 |
| Temas de FC |
V. Soto Insuga, E. González Alguacil, J.J. García Peñas
Unidad de Neuropediatría del Hospital Infantil Universitario Niño Jesús, Madrid
| Resumen
La discapacidad intelectual se define como: aquel trastorno que se inicia antes de los 18 años, caracterizado por una limitación en el funcionamiento intelectual (cociente intelectual bajo) y en el comportamiento adaptativo en las áreas conceptual, social y práctica. En menores de 5 años, al ser las pruebas de inteligencia poco fiables, se debe hablar de retraso global del desarrollo o retraso psicomotor que implica un retraso en, al menos, dos áreas de desarrollo: motricidad fina-gruesa, lenguaje, sociabilidad, cognición y actividades de la vida diaria. Su identificación debe ser precoz, de cara a iniciar tratamientos eficaces específicos. Es fundamental un estudio etiológico que permita: identificar posibles tratamientos específicos, proporcionar un pronóstico específico, prevenir posibles complicaciones asociadas y esperables, ofrecer un consejo genético y evitar otras pruebas complementarias innecesarias, entre otras ventajas. Actualmente, la capacidad de encontrar una causa específica del retraso del desarrollo ha aumentado gracias a algoritmos diagnósticos secuenciales que incluyen tanto la anamnesis y exploración física, como pruebas complementarias, entre otras: pruebas de neuroimagen, metabólicas y genéticas. |
| Abstract
Intellectual disability is defined as a disorder that begins before 18 years of age, characterized by a limitation in intellectual functioning (low IQ) and in adaptive behavior in the conceptual, social and practical areas. In children under 5 years of age, as intelligence tests are unreliable, one should speak of global developmental delay or psychomotor delay that implies a delay in at least two areas of development: fine-gross motor skills, language, sociability, cognition and activities of daily living. Their identification must be early, in order to start specific effective treatments. An etiological study is essential as it allows: identifying possible specific treatments, providing a specific prognosis, prevention of possible associated and expected complications, offering genetic counseling and avoiding other unnecessary complementary tests, among other advantages. Currently, the ability to find a specific cause of developmental delay has increased thanks to sequential diagnostic algorithms that include both anamnesis and physical examination as well as complementary tests, which include: neuroimaging, metabolic and genetic tests. |
Palabras clave: Retraso global del desarrollo; Retraso psicomotor; Retraso madurativo; Discapacidad intelectual; Genética; Regresión psicomotriz.
Key words: Global delay in development; Psychomotor retardation; Maturation delay; Intellectual disability; Genetics; Psychomotor regression.
Pediatr Integral 2020; XXIV(6): 303 – 315
Detección y manejo del retraso psicomotor en la infancia
“El que no posee el don de maravillarse ni de entusiasmarse, más le valdría estar muerto porque sus ojos están cerrados”
Albert Einstein
Introducción y conceptos
Debido a la poca fiabilidad de las pruebas de inteligencia, en menores de cinco años debemos hablar de retraso global del desarrollo.
La discapacidad intelectual (previamente conocida como “retraso mental”) y el retraso global del desarrollo (también denominado “retraso psicomotor”) se incluyen según la última clasificación DSM-5 (Diagnostic and Statistical Manual of Mental Disorders)(1) dentro del grupo de los “Trastornos del Neurodesarrollo”, que son aquellas alteraciones en el desarrollo neurológico que se inician en la infancia.
El término de discapacidad intelectual (DI) no se basa únicamente en la detección de un cociente intelectual bajo, sino que también implica una alteración en la interacción ambiental y social. De esta forma, la DI se entiende como: aquel trastorno que se inicia antes de los 18 años, caracterizado por una limitación en el funcionamiento intelectual (que debe ser confirmado mediante una evaluación clínica y pruebas de inteligencia estandarizadas individualizadas) y en el comportamiento adaptativo en las áreas conceptual, social y práctica. La gravedad se define fundamentalmente por las alteraciones adaptativas, pero sigue siendo válida la estratificación dependiendo del cociente intelectual (CIT), que es un instrumento artificial creado por Binet en 1905, en respuesta al encargo que hizo el Ministerio Francés de diseñar un método objetivo para identificar aquellos alumnos que podían requerir una enseñanza especial. Se estableció un nivel teórico que correspondería al valor medio de la población, al que se le asignó una puntuación típica de 100 y se admitió que la inteligencia tiene una distribución normal entre la población (campana de Gauss). De esta forma, se establecen diferentes grados de gravedad: funcionamiento intelectual límite (CIT 70-85), DI leve (CIT 50-70), DI moderada (CIT 35-50), DI grave (CIT 20-35) y DI profunda (CI<20).
En menores de 5 años, las pruebas de inteligencia son poco fiables, tanto por el elevado porcentaje de falsos positivos como negativos; por lo que, por debajo de esta edad, se habla de retraso global del desarrollo (RGD), que implica un retraso en, al menos, dos áreas de desarrollo: motricidad fina-gruesa, lenguaje, sociabilidad, cognición y actividades de la vida diaria(2) (Fig. 1).
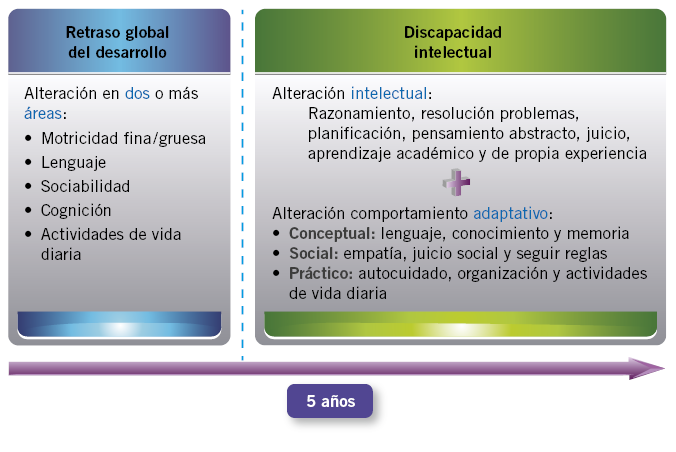
Figura 1. Retraso global del desarrollo y discapacidad intelectual.
Prevalencia
La prevalencia de RGD/DI en niños es del 1-3%.
El estudio de niños con sospecha de RGD es uno de los principales motivos de consulta en Neuropediatría. Actualmente, se estima que la incidencia de RGD en niños menores de 5 años es de 1-3%(3).
De forma similar, los estudios de incidencia de DI, aunque varían dependiendo de los criterios diagnósticos y los métodos de valoración empleados, estiman una incidencia de 1-3%, lo que sugiere que seguramente representen una misma población. Los principales estudios de prevalencia se han realizado en población escandinava, entre los cuales destaca el realizado por Strömme y cols. en 30.037 niños nacidos entre 1980-1985 en Noruega, en el que objetivaron que el 0,62% presentaba DI, siendo leve, moderado, grave y profundo en 0,35%, 0,15%, 0,04% y 0,08%, respectivamente(4). Cifras algo más elevadas se reportaron en el estudio de Murphy et al., en población infantil en Atlanta, en el que refirieron una incidencia de 1,2%(5). Estas cifras sitúan el RGD/DI como una patología frecuente, muy alejada de la frecuencia del concepto de enfermedades poco frecuentes, definidas como: aquellas con que afectan a <0,05% de una población.
¿Retraso madurativo o retraso global del desarrollo?
El concepto de retraso madurativo debe entenderse como una situación no definitiva en la que debemos iniciar una búsqueda etiológica y una estimulación específica, sin retrasar su abordaje.
El concepto ampliamente utilizado de “retraso madurativo” además de no estar recogido en las clasificaciones DSM-V ni CIE 11, debe usarse con cautela y es necesario ser explicado a los padres, de cara a evitar la sensación de banalización de ser “un problema que va a desaparecer con el tiempo” lo que implica, en muchas ocasiones, un retraso en el diagnóstico y en el inicio de tratamiento. es un término que utilizamos para referirnos a una “foto fija” del neurodesarrollo de un niño, que en ese momento se aparta de los límites de la normalidad. De este modo, un niño en el que se detecta un retraso en el desarrollo puede significar:
1. Retraso global del desarrollo.
2. Variante normal del desarrollo que implica una normalización posterior.
3. Inadecuada estimulación.
4. Retraso derivado de una enfermedad crónica extraneurológica, como puede ser: desnutrición, cardiopatías congénitas, enfermedad celiaca o déficits neurosensoriales, entre otros.
5. Primera manifestación de un trastorno motor crónico no progresivo (parálisis cerebral), de una discapacidad intelectual futura o de otros trastornos de neurodesarrollo como: trastorno del espectro autista (TEA), trastorno del lenguaje, trastorno de la coordinación motora o trastorno por déficit de atención e hiperactividad (TDAH), entre otros(6).
Por tanto, el concepto de retraso madurativo debe entenderse como una situación en la que debemos iniciar una búsqueda etiológica y una estimulación específica, sin retrasar su abordaje.
Etiología
Son muchas las causas que justifican la búsqueda etiológica en los niños con RGD/DI.
Numerosas etiologías pueden ocasionar un RGD/DI (Tabla I).

Cada vez se reconoce más una alteración genética como causa de la alteración en el desarrollo(7). Aunque en los últimos años estamos siendo capaces de detectar RGD/DI de causa monogénica, existe una creciente complejidad a la hora de interpretar el resultado de las pruebas genéticas, debido a factores como: herencia poligénica, expresividad variable o factores de modificación epigenéticos(8).
Se estima, según las diferentes series, que en un 23-78% de los pacientes con RGD/DI no se encuentra una causa definida. Esto es variable según el área geográfica y el grado de severidad del retraso, de manera que en aquellos niños con DI grave, se suele encontrar una causa en hasta el 80% de las ocasiones(9). El avance en las técnicas de neuroimagen y fundamentalmente genéticas, ha permitido aumentar la posibilidad de encontrar una etiología definida. En un estudio reciente de Han y cols. en 75 niños con RGD/DI, aplicando diferentes técnicas genéticas (array CGH, estudio de X frágil y panel de genes de RGD/DI) y resonancia cerebral, se consiguió encontrar una causa identificable en el 71% de los pacientes(10).
Son muchos los motivos que justifican una búsqueda etiológica del RGD/DI(11), entre los que se incluyen: identificar posibles tratamientos específicos (como la dieta cetogénica en la deficiencia del transportador de glucosa cerebral o la vitamina B6 en los trastornos de la piridoxina), proporcionar un pronóstico específico según etiología, prevenir posibles complicaciones asociadas y esperables, ofrecer un consejo genético, evitar otras pruebas complementarias innecesarias (lo que es definido por muchos padres como una “odisea diagnóstica”), mejorar el conocimiento de la fisiopatología de la enfermedad, posibilidad de contacto con otras familias con la misma patología y, en muchos casos, evitar sentimientos de culpabilidad de los padres(12).
Aproximación diagnóstica
Mediante una anamnesis y exploración física adecuada, así como con el uso racional de pruebas complementarias (resonancia cerebral, estudios metabólicos y genéticos), es posible encontrar la causa en un alto porcentaje de pacientes con RGD/DI.
Importancia de una detección/diagnóstico precoz
El diagnóstico de RGD debe ser precoz. Son los pediatras de Atención Primaria los principales médicos implicados en esta detección(13).
Es importante alertarse ante un retraso del desarrollo psicomotor, no banalizar el problema. Debemos transmitir a los padres que la detección de un trastorno del neurodesarrollo no es “etiquetar” al niño, sino una identificación de necesidad de estimulación; es decir, un diagnóstico provisional que nos servirá para realizar una intervención individualizada lo antes posible.
Generalmente, los padres son los primeros en detectar los problemas del neurodesarrollo (hasta en el 60%) y se debe aprovechar cualquier visita (de seguimiento, rutinaria o por enfermedad) para consultar a los padres por las preocupaciones o problemas sobre el desarrollo de sus hijos(14).
Debemos atender a los signos de alarma del desarrollo que nos indican una anomalía en el neurodesarrollo (Fig. 2).
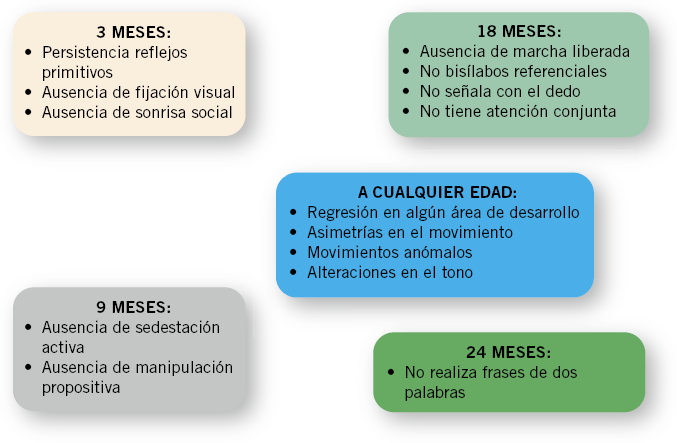
Figura 2. Signos de alarma en el desarrollo psicomotor.
Los hitos del desarrollo psicomotor se adquieren dentro de un amplio rango de normalidad (Tabla II).

Generalmente, suele ser útil, no solo la impresión global clínica, sino el uso de escalas de desarrollo. En este sentido, la más utilizada y validada en nuestro medio es la de Haizea-Llevant, resultado de un estudio en 2.519 niños de 0-5 años de Cataluña y País Vasco. Comprende 97 ítems distribuidos en áreas de: socialización, lenguaje, manipulación y postural, en los que podremos identificar si el niño ha adquirido ese ítem del desarrollo de forma adecuada a su edad(15) (Fig. 3).
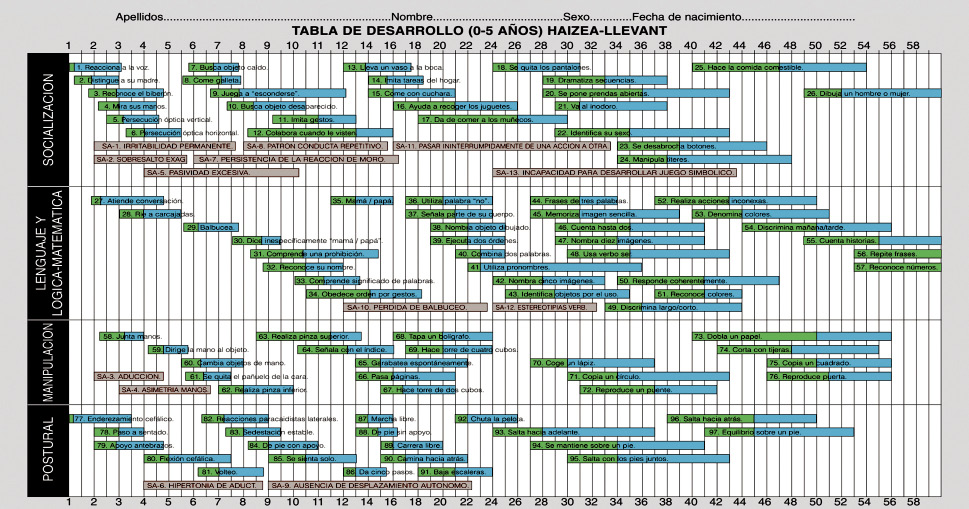
Figura 3. Tabla de desarrollo (0-5 años) Haizea-Llevant.
Asimismo, son de gran utilidad otras herramientas específicas y fáciles de aplicar para el cribado de determinados problemas de aprendizaje como: la M-CHAT (Modified checklist for Autism), para la identificación de síntomas de TEA en niños de 16-30 meses; o la CSBS-CS (Escala de Conducta Comunicativa y Simbólica), para identificar alteraciones en las áreas del lenguaje o sociabilidad.
No hay que olvidar que existen variantes de la normalidad del desarrollo, que no suponen un hallazgo patológico y que debemos conocer como son, entre otras: pinza entre dedo pulgar y medio, no realizar gateo, desplazamiento sentado sobre nalgas o apoyando una rodilla y el pie de la otra extremidad o rodar sobre sí mismo, marcha de puntillas los dos primeros años de vida, tartamudeo fisiológico entre los 2-4 años o dislalias hasta los 4 años de edad(16).
En la evaluación será primordial diferenciar entre un retraso global del desarrollo estático y una encefalopatía progresiva (apartado del que nos ocuparemos posteriormente), que requerirá una valoración preferente por un servicio de Neuropediatría. A veces, puede ser difícil de distinguir una regresión en enfermedades lentamente progresivas, de las cuales muchas veces existe una “meseta prolongada”(17).
Confirmación diagnóstica
Para un diagnóstico definitivo de DI/RGD, serán necesarias valoraciones neuropsicológicas realizadas por personal especializado, que incluyan valoraciones cognitivas (cociente intelectual a partir de los 5 años y cociente de desarrollo en menores), así como de otras áreas de aprendizaje y conducta(18).
A partir de los 5 años, se deben usar test de inteligencia, como las escalas Wechsler. En menores de 5 años, hay escalas que permiten objetivar el desarrollo de un determinado niño con arreglo a estándares de población general, desglosando diferentes áreas (postural-motriz, cognitiva y comunicativa), siendo unas de las más utilizadas: Brunet-Lezine, Bayley o Merril-Palmer, en las que se miden edad de desarrollo y cociente de desarrollo. No existe una correlación fiable con lo que, en referencia a edades posteriores, se conoce como “cociente intelectual”, siendo estas escalas únicamente un elemento auxiliar, que no pueden sustituir a un diagnóstico clínico completo(6).
Diagnóstico etiológico
Anamnesis y exploración física
Una adecuada anamnesis y exploración física permite el diagnóstico de un RGD/DI en el 12,5-38% de los casos y aporta claves fundamentales para una adecuada investigación en hasta el 65% de los casos(19).
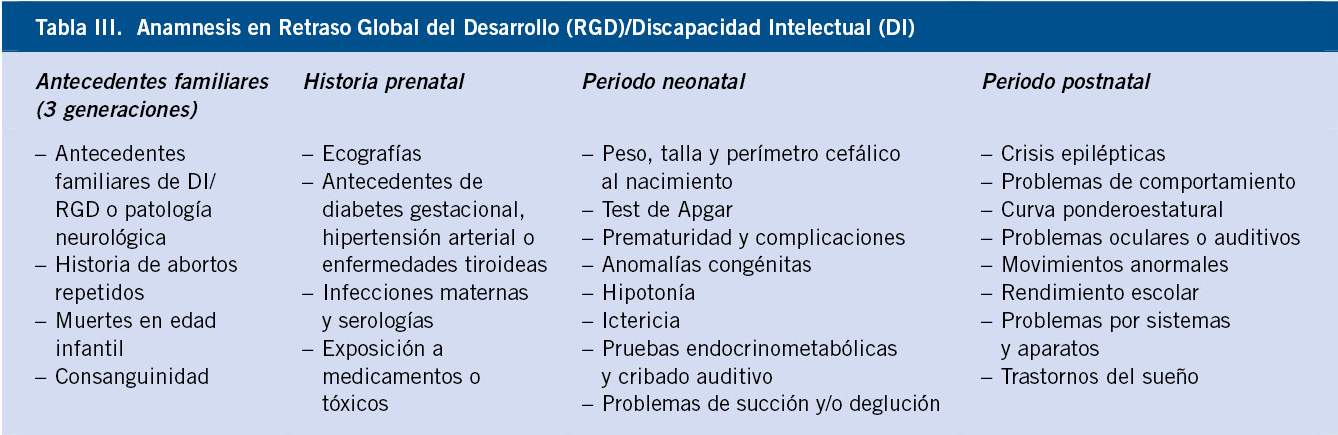
• Anamnesis: deberá incluir una historia familiar completa (3 generaciones) y un relato completo de los antecedentes perinatales, evaluando los factores de riesgo conocidos (infecciones, hemorragias, alcohol materno, drogas, patología del parto, etc.), así como el parto y el periodo neonatal. Se recogerán los hitos fundamentales de las principales áreas del desarrollo psicomotor (motor grueso, fino, lenguaje y sociabilidad), definiendo la presencia o ausencia de regresión psicomotriz. Asimismo, es fundamental recoger las alteraciones conductuales de estos pacientes, que en muchos casos son específicas de determinadas enfermedades (fenotipos neuro-conductuales).
Se valorará la posibilidad de trastornos asociados y se buscarán activamente claves diagnósticas que nos permitan orientar diferentes etiologías (Tabla III).
• Exploración física: en muchos casos nos va a dar la clave etiológica. Debemos realizar una exploración neurológica sistemática, sin olvidarnos de la exploración general pediátrica. De hecho, el examen de la piel, los percentiles de peso-talla-perímetro cefálico, alteraciones en genitales, la presencia de malformaciones menores o visceromegalias pueden ser claves de gran valor orientativo, o incluso diagnósticas (claves diagnósticas).
La exploración neurológica recogerá ordenadamente las posibles alteraciones del psiquismo, motricidad, equilibrio, vías piramidales o sistema nervioso periférico. En el caso de la exploración física del lactante, que es diferente respecto del escolar o adolescente, debemos realizar una exploración que incluya: la valoración del tono, del desarrollo postural en decúbito supino, decúbito prono, sedestación y bipedestación, sin olvidar reacciones posturales, reflejos primitivos, reflejos de percepción y patrón de movimiento (Tabla IV).

Es fundamental realizar exploraciones seriadas en el seguimiento de un niño con RGD/DI, de cara a poder identificar claves diagnósticas que no estuvieran presentes en una primera exploración (p. ej., las alteraciones cutáneas y oculares en la ataxia telangiectasia o el angiofibroma facial en el complejo esclerosis tuberosa).
Tras la anamnesis y la exploración pediátrica-neurológica, podemos encontrarnos antes dos escenarios:
• Indicadores de un síndrome reconocible: pacientes catalogados de RGD/DI tienen determinados rasgos faciales o patrón de malformaciones congénitas que se agrupan en una asociación reconocible (cuadro sindrómico), como un síndrome concreto o un proceso debido a una causa específica. Estas claves diagnósticas permiten confirmar la etiología mediante una prueba específica. Es necesario reconocer las principales y más frecuentes claves diagnósticas (Tabla V).

• Aproximación diagnóstica en retraso global del desarrollo/incapacidad intelectual estático no sindrómico (Fig. 4):
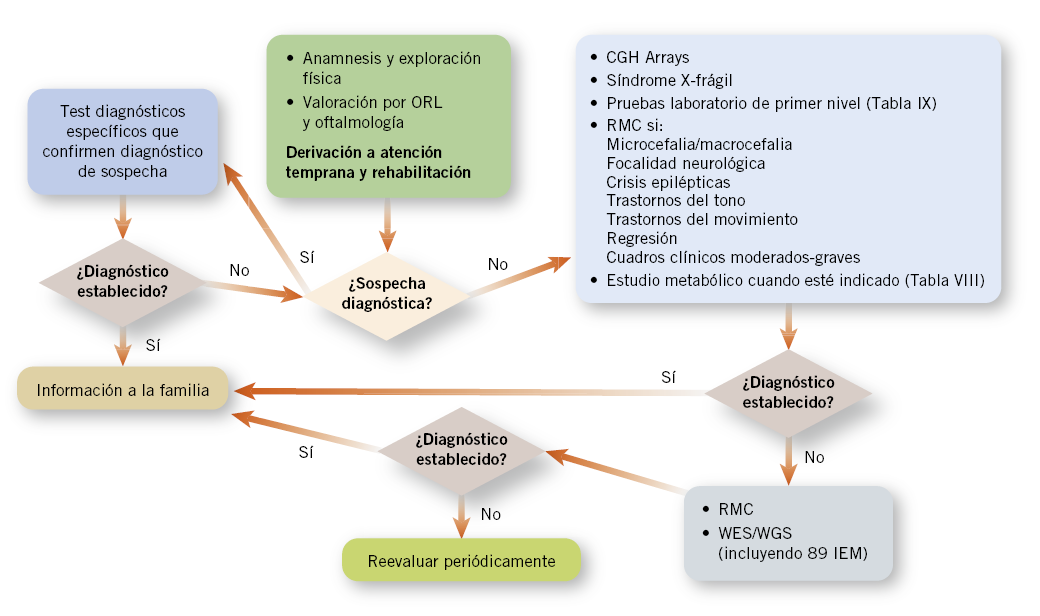
Figura 4. Algoritmo de aproximación diagnóstica ante un Retraso Global del Desarrollo/Discapacidad Intelectual estático, no sindrómico.
RMC: resonancia magnetica craneal; WES: secuenciación de exoma completo. WGS: secuenciación de genoma completo. IEM: errores innatos del metabolismo.
a) Evaluación sensorial: las principales guías diagnósticas recomiendan que todos los niños con RGD/DI deberían tener una evaluación de la vista y del oído por parte de los especialistas indicados. La alteración en uno de estos sentidos podría contribuir al retraso, o podría ayudarnos a una aproximación diagnóstica(9,20).
b) Estudios genéticos (Tabla VI):

– CGH-Arrays: desde el año 2000 existe un consenso generalizado de que los CGH-Arrays deben ser la prueba genética de primer nivel para cualquier paciente con RGD o DI de causa inexplicable(16,21). La tecnología de microarrays permite la detección de deleciones y duplicaciones patogénicas.
Se estima que esta prueba permite un diagnóstico en el 10% de los niños con RGD/DI y hay autores que afirman que el rendimiento diagnóstico puede alcanzar el 15-20% en aquellos pacientes con: dismorfias, anomalías congénitas o síntomas neurológicos(22-23).
Se pueden obtener tres resultados tras realizar un estudio de microarrays:
1. Un resultado normal, es decir, que no se han encontrado variantes que justifiquen la clínica.
2. Un resultado con una variante patológica conocida y causante de la enfermedad.
3. Un resultado con una variante de significado incierto (VUS), que implica que su presencia no explica completamente la clínica del paciente y que no debería implicar interrumpir la búsqueda etiológica. Estos hallazgos deben ser interpretados por genetistas y, en ocasiones, requieren un seguimiento y re-evaluación periódica que nos permita conocer su patogenicidad.
– Estudio de fragilidad del cromosoma X: la repetición de trinucleótidos FMR1 para analizar síndrome de X frágil también se recomienda como prueba de primer nivel ante pacientes con RGD y DI inespecífica, ya que es la causa monogénica más frecuente (prevalencia entre 1/4.000-5.000 nacimientos, pudiendo variar en función de la zona geográfica en la que se realice el estudio). El fenotipo es variable, debutando con retraso del desarrollo psicomotor. Asocia trastornos de conducta y del aprendizaje. Los rasgos físicos son sutiles en niños pequeños, y en niños mayores pueden incluir: cara estrecha y alargada, frente y pabellones auriculares grandes y de baja implantación, hiperlaxitud articular en los dedos, pies planos y macroorquidia en varones postpuberales. Este trastorno no es exclusivo en varones, aunque en mujeres la clínica suele ser más leve(20).
– Cariotipo: en los últimos estudios, la indicación es realizarlo únicamente ante la sospecha clínica de: aneuploidía (síndrome Turner, síndrome Down, síndrome Patau, síndrome Edwards…), antecedentes familiares de reordenamientos cromosómicos o abortos espontáneos múltiples. La rentabilidad de esta prueba sería de alrededor de un 4% (2,9-11,7%)(16).
– Secuenciación de exoma completo (WES) o de genoma completo (WGS): permite el análisis de regiones codificantes para genes conocidos y la identificación de mutaciones causales en hasta un 40% de los pacientes con DI grave(11). En los últimos metanálisis, que incluyen 37 estudios de niños con RGD/DI, la utilidad diagnóstica fue mayor en WGS/WES con respecto a CGH-arrays(24), y hay autores que la recomiendan realizar en primer nivel ante RGD/DI de causa desconocida (incluyendo a ambos progenitores, cuando sea posible y con análisis de miembros de la familia afectados múltiples, cuando esté indicado)(25).
– Estudios moleculares tradicionales:
1. FISH (Hibridación in situ Fluorescente): marcaje con fluorescencia de una sonda de DNA complementaria a la región de interés. El diagnóstico mediante FISH será por sospecha clínica, en la que se seleccionará la sonda del locus específico de la región a estudiar. Alrededor de un 2-10% de los niños con RGD/DI de causa desconocida presentan reordenamientos submicroscópicos(26), como por ejemplo: 5p-, 4p-, CATCH-22…
2. MLPA (Multiple Ligation-dependant Probe Amplification): método cuantitativo muy fiable que se basa en la hibridación de sondas específicas a una región de interés del ADN y su posterior ligación y amplificación. Esta técnica permite evaluar simultáneamente hasta 45 secuencias concretas de ADN en un mismo ensayo, detectando cambios en la dosis (deleciones y/o duplicaciones) de uno o varios exones de un gen o de regiones específicas. Es de utilidad también ante una sospecha clínica con una región a estudio, y previamente solía aplicarse a las regiones subteloméricas.
Las nuevas técnicas genómicas (microarrays y WES/WGS) han reemplazado en la mayoría de los casos a estas técnicas clásicas, aunque en ocasiones, suelen ser complementarias y útiles en determinados casos.
c) Estudios metabólicos: los errores innatos del metabolismo (IEM) constituyen una clase de trastornos que involucran procesos bioquímicos o celulares, causados por la disfunción de una enzima normalmente codificada por un gen. Muchos de estos IEM afectan al sistema nervioso central. Durante la infancia, se presentan como trastorno del desarrollo intelectual, pero en la mayoría de los casos, asocian además otra sintomatología.
La realización de pruebas metabólicas para la búsqueda de IEM en niños con RGD/DI tiene una rentabilidad baja estimada en 0,2-4,6%, dependiendo de la aparición de signos y síntomas que hagan sospechar una enfermedad metabólica(16). La importancia del estudio de trastornos metabólicos radica en que en algunos se podrían iniciar tratamientos específicos(27). En 2018, se identificaron 89 IEM que presentan RGD/DI como característica fenotípica predominante y que disponen de una terapia disponible que mejoraría el pronóstico: trastorno de los aminoácidos (n=12), colesterol y ácidos biliares (n=3), creatina (n=3), ácidos grasos (n=1), transporte de glucosa a nivel cerebral (n=2), hiperhomocisteína (n=7), lisosomas (n=12), metales (n=5), mitocondrias (n=2), neurotransmisores (n=8), ácidos orgánicos (n=19), peroxisomas (n=1), purinas y pirimidinas (n=3), ciclo de la urea (n=8), vitaminas y cofactores (n=10).
Debido a la baja evidencia disponible de las pruebas más rentables en el estudio de niños con RGD/DI, existen muy diferentes protocolos de estudio(9,20,27-29) que varían dependiendo del centro y medios disponibles. En nuestro centro, debido a las pruebas endocrino-metabólicas realizadas como screening en el recién nacido (en las que se incluyen algunas enfermedades del metabolismo de los ácidos orgánicos, de aminoácidos y de ácidos grasos), así como la disponibilidad para realizar resonancia magnética cerebral con espectroscopia, sugerimos realizar estudio metabólico como pruebas de primer nivel (Tabla VII) en los siguientes casos:

• Signos y síntomas compatibles con IEM (Tabla VIII).

• No realizado screening metabólico al nacimiento.
En los demás casos, solicitaremos, como primer nivel, parámetros de un estudio metabólico básico (Tabla IX).

En el caso en que se realiza una secuenciación del exoma, sería recomendable que se incluyeran los genes responsables de los IEM “tratables”.
d) Neuroimagen:
• Resonancia magnética cerebral (RMC): en los últimos estudios, en niños con RGD/DI aislado, la realización de RMC encuentra hallazgos hasta en un 30-40%, la mayoría inespecíficas (más frecuentes, lesiones en sustancia blanca)(31), ayudando únicamente al diagnóstico etiológico entre un 0,2-2,2%, cuando no hay otros síntomas asociados(16).
La RMC se realizará como prueba de primer nivel ante niños con RGD/DI que asocien: historia de asfixia intraparto, microcefalia o macrocefalia, focalidad neurológica, crisis epilépticas, trastornos del tono o movimiento (espasticidad, ataxia o distonía) o regresión. Algunos autores también la recomiendan en cuadros clínicos moderados-graves30.
En ocasiones, especialmente si hay regresión o crisis epilépticas, podría ser útil usar técnicas de espectroscopia (RMS) que permiten determinar la concentración cerebral de determinados metabolitos como: la colina, lactato o creatina.
Diagnóstico diferencial
La mayoría de los trastornos del neurodesarrollo se pueden presentar de forma conjunta y no ser excluyentes con presentar RGD/DI, como veremos más adelante en el apartado de comorbilidades(2). De todas formas, según la edad del paciente, deberemos diferenciarlo de otras patologías:
• Menores de dos años: diferenciarlo de cuadros de hipotonía, cuando el problema es únicamente motor, o de TEA, cuando la afectación fundamental es en el lenguaje y en la interacción social recíproca.
• Edad escolar: habrá que diferenciarlo de trastornos específicos de aprendizaje como: el trastorno del lenguaje (afectación en área de comunicación expresiva y/o comprensiva), TDAH (cuando es un trastorno de la atención, inquietud motriz e impulsividad), dislexia (afectación en lectura o escritura) o trastorno de la coordinación motora (cuando el problema es fundamentalmente en la adquisición de habilidades motoras).
• Adolescentes: habrá que diferenciarlo de cuadros de TEA, TDAH o trastornos de conducta(31).
Comorbilidades
En el manejo de estos niños es fundamental identificar la presencia de comorbilidades, presentes hasta en el 50% de los casos y que, en muchas ocasiones, son la principal causa de alteración de la calidad de vida en niños y sus familiares.
Existe una amplia variabilidad clínica entre los pacientes con RGD/DI que hace “que ninguno de ellos sea igual”, debido fundamentalmente al diferente tipo de trastorno, edad, síntomas propios de la enfermedad subyacente y fundamentalmente a la frecuente y diversa presencia de comorbilidades, presente en, al menos, el 30-50% de los mismos. Es fundamental identificar los trastornos comórbidos que presentan, que generalmente son los síntomas que más limitan la calidad de vida del paciente y sus familiares, de cara a iniciar tratamientos específicos eficaces. En muchas ocasiones, estas comorbilidades aportan claves diagnósticas que nos permiten identificar la etiología del trastorno.
Las comorbilidades más frecuentes que deben descartarse en niños con RGD/DI son:
• Trastornos del sueño: un estudio en niños con RGD/DI en la Comunidad Valenciana, objetivó que un tercio de ellos sufría de insomnio, datos comparables a la prevalencia de alteraciones de sueño estimada en numerosos estudios que la sitúan como la comorbilidad más frecuente en esta población: 44% (rango de 10-86%)(32). Existe una relación bidireccional entre sueño y cognición, de forma que aquellos niños con RGD/DI tendrán más frecuentes problemas de sueño, mientras que aquellos con peor calidad de sueño presentarán más alteraciones cognitivas y de aprendizaje. Las alteraciones en el sueño que pueden presentar son numerosas, dependiendo de la gravedad y tipo de trastorno (p. ej.: el síndrome de apnea-hipopnea del sueño está presente en el 30-55% de los niños con síndrome de Down) y varía desde el insomnio de mantenimiento y conciliación hasta el insomnio conductual por mala adquisición y consolidación de hábitos nocturnos(33).
• Epilepsia: el 22% lo presentan (rango según diferentes estudios: 5-35%). Esta relación es más frecuente en determinados trastornos (p. ej., síndrome de Angelman o síndrome de Down) y cuanto mayor es el grado de discapacidad intelectual(34).
• Trastorno del espectro autista: el 10% de los niños con RGD/DI lo presentan (rango según diferentes estudios: 4-33%). Esta relación es más frecuente en determinadas patologías (p. ej.: complejo esclerosis tuberosa, síndrome de Angelman o X-frágil) y en aquellos trastornos cognitivos de mayor gravedad(35).
• Trastorno por déficit de atención e hiperactividad: el 9,5% lo presentan (rango según diferentes estudios: 5-30%). Ambos trastornos comparten unas bases fisiopatogénicas similares como son: las alteraciones en las funciones ejecutivas y en auto-regulación del aprendizaje, por lo que no son entidades excluyentes. De esta forma, un niño con RGD/DI y que además presente síntomas de inatención, inquietud motriz o impulsividad que le interfieren con su funcionamiento diario, puede beneficiarse de un tratamiento específico dirigido al TDAH(36).
• Entre el 30-50% de los niños con RGD/DI presentan también trastornos psiquiátricos. Es muy variable dependiendo del tipo y gravedad del trastorno, aunque aparentemente aquellos niños con mejor competencia social y habilidades sociales tienen menos probabilidades de presentar síntomas psiquiátricos(37). En muchas ocasiones, son difíciles de diagnosticar debido a la dificultad que tienen estos niños para expresarlos, con la consecuente demora en su diagnóstico y tratamiento. Los trastornos psiquiátricos más frecuentemente asociados son: trastorno oposicionista desafiante (12%), alteraciones del estado de ánimo (10%, sin olvidar que esta población es más vulnerable a sufrir situaciones de acoso), psicosis (2-10%), así como síntomas obsesivos-compulsivos o trastornos de la conducta alimentaria(31).
• Trastornos motores como la parálisis cerebral, presente en el 19% de los pacientes (rango según diferente estudios: 3-33%), o las estereotipias en el 5-25% de los pacientes que, en ocasiones, pueden ser muy características del trastorno subyacente, como la de lavado de manos del síndrome de Rett o la del autoabrazo del síndrome de Smith-Magenis(38).
• Déficits neurosensoriales, tanto visuales (2-26%) como auditivos 4,5% (rango: 0-7%).
• Es más frecuente en esta población que padezcan otros síntomas médicos como: alteraciones gastointestinales (eje intestino-cerebro), que comprenden desde el reflujo gastro-esofágico o el estreñimiento hasta la disfagia; dolor, que está presente en el 75-95% de los pacientes con parálisis cerebral en algún momento del desarrollo; o problemas pulmonares.
• En frecuentes ocasiones, dependiendo fundamentalmente del tipo de trastorno, deben buscarse y descartarse alteraciones en otros sistemas como: genito-urinario, cardíaco o endocrinológico (6%), así como malformaciones del sistema nervioso central asociadas (5%)(2).
Manejo y tratamiento
Desde Atención Primaria y desde Atención Especializada, debemos aconsejar a los familiares de los pacientes con RGD/DI el inicio de terapias precoces y eficaces, al mismo tiempo que desaconsejar tratamientos alternativos carentes de evidencia científica (pseudociencia).
Es fundamental un diagnóstico precoz que permita el inicio de una terapia de estimulación, que ha demostrado que mejorará el pronóstico evolutivo(36). Esta terapia no debe demorarse hasta conseguir un diagnóstico etiológico y debe realizarse por un equipo multidisciplinar. En nuestro medio, generalmente se desarrolla en los Servicios de Rehabilitación y Centros de Atención Temprana, que incluyen terapias de: fisioterapia, lenguaje, comunicación y terapia ocupacional, entre otras.
Asimismo, se considera fundamental una correcta identificación de las comorbilidades, para iniciar tratamientos específicos por equipos multidisciplinares, entre los que se incluyen: neurólogos, rehabilitadores, psiquiatras y, especialmente, pediatras de Atención Primaria.
En los últimos años, fundamentalmente gracias al desarrollo de la genética, hemos podido identificar genes causantes de muchos pacientes con RGD/DI. Todo esto está permitiendo el desarrollo de terapias, tanto génicas como de precisión, que permiten corregir el defecto fisiopatológico, con el objetivo de modificar la historia natural de la enfermedad(36).
Son muchos los tratamientos que han demostrado su eficacia, y estamos asistiendo al desarrollo esperanzador de una medicina de precisión.
Por este motivo, es necesario aconsejar a los pacientes y sus familias evitar las “terapias alternativas”, conociéndose así: aquellos tratamientos en los que no se ha evaluado su eficacia mediante una investigación científica basada en las reglas aceptadas de evidencia (método científico). Se basan en una simplificación de un trastorno con una fisiopatología compleja, por ejemplo: creer que un tratamiento optométrico que permita mejorar las alteraciones de motilidad ocular va a suponer una mejoría significativa en el problema de aprendizaje o de neurodesarrollo de un paciente con RGD/DI.
Los familiares de estos pacientes son una población vulnerable y es frecuente que acaben con el sentimiento de: “no importa probar algo nuevo, si tampoco te perjudica”, cuando la mayoría de estas terapias suponen un efecto nocivo para el paciente y sus familiares, sobre todo, a nivel emocional y económico, suponiendo además una mayor sobrecarga al cuidador. De este modo, debemos recomendar que estos pacientes eviten estas terapias, entre las que se incluyen aquellas que se basan en sustancias supuestamente terapéuticas (homeopatía, dietas específicas, inyección de células madre fuera de los ensayos clínicos rigurosos o administración no indicada de hormona de crecimiento) o aquellas que se fundamentan en el uso de técnicas específicas (optometría, electroterapia, lentes de colores, integración auditiva, métodos de patrones de movimiento u oxígeno hiperbárico, entre otros).
Regresión psicomotriz
La presencia de regresión psicomotriz en cualquier momento del desarrollo implica la necesidad de una valoración neurológica de forma precoz.
Es importante diferenciar en niños con RGD si el curso es progresivo, y esto se pone de manifiesto por tres datos en la historia clínica(39):
1. Existencia de un intervalo libre con desarrollo psicomotor normal.
2. Pérdida de adquisiciones ya alcanzadas (pérdida de habilidades motoras, del lenguaje, alteraciones de la visión, etc.), un hecho difícil de discernir en algunos casos.
3. Aparición de nuevos signos neurológicos (movimientos involuntarios, ataxia, crisis, piramidalismo, nistagmus, etc.
En estos casos, cuando se sospeche que el curso del retraso es progresivo, tendremos que pensar en encefalopatías heredodegenerativas (EHD), enfermedades hereditarias que afectan predominantemente al sistema nervioso central, cuyo curso natural es progresivo, y suelen manifestarse a lo largo de la infancia.
Ante una clínica de regresión, no hay que obviar enfermedades neurológicas como: tumores, infecciones (inmunodeficiencia adquirida, panencefalitis esclerosante subaguda, encefalitis), enfermedades autoinmunes (esclerosis múltiple, encefalitis autoinmunes), neuromusculares (atrofia muscular espinal), neurocutáneas, hidrocefalias o trastornos vasculares (como enfermedad de Moya-Moya), que cuando muestran un curso progresivo pueden simular una EHD.
El diagnóstico de estas enfermedades se basa en las claves clínicas de la anamnesis y exploración física; así como, ciertas pruebas complementarias (neuroimagen y estudios bioquímicos, enzimáticos, neurofisiológicos o moleculares) (Fig. 5).
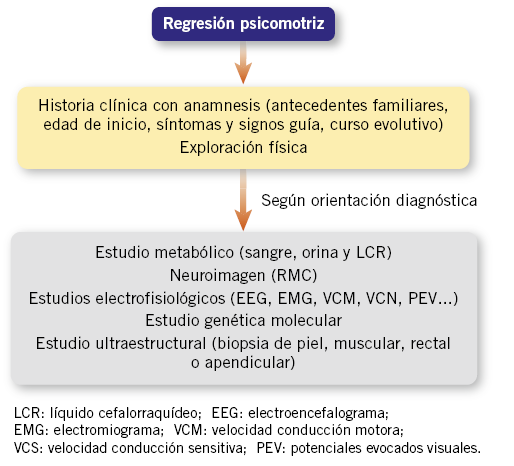
Figura 5. Algoritmo regresión psicomotriz.
Función del pediatra de Atención Primaria
El pediatra de Atención Primaria es la figura principal para el manejo de niños con RGD/DI por diferentes motivos(20):
• Debe ser el médico encargado de detectarlo y, en ese caso, derivarlo a un Servicio de Neuropediatría para su confirmación, estudio etiológico y de comorbilidades.
• Tratamiento de las muchas comorbilidades que presentan estos pacientes, como son: alteraciones de sueño, conducta, aprendizaje y de los múltiples problemas médicos asociados.
• Apoyo y detección de sobrecarga en el cuidador.
• Aconsejar sobre las terapias más eficaces, evitando las “terapias alternativas” pseudocientíficas.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.* American Psychiatric Association [APA] (2013) Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association.
2.*** Pallarés A. Retraso global del desarrollo. En: Narbona, Artigas-pallarés. Trastornos del neurodesarrollo.
3.** Shea S. Mental retardation in children ages 6 to 16. Semin Pediatr Neurol. 2006;13: 262-70.
4.* Strømme P, Valvatne K. Mental retardation in Norway: prevalence and sub-classification in a cohort of 30037 children born between 1980 and 1985. Acta Paediatr. 1998; 87: 291-6.
5.* Murphy CC, Yeargin-Allsopp M, Decoufle P, Drews CD. The administrative prevalence of mental retardation in 10-year-old children in metropolitan Atlanta, 1985 through 1987. Am J Public Health. 1995; 85: 319-23.
6.*** Narbona J, Schlumberger E. Retraso psicomotor. En: Delgado Rubio A, ed. Protocolos Diagnósticos y Terapéuticos en Pediatría: Genética-Dismorfología, Neurología. Asociación Española de Pediatría; 2008.
7.** Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016; 17: 9-18.
8.* Siu MT, Weksberg R. Epigenetics of autism spectrum disorder. Adv Exp Med Biol. 2017; 978: 63-90.
9.*** Moeschler JB, Shevell M; Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014; 134: 903-18.
10.* Han JY, Jang W, Park J, Kim M, Kim Y, Lee IG. Diagnostic Approach with Genetic Tests for Global Developmental Delay and/or Intellectual Disability: Single Tertiary Center Experience. Ann Hum Genet. 2019; 83: 115-23.
11.*** Blesson A, Cohen JS. Genetic Counseling in Neurodevelopmental Disorders. Cold Spring Harb Perspect Med. 2020; 10.
12.* Rosenthal ET, Biesecker LG, Biesecker BB. Parental attitudes toward a diagnosis in children with unidentified multiple congenital anomaly syndromes. AmJ Med Genet. 2001; 103: 106-14.
13.* López Pisón J, Mongue Galindo L. Evaluación y manejo del niño con retraso psicomotor. Rev Pediatr Aten Primaria Supl. 2011; 20: 131-44.
14.** Galbe Sánchez-Ventura, J. Detección precoz de los trastornos del desarrollo. En: Recomendaciones PrevInfad / PAPPS [en línea]. Actualizado en diciembre de 2017. Disponible en: http://previnfad.aepap.org/monografia/trastornos-desarrollo.
15.* Tabla de desarrollo de Haizea Llevant (0-5 años) Gobierno de la Rioja. Consejería de Salud, Consumo y Bienestar Social. Fernández Álvarez E. El desarrollo psicomotor de 1.702 niños de 0 a 24 meses de edad. (Tesis doctoral). Universidad de Barcelona 1988. “Tabla de desarrollo psicomotor”. En: Estudio Haizea Llevant. Servicio Central de Publicaciones. Gobierno vasco eds. Vitoria. 1991. Escala de desarrollo validada en población española.
16.** Fernández-Mayoralas D, Fernández-Jaén A. Detección y manejo del retraso psicomotor en la infancia. Pediatr Integral. 2015; XIX (8): 532-9.
17.** Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011; 77: 1629-35.
18.** Brown KA, Parikh S, Patel DR. Understanding basic concepts of developmental diagnosis in children. Transl Pediatr. 2020; 9: S9-S22.
19.** Jiménez-Gómez A, Standridge SM. A refined approach to evaluating global developmental delay for the international medical community. Pediatr Neurol. 2014; 51: 198-206.
20.** Mithyantha R, Kneen R, McCann E, Gladstone M. Current evidence-based recommendations on investigating children with global developmental delay. Arch Dis Child. 2017; 102: 1071-6.
21.* Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010; 86: 749-64.
22.* Miclea D, Peca L, Cuzmici Z, Pop IV. Genetic testing in patients with global developmental delay / intellectual disabilities. A review. Clujul Med. 2015; 88: 288-92.
23.* Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med (Lond). 2017; 17: 558-61.
24.** Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018; 3: 16.
25.*** Srivastava S, Cohen JS, Vernon H, Barañano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014; 76: 473-83.
26.* Bishop R. Applications of fluorescence in situ hybridization (FISH) in detecting genetic aberrations of medical significance. Biosci Horizons. 2010; 3: 85-95.
27.** Van Karnebeek CD, Stockler-Ipsiroglu S. Early identification of treatable inborn errors of metabolism in children with intellectual disability: The Treatable Intellectual Disability Endeavor protocol in British Columbia. Paediatr Child Health. 2014;19: 469-71.
28.* Silove N, Collins F, Ellaway C. Update on the investigation of children with delayed development. J Paediatr Child Health. 2013; 49: 519-25.
29.** O’Byrne JJ, Lynch SA, Treacy EP, King MD, Betts DR, Mayne PD, et al. Unexplained developmental delay/learning disability: guidelines for best practice protocol for first line assessment and genetic/metabolic/radiological investigations. Ir J Med Sci. 2016; 185: 241-8.
30.* Murias K, Moir A, Myers KA, Liu I, Wei XC. Systematic review of MRI findings in children with developmental delay or cognitive impairment. Brain Dev. 2017; 39: 644-55.
31.** Matson JL, Sipes M, Horovitz M, Worley JA, Shoemaker ME, Kozlowski AM. Behaviors and corresponding functions addressed via functional assessment. Res Dev Disabil. 2011; 32: 625-9.
32.* Vila T, Beseler B, Benac M, Cardona C, Pascual MJ, Lozano I, et al. Trastornos del sueño en niños y adolescentes con incapacidad psíquica. Análisis comparativo entre alumnos escolarizados en centros ordinarios y centros de educación especial de la Comunidad Valenciana. An Pediatr (Barc). 2008; 69: 335-41.
33.* Soto-Insuga V. Problemática del sueño en los niños con trastornos neurológicos. Rev Neurol. 2015; 60: 39-53.
34.** Van Ool JS, Snoeijen-Schouwenaars FM, Tan IY, Jurgen Schelhaas H, Aldenkamp AP, Hendriksen JGM. Challenging behavior in adults with epilepsy and intellectual disability: An analysis of epilepsy characteristics. Epilepsy Behav. 2018; 86: 72-8.
35.** Oeseburg B, Dijkstra GJ, Groothoff JW, Reijneveld S, Jansen DEM. Prevalence of Chronic Health Conditions in Children With Intellectual Disability: A Systematic Literature Review. 2011.
36.* Mulas F, Rojas M. Intellectual developmental disability overlapping with autism spectrum disorder and attention deficit-hyperactivity disorder. Medicina (B Aires). 2018; 78: 63-8.
37.* Glasson EJ, Buckley N, Chen W, Leonard H, Epstein A, Skoss R, Jacoby P, et al. Systematic Review and Meta-Analysis: Mental Health in Children With Neurogenetic Disorders Associated With Intellectual Disability . J Am Acad Child Adolesc Psychiatry. 2020.
38.* Whitney DG, Warschausky SA, Ng S, Hurvitz EA, Kamdar NS, Peterson MD, et al. Prevalence of Mental Health Disorders Among Adults With Cerebral Palsy: A Cross-sectional Analysis. Ann Intern Med. 2019; 171: 328-33.
39.*** González Gutiérrez-Solana L, García Peñas JJ, López Marín L, Lara J. Involución psicomotriz. Protocolos Neurología AEPED.
Bibliografía recomendada
- Pallarés A. Retraso global del desarrollo. En: Narbona, Artigas-Pallarés. Trastornos del neurodesarrollo.
Monografía fundamental para comprender una visión global de los síntomas, diagnóstico y evaluación de los niños con RGD/DI.
- Narbona J, Schlumberger E. Retraso psicomotor. En: Delgado Rubio A, ed. Protocolos Diagnósticos y Terapéuticos en Pediatría: Genética-Dismorfología, Neurología. Asociación Española de Pediatría; 2008.
Artículo que describe los distintos escenarios ante los que nos podemos encontrar, cuando realizamos el diagnóstico de retraso psicomotor en un niño.
- Moeschler JB, Shevell M; Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014; 134: 903-18.
Guía más reciente publicada sobre el manejo y evaluación de niños con RGD/DI.
- Srivastava S, Cohen JS, Vernon H, Barañano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014; 76: 473-83.
Artículo que muestra la importancia y complejidad de la genética dentro de la evaluación de niños con RGD/DI.
| Caso clínico |
|
Niña de 13 meses de edad, remitida por el pediatra de Atención Primaria por retraso psicomotor. Los padres describen un desarrollo psicomotor normal hasta los 7-8 meses de edad, momento en el que observan un estancamiento, con pérdida de bisílabos referenciales adquiridos, menor contacto visual e interacción, asociando también un descenso en el percentil de peso y de perímetro cefálico y mayor torpeza motora. Los ítems del desarrollo psicomotor son: inicio de sonrisa social al mes, sostén cefálico a los 3 meses, volteos a los 7-8 meses, sin realizar sedestación activa. En relación al lenguaje, bisílabos no referenciales a los 7 meses e inicio de bisílabos referenciales a los 11 meses, con aparente pérdida de estos “mamá” y “papá”. No presentaba antecedentes personales ni familiares de interés. En la exploración neurológica destacaba una microcefalia con un peso también menor de percentil 3, escaso contacto visual y leve retraso postural. Aunque se recomienda una valoración neurológica e inicio de estimulación, los familiares no vuelven a consultar hasta los 24 meses, cuando refieren aumento de las estereotipias y aparición de nuevas, esta vez impresionando de manierismos mano-mano y manierismos mano-boca, perdiendo el uso intencional de la mano. En la exploración, destaca aparición de temblor intencional distal bilateral y una marcha atáxica, así como una mayor microcefalia (percentil <3).
|
