular(21).
En función del momento de producción de la torsión testicular neonatal, se distinguen dos entidades diferenciadas:
1. Torsión prenatal intraútero, de curso prolongado y evolucionada al diagnóstico (80 %). No se considera, por lo tanto, un cuadro clínico urgente. Puede detectarse en el momento del parto o durante las primeras exploraciones del recién nacido. Suele presentarse como una tumoración inguinal o escrotal de consistencia dura, más o menos dolorosa y con signos inflamatorios o no, en función del tiempo evolutivo. En los casos de mayor tiempo de evolución, en los que la torsión se ha producido durante los meses de gestación lejanos al parto, se diagnostica con frecuencia como criptorquidia, al no palparse el testículo ya atrófico.
2. Torsión postnatal (Fig. 5): ocurre después del nacimiento y se manifiesta como un cuadro de irritabilidad, dolor escrotal agudo, con eritema y tumefacción escrotal, en un recién nacido en el que las exploraciones previas detectaban un testículo normal. Su tratamiento es urgente.
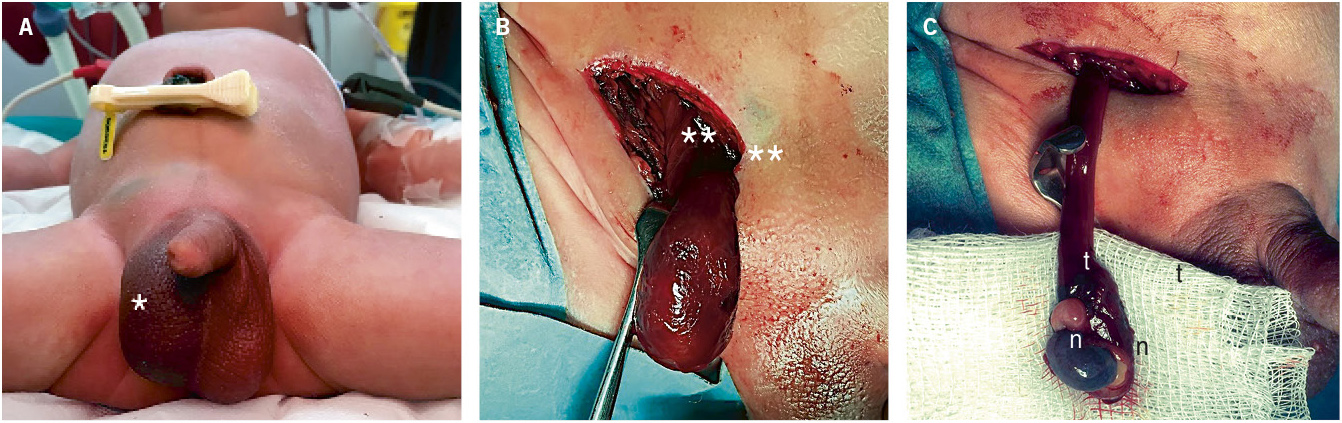
Figura 5. Torsión testicular neonatal. A. Paciente de 10 días de vida, valorado por irritabilidad, dolor a la palpación, tumefacción e induración escrotal (*). B. Torsión extravaginal del testículo y túnica vaginal sobre el cordón espermático, confirmada mediante exploración inguinal (**). C. Necrosis testicular (n) y trombosis vascular (t) del cordón espermático tras detorsión, que requirió orquiectomía. Fuente: propiedad de la autora.
Epididimitis, orquitis y orquiepididimitis
Se trata de la inflamación del epidídimo, testículo o ambos, respectivamente. La orquitis aparece habitualmente asociada a epididimitis por evolución de esta. Cuando se presenta de forma aislada, se produce como consecuencia de la diseminación hematógena de una infección bacteriana o secundaria a una infección viral, como la parotiditis (orquitis urliana), infecciones por adenovirus, enterovirus, influenza o parainfluenza(4). Con una incidencia anual aproximada de 1,2/1.000 varones, la epididimitis/orquiepididimitis es la 2ª causa de escroto agudo en la infancia (20-30 %). En muchos pacientes, sobre todo en los prepúberes, el origen es desconocido, pero en aquellos casos en los que aparecen episodios de repetición, se debe investigar la existencia de malformaciones o trastornos funcionales urológicos. El reflujo vesicoureteral, los uréteres ectópicos o la vejiga neurógena, así como las pruebas diagnósticas que supongan la manipulación de la vía urinaria (sondaje, cistografía o cistoscopia) suponen factores de riesgo a tener en cuenta en estos casos. Los gérmenes más frecuentemente aislados a esta edad son: E. Coli, Mycoplasma pneumoniae, enterococos, enterovirus o adenovirus. En los adolescentes sexualmente activos, los cuadros de epididimitis, secundarios a infecciones de transmisión sexual (ETS), son la causa más frecuente, siendo Chlamydia trachomatis, N. gonorrhoeae, E. Coli y los virus, los microorganismos causantes más habituales(22,23).
Torsión de apéndices testiculares y de epidídimo
La torsión del apéndice testicular (hidátide de Morgagni) es la causa más frecuente de escroto agudo. Su tratamiento es médico, siendo necesaria la resección quirúrgica solo en los casos recidivantes o los refractarios al tratamiento médico.
Es la primera causa de escroto agudo (45 %), y se presenta más habitualmente en los niños entre los 7 y 12 años de edad.
Se produce por la torsión e inflamación secundaria de los apéndices testiculares o epididimarios (remanentes de los conductos de Müller y Wolff), localizados en el polo superior del testículo (hidátide de Morgagni) o en el epidídimo.
Otras causas de escroto agudo
Suponen el 10-15 % de los casos de escroto agudo.
• Traumatismo testicular: los traumatismos testiculares de alto impacto pueden ocasionar complicaciones con repercusión sobre la viabilidad del testículo, por lo que es necesaria la valoración urgente con ecografía testicular. Además de la producción de torsión testicular secundaria al traumatismo, la visualización de hematocele (hematoma en la túnica vaginal), hematoma intratesticular o la disrupción de la túnica albugínea con ruptura del testículo, requieren la exploración quirúrgica para descomprimir el parénquima testicular y evitar la necrosis testicular.
• Hernia incarcerada: se puede manifestar como escroto agudo, como consecuencia del dolor irradiado desde la región inguinal. En ocasiones, puede palparse el contenido intestinal de la hernia en el escroto.
• Púrpura de Schönlein-Henoch: vasculitis sistémica que puede asociarse a dolor articular, dolor abdominal, afectación renal, hemorragia digestiva y, de forma ocasional, a dolor escrotal, tanto agudo como insidioso.
• Tumor testicular: aunque el modo de presentación más habitual del cáncer testicular es la aparición de una masa testicular indolora, la hemorragia intratumoral, puede originar un cuadro de dolor agudo testicular y/o escrotal, asociado a cambios inflamatorios.
• Edema escrotal idiopático: inflamación escrotal, de corta evolución, con tumefacción de los tejidos blandos y eritema local. De forma característica, el paciente presenta buen estado general y, si refiere dolor, es de poca intensidad. Es bilateral en >50 % de los casos, y recurrente en el 10 % de los pacientes. El edema puede extenderse al periné, región inguinal o al pene. Los hallazgos ecográficos muestran, de forma característica, hipervascularización y engrosamiento hipoecoico del saco escrotal, sin alteraciones a nivel del testículo.
Diagnóstico de los cuadros clínicos de escroto agudo(4,19,22)
El diagnóstico de escroto agudo debe basarse en la anamnesis y exploración física. Ante situaciones sospechosas de torsión testicular, la actitud indicada es la exploración quirúrgica, sin la realización de pruebas complementarias que retrasen su tratamiento precoz.
Anamnesis y exploración física
Se consideran fundamentales en el diagnóstico del escroto agudo, ya que existen datos y signos clínicos característicos de cada una de las entidades clínicas, que nos orientan al diagnóstico. La exploración física se basa en la inspección y palpación testicular, escrotal y bilateral. Se debe prestar atención a la posición del testículo, la intensidad y localización del dolor, la presencia de signos inflamatorios, y la valoración del reflejo cremastérico. Con frecuencia, suele observarse hidrocele reactivo, más característico de la torsión testicular, la epididimitis evolucionada o de los traumatismos testiculares.
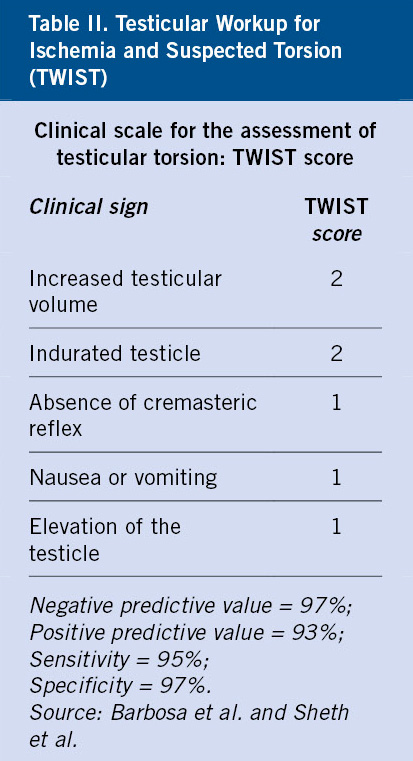
En la valoración clínica del escroto agudo, se ha desarrollado el score TWIST (Testicular Workup for Ischemia and Suspected Torsion) para la identificación de los pacientes con riesgo de presentar un cuadro de torsión testicular. En esta escala se valoran: la inflamación y consistencia testicular, el reflejo cremastérico, la posición del testículo y la presencia o no de náuseas o vómitos (Tabla II). Con unos valores que van desde el 0 al 7 (máxima probabilidad de TT), se ha validado como una escala fiable y útil en el diagnóstico de torsión testicular, dada su sensibilidad y especificidad(19).

Además, en la anamnesis es importante investigar sobre determinados antecedentes que pueden predisponer a episodios de escroto agudo, entre los que destacan:
• Antecedente de maldescenso testicular; traumatismo escrotal/actividad física intensa: torsión testicular.
• Actividad sexual; malformaciones, anomalías funcionales urológicas; manipulación de la vía urinaria; episodios infecciosos previos o enfermedades sistémicas (vasculitis): epididimitis/orquiepididimitis.
Entre los datos clínicos a tener en cuenta, destacamos:
• Edad: factor importante en el diagnóstico diferencial de escroto agudo, ya que la frecuencia de aparición es diferente según las diferentes patologías. De este modo, la torsión testicular, con un patrón de distribución bimodal, es más frecuente en la adolescencia, periodo en el que son más prevalentes también los episodios de orqui-epididimitis. La torsión de hidátide testicular se presenta con más frecuencia en la etapa prepuberal.
• Dolor: intensidad, duración y asociación a otros síntomas. La torsión testicular se manifiesta de forma típica con dolor intenso, de inicio brusco, de pocas horas de evolución. Además, el paciente presenta mal estado general y puede asociar otros síntomas, como náuseas o vómitos.
En el caso de la torsión de hidátides testiculares o de la epididimitis, el dolor, localizado en el polo superior del testículo, sigue un curso más gradual, no es tan intenso y, de forma típica, el paciente mantiene un buen estado general. En el caso de la torsión de apéndice testicular, el dolor, es muy delimitado en un punto del polo superior testicular.
Además, tanto en la torsión testicular como en la epididimitis, puede existir dolor irradiado a la región inguinal.
No hay que olvidar que los cuadros de torsión testicular neonatal evolucionada, pueden cursar sin dolor asociado(21).
• Fiebre: no es un síntoma frecuente en el escroto agudo. Aparece en <20 % de los cuadros de epididimitis-orquiepididimitis. Una excepción, son los cuadros de orquitis de origen viral en los que sí es frecuente el antecedente de síndrome febril, unos días previos al episodio de escroto agudo.
• Síntomas urinarios: la existencia de sintomatología, como disuria, polaquiuria o tenesmo vesical, debe orientarnos hacia la presencia de epididimitis. Si, además, existe clínica de uretritis, debemos sospechar epididimitis secundaria a ETS en los adolescentes.
En relación a la exploración física, son destacables los siguientes datos en algunas entidades:
• En la torsión testicular son muy evidentes el eritema y la tumefacción escrotal, junto al aumento de la consistencia del testículo, que es muy doloroso a la palpación (Fig. 3). Este aparece “fijo”, horizontalizado y ascendido, con el epidídimo en posición anterior: “Signo de Gouverneur”. Además, el reflejo cremastérico suele estar abolido; aunque su presencia no descarta la existencia de torsión testicular. Es importante recordar también, que el reflejo cremastérico puede estar ausente en los pacientes menores de 6 meses de edad de forma fisiológica.
En los pacientes con torsión de un testículo no descendido, se palpa el testículo doloroso en la región inguinal con signos inflamatorios locales y escroto vacío ipsilateral.
• En la epididimitis, el dolor es máximo a la palpación en la zona correspondiente al epidídimo, que parece engrosado. Puede extenderse al resto del testículo en las orquiepididimitis. El testículo es móvil dentro de la bolsa escrotal, está normoposicionado y mantiene el reflejo cremastérico, aunque este último puede ser difícil de valorar en los casos con gran inflamación. Signo de Prehn: el dolor se alivia con la elevación del testículo (al contrario de lo que ocurre en la torsión testicular: el dolor no disminuye o incluso aumenta con la elevación testicular).
• En la torsión de hidátide testicular, el dolor se localiza en el polo superior del testículo. Pueden asociarse o no eritema y calor en la piel escrotal en grado variable. Es característico el signo del “punto azul”, que se observa en el polo superior del testículo al realizar la transiluminación escrotal, y que se corresponde con la necrosis o congestión del apéndice torsionado. La existencia del punto azul (10-23 % de los casos) es un signo característico de la torsión de hidátide, pero su ausencia (cuando hay inflamación de la hidátide, pero no se ha producido la necrosis) no la descarta.
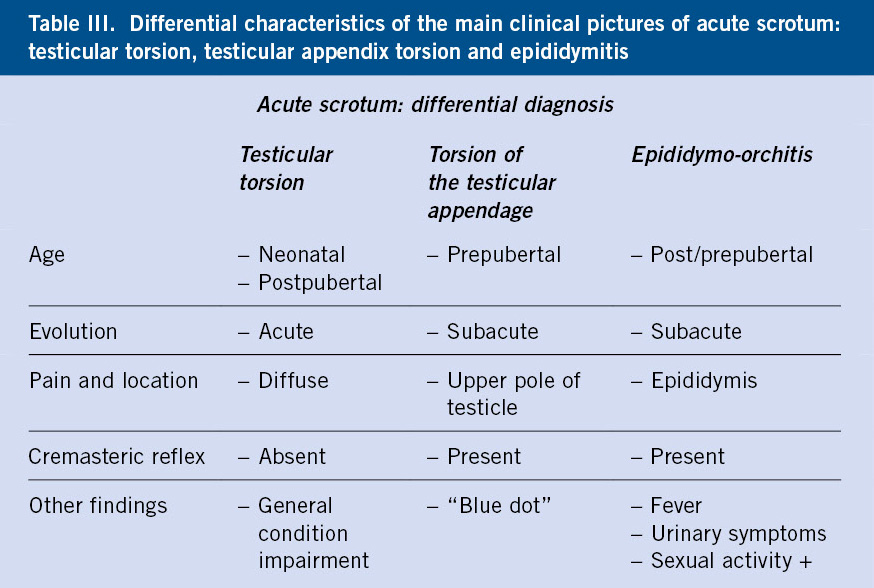
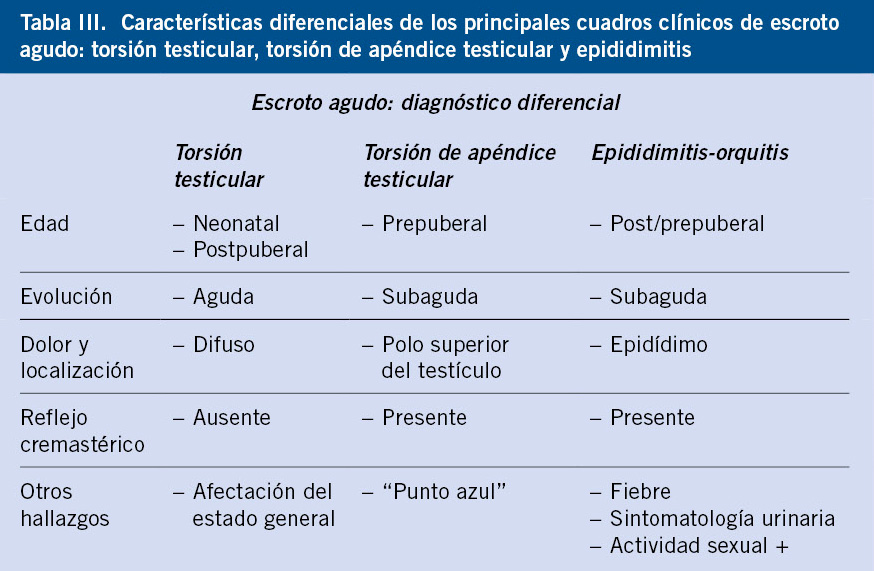
En la tabla III, se resumen los datos clínicos más característicos de la torsión testicular, torsión de hidátide y epididimitis, en el diagnóstico diferencial de los cuadros de escroto agudo.
Pruebas complementarias
El primer objetivo en el diagnóstico del escroto agudo es confirmar o descartar si estamos ante un cuadro de torsión testicular y, ante esta situación, el paciente debe ser valorado por un especialista en Cirugía Pediátrica.
La ecografía asociada a doppler testicular es la prueba de imagen de elección en el diagnóstico del escroto agudo (Fig. 6), pero hay que tener cuenta que, ante la alta sospecha diagnóstica de torsión testicular, la actitud a seguir es la exploración quirúrgica urgente, única medida diagnóstica de certeza, que también será terapéutica en el caso de que se confirme la torsión testicular. Es importante insistir en que, aunque la ecografía es la técnica gold standard en el diagnóstico de la patología testicular, nunca se debe retrasar el tratamiento quirúrgico en los cuadros que sugieran TT, a la espera de su realización. Además, a pesar de que el flujo arterial está disminuido o ausente en el testículo torsionado, en las fases iniciales este puede estar preservado, y se puede asociar también hipervascularización de las cubiertas testiculares por la congestión venosa y el propio proceso inflamatorio, lo que puede dar lugar a fallos (falsos negativos) en el diagnóstico de TT. Es por esto también, que los hallazgos ecográficos no deben cambiar nunca la indicación de la exploración quirúrgica urgente ante la sospecha de torsión testicular, a pesar de que el estudio doppler informe sobre la presencia de flujo testicular.
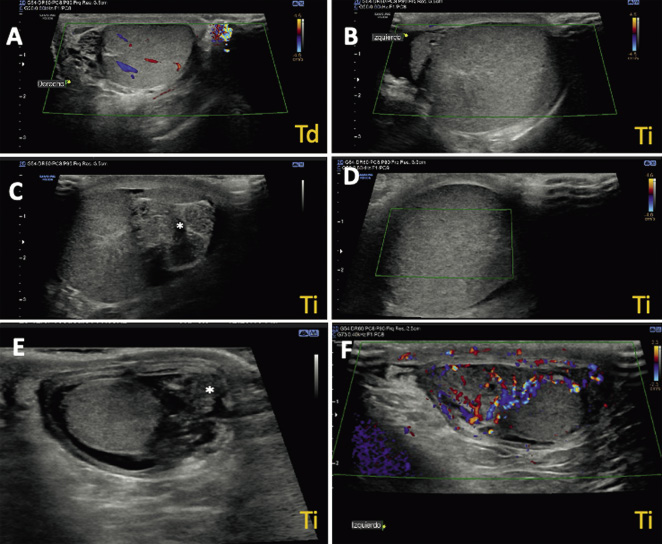
Figura 6. Ecografía-doppler testicular bilateral en torsión testicular izquierda (A, B, C y D) y epididimitis (E y F). A. Testículo derecho (Td) de aspecto ecográfico normal, con preservación de flujo arterial. B, D. Testículo izquierdo (Ti) con signos de torsión testicular: aumento de tamaño y ausencia de flujo en estudio Doppler. C. Testículo izquierdo (Ti) en el que se aprecia la vuelta del cordón espermático (asterisco). E. Testículo izquierdo con epididimitis. Marcado con asterisco se muestra el engrosamiento del epidídimo. F. Hipervascularización de las cubiertas del testículo izquierdo con epididimitis. Fuente: propiedad de la autora.
La ecografía testicular bilateral permite valorar además, la simetría en relación al tamaño, localización y aspecto del parénquima testicular, engrosamiento y posición del cordón espermático, engrosamiento de cubiertas testiculares y de la bolsa escrotal, o tamaño y signos inflamatorios de los apéndices testiculares o del epidídimo.
No se debe olvidar también, que la ecografía-doppler puede ofrecer falsos diagnósticos positivos de torsión testicular en situaciones, como grandes hidroceles a tensión, hernias inguinoescrotales o hematomas testiculares de gran tamaño, que dificulten la vascularización testicular.
El análisis de orina se debe realizar en los cuadros de epididimitis y orquiepididimitis. Si se observa leucocituria o nitritos positivos, o el paciente presenta sintomatología urinaria clara, se realizará cultivo con antibiograma para validar el tratamiento antibiótico empírico pautado en un inicio. Es importante señalar que la positividad del análisis y cultivo de orina se obtiene en un bajo porcentaje de pacientes con epididimitis y, al contrario, la normalidad de los resultados de estas pruebas no excluye el diagnóstico de epididimitis. Del mismo modo, un examen de orina patológico no excluye la torsión testicular. Está indicada, adicionalmente, la recogida de exudado uretral para estudio microbiológico en los adolescentes con vida sexual activa, y sospecha de epididimitis como enfermedad de transmisión sexual (ETS).
Tratamiento del escroto agudo
La torsión testicular es una urgencia quirúrgica que requiere un tratamiento quirúrgico precoz para garantizar la viabilidad del testículo. El grado de torsión y el tiempo de evolución son los dos factores principales en la predicción del daño testicular. Por el contrario, en la torsión de hidátide de Morgagni, el tratamiento indicado es médico, con reposo y fármacos antiinflamatorios.
La torsión testicular es una urgencia quirúrgica, que requiere un tratamiento precoz para preservar la viabilidad del testículo. Los dos factores determinantes del daño testicular son el grado de torsión y el tiempo de evolución, siendo más probable la recuperación del parénquima testicular, cuando la detorsión se realiza en las primeras 4-8 horas de evolución. En los casos de torsión completa, el riesgo de necrosis testicular es muy elevado con un tiempo de evolución corto (4 h), y cuando la torsión es incompleta, existe la posibilidad de que el testículo permanezca viable, con cursos evolutivos de hasta 12 horas(4,18,19).
La intervención quirúrgica consiste en la detorsión manual del testículo y fijación del mismo en el escroto: orquidopexia, si se confirma la viabilidad del mismo durante la exploración quirúrgica. En la actualidad, se recomienda la orquidopexia contralateral en el mismo acto, dado el riesgo de torsión futura en ese lado(4).
Si el testículo está necrosado, está indicada la extirpación del testículo: orquiectomía, y fijación del testículo contralateral en el escroto. Para el tratamiento de la torsión intravaginal, el abordaje se realiza mediante una incisión escrotal, mientras que en el caso de la torsión extravaginal, este se lleva a cabo por vía inguinal.
Se puede intentar la detorsión manual antes de la intervención quirúrgica, mediante la rotación externa del testículo (maniobra similar a la apertura de un libro) si no se observa resistencia a la misma o aumento del dolor. En los casos en los que la maniobra consigue la detorsión del testículo, el dolor desaparece. En esta situación, debe indicarse también la exploración quirúrgica urgente ante la posibilidad de que permanezca aún una torsión parcial.
Los pacientes con torsión intermitente: sufren episodios de torsión testicular con resolución espontánea tras segundos o minutos de inicio del cuadro. La exploración testicular y la ecografía pueden ser normales en el momento de la valoración clínica, si el cuadro se ha resuelto. Está indicado el seguimiento, y el tratamiento quirúrgico mediante orquidopexia para evitar futuros episodios.
En los casos de torsión neonatal, está indicada la exploración quirúrgica y orquidopexia bilaterales debido a la posibilidad de torsión bilateral hasta en el 20 % de los casos.
En el caso de la torsión prenatal, no existe consenso en relación al momento de la intervención quirúrgica y la exploración quirúrgica del lado contralateral. Aunque el testículo afectado está necrosado en la práctica totalidad de las ocasiones y, por lo tanto, no supone una urgencia quirúrgica, el objetivo del tratamiento es la fijación escrotal del testículo contralateral en prevención de torsiones futuras. Teniendo en cuenta también, la posibilidad de casos de torsión bilateral sincrónica no diagnosticada, en general se recomienda el tratamiento quirúrgico preferente, cuando el paciente esté estable y no existen contraindicaciones clínicas ni anestésicas para llevarlo a cabo(21).
Seguimiento y evolución de la torsión testicular
En general, el riesgo de recurrencia de torsión testicular es < 5 %, y puede aparecer años después del primer episodio. Por otra parte, a pesar de una detorsión adecuada y la apariencia de viabilidad testicular durante el procedimiento quirúrgico del episodio agudo, algunos pacientes desarrollan atrofia testicular con el tiempo, lo que puede afectar a su fertilidad futura. Esta situación puede deberse, tanto al daño directo del parénquima testicular por la isquemia durante la torsión, como por la rápida reabsorción de radicales libres en el testículo durante la fase de reperfusión post-isquemia. Estos cambios podrían llevar a implicaciones sobre la fertilidad futura, más frecuentes en los casos de torsión testicular grave y evolucionada(4,19).
En la epididimitis/orquiepididimitis, el tratamiento de elección es médico mediante reposo y fármacos antiinflamatorios, y no está indicada la antibioterapia de forma generalizada, a no ser que exista sospecha de infección de orina tras los resultados del examen y cultivo de orina(4). En estos casos, nosotros proponemos iniciar tratamiento antibiótico empírico según las recomendaciones actuales publicadas por la Asociación Española de Pediatría de Atención Primaria(23) (Guía ABE: https://www.guia-abe.es/).
Se instaura de este modo, tratamiento inicial con cobertura para Gram + y Gram -, con cefalosporinas de 2ª/3ª generación (cefuroxima: 20-30 mg/kg/día; cefixima: 8 mg/kg/día), amoxicilina-clavulánico (50 mg/kg/día) o quinolonas en los niños más mayores (ciprofloxacino: 20-40 mg/kg/día; levofloxacino: 500 mg/día), durante 7-10 días. Dichos tratamientos se reevaluarán con los resultados del cultivo y antibiograma.
En la torsión de hidátide testicular, también se establece el tratamiento médico con reposo y antiinflamatorios como primera opción. Habitualmente, la mejoría es progresiva hasta la resolución del cuadro clínico a los 7-10 días. Solo se indica el tratamiento quirúrgico para la extirpación del apéndice testicular/epididimario, en los casos de dolor refractario al tratamiento médico o en aquellos pacientes con episodios recidivantes. No está indicada la exploración quirúrgica del testículo contralateral(22).
En los pacientes con traumatismo testicular está indicado el reposo y tratamiento analgésico y antiinflamatorio. En los casos de ruptura, hematoma compresivo o sospecha de torsión testicular, está indicada la exploración quirúrgica.
El edema escrotal idiopático no requiere tratamiento habitualmente, salvo en los pacientes que cursen con molestia dolorosa local, que se tratarán con tratamiento antiinflamatorio(24).
Valoración del escroto agudo por el pediatra: aspectos importantes a considerar
• El principal objetivo del pediatra en la valoración del paciente con dolor testicular agudo debe ser la identificación de síntomas y signos que puedan sugerir la existencia de torsión testicular. Ante su mínima sospecha, se debe indicar la valoración urgente por Cirugía Pediátrica.
• El diagnóstico de escroto agudo es clínico y no está indicada la realización de pruebas complementarias que puedan retrasar el tratamiento quirúrgico precoz de un posible caso de torsión de testículo.
• La ecografía escrotal y testicular con estudio doppler, es la prueba de imagen de elección en la valoración de la patología escrotal y testicular. En los cuadros de escroto agudo, ayuda a la valoración del flujo y parénquima testicular, grado de inflamación en los casos de epididimitis severas o detección de un apéndice inflamado en la torsión de apéndices testiculares/epididimarios.
• En los cuadros de epididimitis en los niños prepúberes, el tratamiento antibiótico sistemático no está justificado, salvo en el caso de sintomatología urinaria, examen de orina patológico o antecedentes que hagan sospechar la presencia de infección de orina. En la mayoría de los casos, en los que no se dan estas circunstancias, se indica tratamiento sintomático con reposo, fármacos antiinflamatorios y analgésicos.
La epididimitis en el adolescente con vida sexual activa, debe tratarse de forma empírica con pautas de antibioterapia que cubran N. Gonorrhoeae y Chlamydia trachomatis, para cubrir infecciones de transmisión sexual.
En los casos de orquitis/epididimitis con gran componente inflamatorio, evolución tórpida a pesar del tratamiento médico, o aparición de sintomatología general, está indicada la valoración por el servicio de Cirugía, quien valorará la indicación de tratamiento antibiótico intravenoso y la realización de ecografía testicular para el estudio de la afectación del parénquima testicular.
• En los pacientes con cuadros claros de torsión de hidátide testicular, se indicará reposo y tratamiento sintomático con fármacos antiinflamatorios por vía oral. El tratamiento quirúrgico está indicado en aquellos casos de dolor persistente refractario al tratamiento médico o en los recidivantes.
• El traumatismo testicular moderado-severo debe valorarse mediante la realización de ecografía testicular y atención urgente por el servicio de Cirugía, para la valoración de la afectación parenquimatosa y/o posibles complicaciones que requieran tratamiento quirúrgico, como ruptura, hematoma a tensión o torsión testicular. La misma actitud debe seguirse en los casos de tumoración testicular, con el objetivo de descartar la existencia de un posible tumor testicular.
Otras causas de patología escrotal
Hidrocele(4)
Se define como el acúmulo de contenido líquido entre las capas visceral y parietal de la túnica vaginal testicular. En la exploración física se identifica como un aumento del volumen escrotal, con transiluminación positiva e indolora (cuando no se asocia a procesos inflamatorios testiculares). Puede ser primario, malformativo o por hipersecreción, o secundario a otros procesos inflamatorios del testículo, como torsión testicular, orqui-epididimitis o traumatismo testicular. También, se puede producir tras las operaciones de varicocele por ligadura de vasos linfáticos durante la intervención.
En este apartado nos referiremos al hidrocele primario, que se clasifica en:
1. Hidrocele congénito: aparece en la infancia, y se produce por la persistencia del proceso vaginal (cpvp).
2. Hidrocele del tipo adulto: generado por el acúmulo de líquido por hipersecreción de la mucosa de la túnica vaginal y el desequilibrio entre esta producción y su reabsorción. Se debe sospechar cuando se presenta en torno a la adolescencia, en pacientes sin historia de hidrocele o hernia inguinal durante la infancia.
En el caso del hidrocele congénito, este puede aparecer como un hidrocele comunicante: en el que el proceso vaginal permanece abierto en todo su trayecto y, por lo tanto, es oscilante, siendo más evidente al final del día. En otros casos, el cpvp se oblitera en algún punto, dando lugar a una tumoración fija, no reductible, visible en la región inguinal (quiste de cordón espermático), o en el escroto, según la localización del cierre del proceso vaginal.
El diagnóstico de hidrocele es clínico y no son necesarias pruebas complementarias adicionales, salvo en los casos en los que se sospeche asociación a otras patologías, como masa testicular o cualquier cuadro inflamatorio testicular.
En los pacientes con hidrocele congénito, se debe esperar a la obliteración espontánea durante el primero o dos primeros años de edad. A partir de este momento, el tratamiento es quirúrgico y consiste en la ligadura y sección del proceso vaginal mediante un abordaje inguinal, procedimiento denominado herniorrafia, ya que es el mismo que se realiza en el tratamiento de la hernia inguinal. Como abordaje alternativo, se puede indicar la herniorrafia por vía laparoscópica, en función de la experiencia y preferencias del cirujano.
Es importante recordar que el hidrocele no provoca daño testicular, por lo que la intervención quirúrgica se programa a partir del año de edad, sin carácter de urgencia.
Como excepción, en los casos en los que se sospeche la existencia de hernia inguinal concomitante, el tratamiento quirúrgico se indica en el momento del diagnóstico.
En el caso del hidrocele escrotal, no comunicante, del tipo adulto, el tratamiento quirúrgico es la hidrocelectomía por vía escrotal: drenaje del hidrocele, mediante apertura y sección de la túnica vaginal.
Varicocele
El varicocele del adolescente suele ser asintomático. Los criterios de tratamiento incluyen la aparición de síntomas (dolor) o la disminución del crecimiento testicular.
Se trata de la dilatación de las venas testiculares en el plexo pampiniforme producida por reflujo venoso. Lo más habitual es su presentación a partir de la pubertad (15-20 % en adolescentes), siendo infrecuente en niños menores de 10 años. El 90 % de los casos aparece en el lado izquierdo, y puede ser bilateral en algunos pacientes(25). La disposición anatómica de la vena espermática izquierda en su unión a la vena renal izquierda en un ángulo de 90º, se ha descrito como una de las causas.
En función de su severidad, se diferencian 3 grados según la Clasificación de Dubin-Amelar:
• Grado I: varicocele palpable con maniobras de Valsalva.
• Grado II: varicocele palpable (sin maniobras de Valsalva).
• Grado III: varicocele visible.
El varicocele subclínico es aquel que se detecta únicamente en la ecografía, de forma incidental, sin hallazgos durante la exploración física.
En los adolescentes, habitualmente el varicocele es asintomático y poco evolucionado, pero es imprescindible un seguimiento clínico y ecográfico cercano para el diagnóstico precoz de síntomas, como dolor testicular o la disminución del volumen testicular, que suponen indicaciones para su tratamiento. Como complicación a largo plazo, se ha descrito que el 20 % de los adolescentes con varicocele puede desarrollar problemas de fertilidad en el futuro, secundarios al hipogonadismo y la disminución de la secreción de testosterona, generados por el daño parenquimatoso del testículo expuesto a una temperatura anormalmente elevada.
Diagnóstico
El diagnóstico se basa en:
• Exploración física: el varicocele se detecta mediante la visualización y/o palpación de venas testiculares dilatadas y tortuosas en posición de bipedestación, que se hacen más evidentes con las maniobras de Valsalva.
Siempre se debe evaluar el tamaño de ambos testículos, en la búsqueda de asimetrías por disminución del volumen testicular en el lado del varicocele.
• Ecografía-doppler testicular: confirma la existencia de varicocele y su severidad. Mide el reflujo venoso en el plexo pampiniforme en posición de bipedestación y en decúbito supino; y el tamaño testicular, considerándose signo de hipoplasia cuando el volumen testicular es <20 % con respecto al contralateral.
Se debe asociar la realización de ecografía abdominal en los pacientes con varicocele de presentación más “atípica”, en los que es obligado descartar la existencia de una masa tumoral como causa de varicocele, por compresión de la vena renal o la vena cava inferior. En este sentido, se debe indicar especialmente en pacientes prepúberes o en aquellos casos de varicocele derecho aislado.
Tratamiento
Se ha demostrado que el tratamiento del varicocele mejora el volumen testicular, los parámetros seminales y las posibilidades de fertilidad futura. El éxito del tratamiento en cuanto a la desaparición del varicocele se sitúa en el 85-100 % de los pacientes según las distintas técnicas y la experiencia de cada Centro. Por ello, se han establecido dos criterios principales de tratamiento en el varicocele de la infancia y adolescencia, que son los siguientes:
1. Varicocele asociado a disminución del tamaño testicular (hipotrofia).
2. Varicocele sintomático.
Además, alteraciones en los parámetros de estudio de las muestras de semen, u otras situaciones, como el varicocele palpable bilateral o la asociación de otras patologías con riesgo de fertilidad, podrían valorarse también, como otras indicaciones de tratamiento en el varicocele(4,26-29).
Las opciones de tratamiento incluyen diversas técnicas quirúrgicas de ligadura y sección vascular o la embolización endovascular, como alternativa no quirúrgica. Debido a la falta de estudios en la edad pediátrica, no se han podido confirmar diferencias significativas en los resultados terapéuticos de las distintas técnicas, que dependen, en gran medida, de la experiencia de cada Centro. Por lo tanto, existe controversia aún en relación al mejor método de tratamiento del varicocele en los pacientes pediátricos y adolescentes.
La embolización se realiza mediante un procedimiento endovascular percutáneo, consistente en la introducción de espirales metálicos (coils) o sustancias esclerosantes en las venas espermáticas, a través de la cateterización de una vena principal en la ingle (vena femoral), el cuello (vena yugular interna) o el brazo (vena basílica). Debido a la actuación selectiva sobre los vasos venosos, tiene la ventaja de no desarrollar posteriormente hidrocele por lesión de los vasos linfáticos del cordón espermático, complicación más frecuente del tratamiento quirúrgico. Al ser una técnica poco invasiva, en la mayoría de los pacientes se plantea, además, como una intervención de corta estancia, que no precisa ingreso en la mayoría de los pacientes. En contrapartida, y a pesar de ser un tratamiento eficaz con tasas de éxito en torno al 80-90 %(25,29), su principal desventaja es una mayor tasa de recurrencia (13-15 %) con respecto al tratamiento quirúrgico (5-7 %), que hace necesaria en algunos pacientes, la repetición de la embolización.
El tratamiento quirúrgico consiste en la ligadura y sección de las venas espermáticas dilatadas, mediante un abordaje inguinal, retroperitoneal o laparoscópico. La ligadura de las venas espermáticas puede realizarse de forma selectiva, con preservación de la arteria espermática (técnica de Ivanissevich-abordaje inguinal), o de forma conjunta junto a la arteria espermática (técnica de Paloma-vía retroperitoneal). En el caso del abordaje laparoscópico, se puede realizar cualquiera de las dos opciones. En este último caso, puede optimizarse la técnica mediante la inyección intra-testicular de sustancias colorantes (p. ej., verde de indocianina), que tiñe los vasos linfáticos del cordón espermático, lo que permite su preservación y evita la aparición de hidrocele postquirúrgico.
Las complicaciones del tratamiento en el varicocele son escasas. Como se ha comentado anteriormente, la del hidrocele postquirúrgico es la más frecuente, con tasas de presentación del 5-10 %, que disminuyen significativamente con la preservación de los vasos linfáticos. Otras complicaciones descritas más raras son: la atrofia testicular tras la ligadura de la arteria espermática o la migración del material de embolización a la vena renal con trombosis de la misma.
Quistes de epidídimo y espermatocele (Fig. 7)
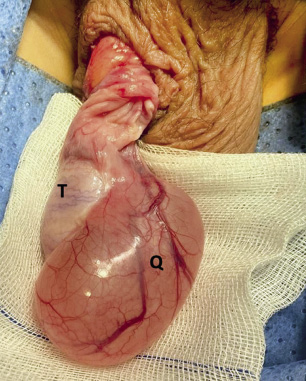
Figura 7. Quiste de epidídimo en paciente de 15 años de edad. La indicación de resección quirúrgica se realizó por su gran tamaño, imposibilidad de diferenciar el testículo del quiste en la exploración física (lo que dificultaba una correcta exploración testicular ante la aparición de otras patologías escrotales), y la presentación de molestias dolorosas con el ejercicio. Fuente: propiedad de la autora.
Los quistes de epidídimo son de etiología benigna, aparecen en cualquier zona del epidídimo y se rellenan de un líquido claro que no contiene espermatozoides. Son más frecuentes en varones prepuberales. Con frecuencia son asintomáticos, de pequeño tamaño y se diagnostican habitualmente de forma casual durante la exploración física o ecográfica testicular realizadas por otros motivos. Su etiología es desconocida en la mayoría de los casos, aunque se han descrito, a veces, antecedentes de traumatismo testicular o epididimitis. Pueden desaparecer espontáneamente.
Deben diferenciarse del espermatocele, indistinguible en la exploración física y, también, manifestado como quiste epididimario, que aparece en las etapas post-puberales y contiene esperma en su interior. Aparecen en la cabeza del epidídimo y, aunque suele ser también asintomático, puede producir dolor en un porcentaje mayor a los quistes epididimarios de la infancia. En la ecografía, puede diferenciarse de estos por tener un contenido con ecos variables y septos en su interior.
El diagnóstico diferencial de los quistes de epidídimo debe realizarse con otras patologías testiculares de distinta naturaleza como: epididimitis, hidrocele, varicocele o tumores testiculares.
La observación es la medida indicada como primera opción, tanto en los quistes de epidídimo como en el espermatocele que aparece en los pacientes asintomáticos, sin dudas diagnósticas o con quistes de pequeño tamaño (<10 mm). Por el contrario, los quistes de epidídimo o espermatoceles, de gran tamaño, o de crecimiento progresivo, con dudas diagnósticas o con dolor asociado, se tratan quirúrgicamente mediante la extirpación completa del quiste, por vía escrotal(30).
Función del pediatra de Atención Primaria
El principal objetivo del pediatra de Atención Primaria en la valoración de los problemas del descenso testicular debe ser su identificación precoz para indicar su seguimiento por parte del cirujano pediátrico en caso de sospecha, duda o confirmación. Esto permitirá el tratamiento en torno al año de edad, evitando así las posibles secuelas que puedan producirse sobre la fertilidad en el futuro. En los casos en los que se identifiquen de forma tardía, también se deberán remitir para su tratamiento antes de la pubertad, minimizando así los potenciales riesgos de malignización testicular.
Para ello, se establece como función primordial, llevar a cabo una adecuada exploración de la región inguinoescrotal, tanto durante las consultas del programa de revisión del niño sano, como en otras que se produzcan por cualquier otro síntoma o signo referidos al testículo, escroto, región inguinal o abdominal. Junto a esto, es muy importante que el pediatra esté familiarizado con el calendario quirúrgico, en relación a las recomendaciones sobre el momento de establecer el diagnóstico de criptorquidia según la edad gestacional o con la diferenciación entre los verdaderos casos de MDT de aquellos de “testículos en ascensor”, que no requieren ningún tipo de tratamiento.
En cuanto a los cuadros de escroto agudo, y ante la mínima sospecha de torsión testicular, la solicitud inmediata de valoración urgente por Cirugía Pediátrica desde la consulta de Atención Primaria, es vital para poder llevar a cabo una exploración quirúrgica urgente y la recuperación del testículo, en el caso de que esta se confirme. En este caso, no se debe solicitar la realización de pruebas complementarias previas, que retrasarían el diagnóstico.
En cuanto al varicocele, el pediatra de Atención Primaria debe considerar un signo de alarma, cuando este se presente en el lado derecho, y solicitar una ecografía abdominal preferente para descartar la existencia de masa abdominal.
En los casos de epididimitis-orquiepididimitis de repetición en pacientes en edad prepuberal, se debe investigar la existencia de trastornos malformativos o funcionales del sistema urinario, que se deben iniciar mediante la realización de ecografía a ese nivel.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de la autora.
1. Barthold JS, González R. The epidemiology of congenital cryptorchidism, testicular ascent and orchiopexy. The Journal of Urology. 2003; 170: 2396-401.
2. Leslie SW, Sajjad H, Villanueva CA. Cryptorchidism. En: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK470270/.
3.*** Shin J, Jeon GW. Comparison of diagnostic and treatment guidelines for undescended testis. Clin Exp Pediatr. 2020; 63: 415-21.
4.*** Radmayr C, Bogaert G, Burgu B, Dogan HS, Nijman JM, Quaedackers J, et al. EAU Guidelines on Paediatric Urology. European Association of Urology. 2022. Disponible en: https://uroweb.org/guidelines/paediatric-urology.
5.** Chedrawe ER, Keefe DT, Romao RLP. Diagnosis, Classification, and Contemporary Management of Undescended Testicles. Urol Clin North Am. 2023; 50: 477-90.
6. Muíños CC. Criptorquidia y patología testículo-escrotal en la edad pediátrica. Pediatr Integral. 2019; XXIII: 271-82. Disponible en: https://www.pediatriaintegral.es/publicacion-2019-09/criptorquidia-y-patologia-testiculo-escrotal-en-la-edad-pediatrica/.
7.** Echeverría Sepúlveda MP, Yankovic Barceló F, López Egaña PJ. The undescended testis in children and adolescents part 2: evaluation and therapeutic approach. Pediatr Surg Int. 2022; 38: 789-99.
8. Hutson JM. Cryptorchidism and Hypospadias. En: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2022. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK279106/.
9.*** Hutson JM, Vikraman J, Li R, Thorup J. Undescended testis: What paediatricians need to know. J Paediatr Child Health. 2017; 53: 1101-4.
10. Boehme P, Degener S, Wirth S, Geis B, Aydin M, Lawrenz K, et al. Multicenter Analysis of Acquired Undescended Testis and its Impact on the Timing of Orchidopexy. J Pediatr. 2020; 223: 170-177.e3.
11. Park KH, Lee JH, Han JJ, Lee SD, Song SY. Histological evidences suggest recommending orchiopexy within the first year of life for children with unilateral inguinal cryptorchid testis. Int J Urol. 2007; 14: 616-21.
12.** Kim JK, Chua ME, Ming JM, Santos JD, Zani-Ruttenstock E, Marson A, et al. A critical review of recent clinical practice guidelines on management of cryptorchidism. Journal of Pediatric Surgery. 2018; 53: 2041-7.
13.** Braga LH, Lorenzo AJ, Romao RLP. Canadian Urological Association-Pediatric Urologists of Canada (CUA-PUC) guideline for the diagnosis, management, and followup of cryptorchidism. CUAJ. 2017; 11: E251-60.
14. Hadziselimovic F. Is Hormonal Treatment of Congenital Undescended Testes Justified A Debate. Sex Dev. 2019; 13: 3-10.
15.** **Dinkelbach L, Lehnick D, Shavit S, Szavay P, Zundel S. Acquired undescended testis: When does the ascent occur? Journal of Pediatric Surgery. 2021; 56: 2027-31.
16. Koch T, Hansen AH, Priskorn L, Petersen JH, Carlsen E, Main KM, et al. A history of cryptorchidism is associated with impaired testicular function in early adulthood: a cross-sectional study of 6376 men from the general population. Human Reproduction. 2020; 35: 1765-80.
17. Pettersson A, Richiardi L, Nordenskjold A, Kaijser M, Akre O. Age at surgery for undescended testis and risk of testicular cancer. N Engl J Med. 2007; 356: 1835-41.
18.*** Bowlin PR, Gatti JM, Murphy JP. Pediatric Testicular Torsion. Surg Clin North Am. 2017; 97: 161-72.
19.*** Shunmugam M, Goldman RD. Testicular torsion in children. Can Fam Physician. 2021; 67: 669-71.
20. Jefferies MT, Cox AC, Gupta A, Proctor A. The management of acute testicular pain in children and adolescents. BMJ. 2015; 350: h1563.
21.** Kylat RI. Perinatal testicular torsion. Arch Pediatr. 2021; 28: 75-9.
22. Huertas AL, Delfa SB. Escroto agudo. Pediatr Integral. 2019; 23: 283-91. Disponible en: https://www.pediatriaintegral.es/publicacion-2019-09/escroto-agudo/
23. Villares Alonso R, Jiménez Jiménez JI. Orquiepididimitis aguda. Guía_ABE. Infecciones en Pediatría. Guía rápida para la selección del tratamiento antimicrobiano empírico. 2019. Disponible en: https://www.guia-abe.es/temas-clinicos-orquiepididimitis-aguda.
24. Santi M, Lava SAG, Simonetti GD, Bianchetti MG, Milani GP. Acute Idiopathic Scrotal Edema: Systematic Literature Review. Eur J Pediatr Surg. 2018; 28: 222-6.
25. Rojas-Ticona J, Fernández Córdoba MS, Argumosa Salazar YM, Marijuán Sahuquillo V, Ramírez Piqueras M, Moratalla Jareño T, et al. Two decades of experience in endovascular treatment of varicocele in pediatric age. Cir Pediatr. 2019; 32: 93-8.
26. Chung JM, Lee SD. Current Issues in Adolescent Varicocele: Pediatric Urological Perspectives. World J Mens Health. 2018; 36: 123-31.
27.** Macey MR, Owen RC, Ross SS, Coward RM. Best practice in the diagnosis and treatment of varicocele in children and adolescents. Ther Adv Urol. 2018; 10: 273-82.
28. Gómez Beltrán O, Garrido Pérez JI, García Ceballos A, Escassi Gil A, Vargas Cruz V, Lasso Betancor CE, et al. Open surgery, laparoscopic Palomo varicocelectomy and embolization in children with varicocele. Cir Pediatr. 2013; 26: 9-12.
29. Wong S, Vigneswaran G, Maclean D, Bryant T, Hacking N, Maher B, et al. 10-year experience of Paediatric varicocele embolization in a tertiary centre with long-term follow-up. J Pediatr Urol. 2022; 18: 113.e1-e6.
30. Boscarelli A, Bellini T. Epididymal cyst in children. Eur J Pediatr. 2021; 180: 2723-9.
Bibliografía recomendada
– Radmayr C, Bogaert G, Burgu B, Dogan HS, Nijman JM, Quaedackers J, et al. EAU Guidelines on Paediatric Urology. European Association of Urology. 2022.
Consenso Europeo sobre los principales aspectos de la clasificación, diagnóstico y tratamiento del maldescenso testicular y la patología escrotal. Actualización del año 2022. Guía diagnóstico-terapéutica de referencia en la atención del paciente con criptorquidia y patología escrotal, esquemática, y validada por la Asociación Europea de Urología (EAU).
– Hutson JM, Vikraman J, Li R, Thorup J. Undescended testis: What paediatricians need to know. J Paediatr Child Health. 2017; 53: 1101-4.
Resumen de las principales claves en el abordaje diagnóstico-terapéutico del paciente con criptorquidia, dirigido a los profesionales de Pediatría.
– Chedrawe ER, Keefe DT, Romao RLP. Diagnosis, Classification, and Contemporary Management of Undescended Testicles. Urol Clin North Am. 2023; 50: 477-90.
Artículo de revisión americano del maldescenso testicular, muy actualizado. A destacar, tabla explicativa de los distintos tipos en función de la prevalencia, la exploración y su naturaleza, congénito o adquirido. Exposición clara y contrastada de las complicaciones y secuelas a largo plazo, descritas.
– Echeverría Sepúlveda MP, Yankovic Barceló F, López Egaña PJ. The undescended testis in children and adolescents part 2: evaluation and therapeutic approach. Pediatr Surg Int. 2022; 38: 789-99.
Revisión de los principales aspectos diagnósticos y de tratamiento del maldescenso testicular. Descripción detallada de los distintos procedimientos quirúrgicos indicados, en función del tipo y evolución del MDT, con imágenes quirúrgicas que ayudan a la comprensión anatómica y del abordaje terapéutico en cada caso.
– Shin J, Jeon GW. Comparison of diagnostic and treatment guidelines for undescended testis. Clin Exp Pediatr. 2020; 63: 415-21.
Revisión actualizada sobre la criptorquidia: etiopatogenia, diagnóstico y tratamiento. Exposición de la clasificación del maldescenso testicular de forma esquemática y muy gráfica, y con relación a la exploración física del paciente. Revisión de los aspectos más relevantes del diagnóstico y tratamiento del MDT y comparación de las principales guías diagnósticas y terapéuticas internacionales: AUA: American Urological Association; BAPS/BAUS: British Association of Pediatric Surgeons/British Association of Urologic Surgeons; CUA: Canadian Urological Association; EAU: European Association of Urology.
– Shunmugam M, Goldman RD. Testicular torsion in children. Can Fam Physician. 2021; 67: 669-71.
Resumen de los principales aspectos del diagnóstico clínico del paciente con escroto agudo. Enfoque en la atención y valoración por el profesional pediatra. Explicación del score TWIST en la valoración inicial del escroto agudo y su relación con el diagnóstico de torsión testicular.
– Bowlin PR, Gatti JM, Murphy JP. Pediatric Testicular Torsion. Surg Clin North Am. 2017; 97: 161-72.
Exposición detallada de los aspectos sobre el diagnóstico, tratamiento y seguimiento de los pacientes con torsión testicular.
– Kylat RI. Perinatal testicular torsion. Arch Pediatr. 2021; 28: 75-9.
Artículo sobre la etiopatogenia, clínica y tratamiento de la torsión de aparición durante el período perinatal, con especial énfasis en las actitudes diagnósticas y terapéuticas a seguir ante su sospecha. Incide en los aspectos controvertidos en cuanto al momento y abordaje quirúrgicos. En la explicación de los factores etiopatogénicos, describe, de forma clara y esquemática, los diferentes tipos de torsión testicular, en función de la edad y mecanismo de producción.
– Macey MR, Owen RC, Ross SS, Coward RM. Best practice in the diagnosis and treatment of varicocele in children and adolescents. Ther Adv Urol. 2018; 10: 273-82.
Exposición de la etiología, clínica, diagnóstico e indicaciones y tipo de tratamiento del varicocele. Revisión de las recomendaciones de las guías europeas y americanas de atención al paciente con patología urológica de la población pediátrica y adolescente.
| Caso clínico |
|
Paciente de 2 meses de edad que acude a la consulta de su pediatra en el contexto de la revisión del niño sano. Sin antecedentes de interés, parto a término, eutócico, con peso al nacer: 3,350 kg. Lactancia materna exclusiva en la actualidad.
En la exploración de los genitales externos, se observa testículo derecho en escroto, de tamaño adecuado para su edad, móvil, sin masas palpables. Sin alteraciones escrotales. En el lado izquierdo se observa escroto vacío, palpándose testículo en región inguinal.
|
|