 |
| Temas de FC |
A. Duat Rodríguez
Sección de Neurología del Hospital Universitario Niño Jesús, Madrid
| Resumen
La neurofibromatosis tipo 1 (NF1), también conocida como enfermedad de von Recklinghausen, es un trastorno autosómico dominante causado por mutaciones en la línea germinal del gen NF1, un gen de supresión tumoral que codifica una proteína dentro de la vía de señalización de Ras. La NF1 es la enfermedad neurocutánea más frecuente, caracterizada por la presencia de: máculas café con leche, efélides axilares e inguinales, nódulos de Lisch en el iris, neurofibromas cutáneos y un riesgo superior de desarrollar tumores. Es una enfermedad multisistémica, cuya expresión clínica es heterogénea y su progresión impredecible. Se diagnostica por la existencia de ciertos criterios clínicos. Algunas características de NF1 pueden estar presentes al nacer, pero la mayoría de las manifestaciones aparecerán con la edad. Existe una dificultad para el diagnóstico clínico en edades tempranas y las pruebas moleculares para identificar mutaciones causales en el gen NF1 son complejas. Se deben considerar otros diagnósticos en pacientes cuyos únicos síntomas son manchas café con leche. En la infancia, su principal diagnóstico diferencial, son las otras rasopatías, grupo de trastornos que se caracterizan por una desregulación en la vía de señalización Ras y un fenotipo parecido al síndrome de Noonan. Precisa un seguimiento multidisciplinar, donde los pediatras pueden desempeñar un importante papel mediante la identificación de signos que pueden llevar a un diagnóstico y con una supervisión adecuada. |
| Abstract
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is an autosomal dominant disorder caused by germline mutations in the NF1 tumour suppressor gene which encodes a protein involved in the Ras signaling pathway. NF1 is the most common neurocutaneous disease, characterised by the presence of café-au-lait macules, axillary and inguinal freckling, iris Lisch nodules, cutaneous neurofibromas and a higher-than-average risk of tumour development. It is a multisystemic disorder, with heterogeneous clinical expression and unpredictable progression. It is diagnosed by the existence of certain clinical criteria. Some features of NF1 can be present at birth, but most manifestations emerge with age. It continues to be difficult to make a clinical diagnosis at early ages and molecular testing to identify causative mutations in NF1 is complex. Other diagnoses should be considered in patients whose only symptoms are café- au-lait spots. Other RASopathies, a group of disorders that are characterized by constitutional dysregulation of the Ras signaling pathway and a phenotype resembling Noonan syndrome, are the main differential diagnosis in childhood. |
Palabras clave: Neurofibromatosis tipo 1; Gen NF1; Glioma de vía óptica; Neurofibromas; Ras/MAPK.
Key words: Neurofibromatosis type 1; NF1 gene; Optic glioma; Neurofibromas; Ras/MAPK.
Pediatr Integral 2020; XXIV (6): 334 – 341
Neurofibromatosis tipo 1
Introducción
La neurofibromatosis tipo 1 (NF1) es una enfermedad multisistémica caracterizada por lesiones pigmentarias, neurofibromas y mayor riesgo de desarrollo tumoral, cuya morbimortalidad está asociada a las complicaciones multisistémicas que pueden aparecer a lo largo de su evolución.
La neurofibromatosis tipo 1 (NF1), también llamada enfermedad de von Recklinghausen, es la enfermedad neurocutánea más frecuente, un grupo heterogéneo de más de 50 enfermedades, generalmente hereditarias, con anomalías en el sistema nervioso y en la piel(1,2). Las enfermedades neurocutáneas son fenotípica y genéticamente diferentes, pero tienen un origen común en el tejido ectodérmico primitivo que da lugar a la piel y al sistema nervioso(1). La NF1 es una enfermedad multisistémica, que se caracteriza principalmente por la presencia de: manchas café con leche (MCCL), efélides axilares e inguinales, nódulos de Lisch en el iris, neurofibromas cutáneos y mayor riesgo de desarrollo tumoral. No obstante tiene una marcada variabilidad clínica y, aunque algunos rasgos se presentan al nacimiento, otros van a manifestarse con la edad(3,4).
Epidemiología
Se calcula que la NF1 tiene una incidencia de 1 cada 2.500 o 3.500 recién nacidos, con una distribución similar en cuanto al sexo(5-7).
Fisiopatología
Se transmite por herencia autosómica dominante, aunque el 50% de casos son esporádicos. Su gen responsable (NF1) se localiza en el cromosoma 17q11.2 y sintetiza una proteína denominada neurofibromina.
Presenta una penetrancia cercana al 100% en la edad adulta(4). Por ello, no se salta generaciones y, en casos familiares, la madre o el padre deben mostrar algún rasgo característico de esta enfermedad. No hay pacientes asintomáticos, no obstante, algunos pueden cumplir únicamente las características cutáneas sin presentar grandes complicaciones a lo largo de su vida. La expresividad es muy variable, incluso en la misma familia, por lo que los rasgos y complicaciones pueden ser distintos entre los familiares(4,8). La tasa de mutación del gen NF1 es muy alta y más del 50% no tienen historia familiar de NF1 y corresponden a mutaciones de novo(3).
El gen NF1 sintetiza una proteína denominada neurofibromina. Esta se expresa de forma ubicua en el organismo, por lo que las complicaciones pueden afectar a prácticamente cualquier parte del cuerpo. La neurofibromina inhibe la vía enzimática de la RAS/MAPK (mitogen activated protein kinase pathway), implicada en funciones de crecimiento celular, proliferación diferenciación y apoptosis(1). En la NF1, esta vía se activa de una manera incontrolada. La NF1 forma parte así, junto a otras enfermedades que regulan esta vía metabólica de las llamadas rasopatías. Estas enfermedades aun siendo diferentes, comparten entre sí ciertos rasgos fenotípicos como: dismorfia facial, alteraciones cardiacas, dermatológicas, gastrointestinales, alteraciones del aprendizaje y predisposición genética al desarrollo de tumores(9).
Existen formas de NF1 restringidas únicamente a un segmento corporal, se trata de NF1 segmentarias o en mosaico, producidas por una mutación somática durante el desarrollo fetal(8,10,11).
Clínica
Las manifestaciones clínicas de la NF1 son extremadamente variables, tanto en función del individuo como de la edad (Fig. 1).

Figura 1. Desarrollo de hallazgos clínicos de neurofibromatosis tipo 1 según la edad en años.
El patrón clínico característico está presente en la gran mayoría de los adultos, aunque en diferente proporción y consiste en: MCCL, efélides axilares e inguinales, neurofibromas periféricos y nódulos de Lisch. Las complicaciones severas afectan a una minoría de pacientes, sin embargo, es lo que va a condicionar la morbimortalidad de esta enfermedad(3).
Manifestaciones no tumorales
Rasgos fenotípicos menores. Existe un grupo de rasgos físicos menores que se encuentran en una gran proporción de pacientes, pero no se incluyen en los criterios diagnósticos. Son hallazgos característicos de la enfermedad, pero de menor importancia, que comparten con otras rasopatías. Descrito inicialmente como una variante llamada neurofibromatosis-Noonan, se observó que está causada mayoritariamente por mutaciones en el gen NF1(12). Los rasgos físicos más habituales son: macrocefalia, talla baja, rasgos faciales sutiles (hipertelorismo con fisuras palpebrales antimongoloides, epicantus, ptosis y orejas en rotación posterior con hélix engrosada) y anomalías torácicas (pectus carinatum y excavatum). La talla baja es común, pero pocos individuos con NF1 tienen una altura de más de 3 desviaciones estándar por debajo de la media.
Manifestaciones cutáneas. Las manchas café con leche (MCCL) se presentan con una frecuencia cercana al 100% (Fig. 2).
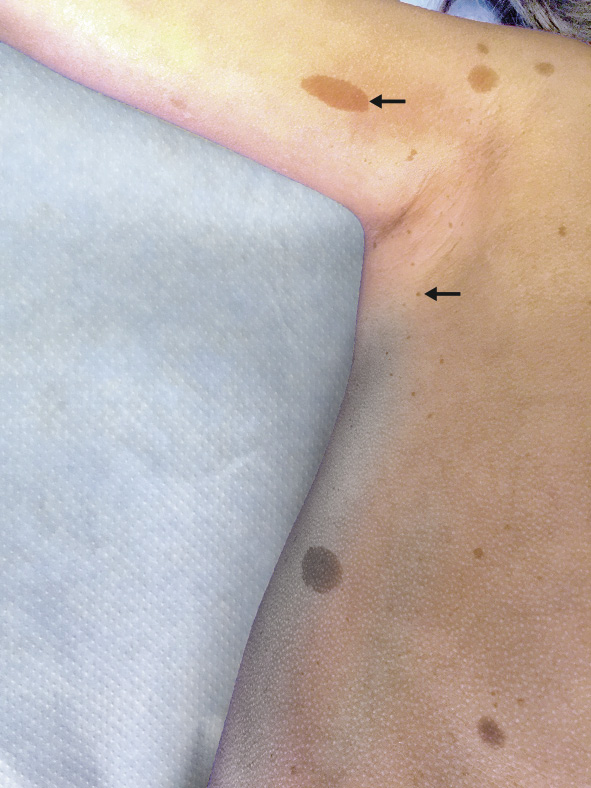
Figura 2. Efélides axilares y manchas café con leche en paciente con neurofibromatosis tipo 1.
Las típicas son de tono homogéneo y contorno liso y regular(10). Alrededor del 10-20% de la población general de raza blanca tiene entre 1 y 3 MCCL, pero la presencia de más de 6 es muy sugestiva de NF1. No existe asociación entre el número de MCCL y la gravedad de la NF1(10). Las efélides o pecas son MCCL de pequeño tamaño de localización característica en pliegues axilares e inguinales que se desarrollan durante la infancia, siendo rara su presentación antes de los 2 años. También pueden observarse en otras localizaciones como la cara, el cuello y el tronco(10). Los nevus anémicos están presentes hasta en el 50%, son máculas pálidas de contorno polilobulado muy sutiles, generalmente en la región preesternal, que se ponen de manifiesto al frotar ligeramente la zona(13,14). Los xantogranulomas juveniles consisten en una pápula anaranjada, solitaria o múltiple, en cualquier localización corporal. Suele presentarse en los dos primeros años de vida en alrededor de 6-8% de los pacientes y regresa al cabo de pocos años(3,14). Otros hallazgos cutáneos como: el prurito, la hiperpigmentación generalizada, la presencia de máculas hipocrómicas o la suavidad de la piel son fácilmente constatables en los pacientes con NF1(14).
Manifestaciones esqueléticas. Las lesiones óseas características de la NF1 son: displasias de huesos largos, displasia del esfenoides, displasias vertebrales y pseudoartrosis. Son congénitas y aparecen solo en una minoría de pacientes afectos de NF1 (7%)(3). Presentan predilección por algunos huesos y su presentación es irregular (unilateral o con la participación solo de algunas vértebras). Por lo general, se presenta en la infancia con arqueamiento de la parte inferior de la pierna. Algunas lesiones tienen una naturaleza progresiva y, en ocasiones, se presentan de forma conjunta e indistinguible de otras lesiones adyacentes, como neurofibromas o lesiones de los tejidos blandos(5). Además, con frecuencia, los pacientes con NF1 sufren escoliosis. Es más frecuente que afecte la región cervical o torácica superior. Generalmente es discreta y poco evolutiva, pero también puede ser distrófica. Esta ocurre a una edad más temprana (6 a 8 años) con progresión rápida y difícil tratamiento, secundaria a neurofibromas plexiformes subyacentes que infiltran las vértebras o acompañarse de diferentes anomalías vertebrales, masas, subluxaciones, dislocaciones o destrucción ósea(5).
Manifestaciones oculares. Los nódulos de Lisch son hamartomas del iris pigmentados y asintomáticos, que aumentan en número y tamaño con la edad, y son observados fácilmente con lámpara de hendidura. Las anormalidades coroideas están presentes en prácticamente el 100% de pacientes con NF1, aunque su presencia es algo inferior en niños (71-78%). No son fáciles de detectar, pues precisan de tomografía de coherencia óptica con luz infrarroja y son difíciles de identificar en niños menores de dos años por falta de colaboración(10). Son proliferaciones concéntricas de células de Schwann y al igual que los nódulos de Lisch, tampoco son patognomónicos de NF1, pudiendo presentarse en sujetos sanos. Un 0,7% de niños con NF1 tienen glaucoma congénito(12).
Manifestaciones neurológicas. Aunque la frecuencia de discapacidad intelectual es solo ligeramente superior a la población general, las alteraciones del aprendizaje, el trastorno por déficit de atención e hiperactividad (TDAH) y el fracaso escolar, suponen el mayor problema evolutivo de los niños con NF1, afectando a más de un 50% de los casos. El perfil neuropsicológico de los niños con NF1 se caracteriza por déficits en: habilidades de percepción (viso-espaciales y viso-perceptivas), en el funcionamiento ejecutivo (planificación y formación de concepto abstracto) y la atención (sostenida y mantenida)(5). Problemas con la función motora, la función ejecutiva, la memoria y el lenguaje también son frecuentes. Aunque el trastorno de espectro autista se estima en estudios epidemiológicos alrededor de un 4%, hasta un 30% tienen problemas en el funcionamiento social(12), presentando síntomas que puntúan en escalas cuantitativas de rasgos o síntomas autistas, con una distribución más severa en el 13,2%. Las malformaciones del sistema nervioso central (SNC) están presentes en aproximadamente un 4-5% de los pacientes con NF1(3). Las más comunes incluyen: la malformación de Arnold Chiari tipo I, las ectasias durales y los mielomeningoceles e hidrocefalia. Además, gran variedad de lesiones cerebrovasculares han sido descritas en la NF1 como: estenosis, aneurismas o síndrome de moyamoya(15). Las hiperseñales en las secuencias T2 de la resonancia magnética (RM) craneal o UBOs (unidentified bright objects), corresponden a áreas de vacuolización intramielínica (Fig. 3).
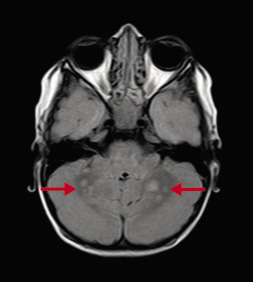
Figura 3. Resonancia craneal. Imagen axial FLAIR ponderadas en T2, corte en fosa posterior. Se observan áreas de señal hiperintensa bilaterales en cerebelo, compatibles con cambios de vacuolización mielínica.
Constituyen un patrón característico que se presenta más comúnmente en: ganglios basales (preferentemente en núcleos pálidos), tálamos, cerebelo, en el tronco cerebral y en la sustancia blanca subcortical. Aparecen en el 80% en niños a partir de los dos o tres años de edad(3). En ocasiones, hay que realizar el diagnóstico diferencial de estas con tumores, principalmente con astrocitomas. No se asocian con déficit focales y, aunque se ha intentado establecer una correlación con las alteraciones cognitivas, no existen estudios definitivos a este respecto(12).
La epilepsia se presenta en aproximadamente un 4-9% de los pacientes con NF1(3,16). Con mayor frecuencia son crisis focales, pero solo puntualmente se relacionan con algún tumor o malformación que lo justifique. No se ha demostrado que exista una relación de las hiperseñales en T2 características de la NF1 con la epileptogénesis(16). Algunos pacientes con NF1 presentan síndromes electro-clínicos bien definidos, principalmente, epilepsia ausencia infantil y síndrome de West(3). Las convulsiones suelen controlarse bien con tratamiento antiepiléptico y, aunque existen casos de fármaco-resistencia, suelen ser de mejor pronóstico que otras epilepsias neurogenéticas(16).
También presentan una mayor prevalencia de: trastornos del sueño(17), retrasos motores(3), polineuropatia difusa(12) y cefaleas, incluida migraña y cefalea secundaria(3,15).
Manifestaciones tumorales
Neurofibromas. La NF1 se asocia con una gran cantidad de tumores benignos y malignos. Entre ellos, los neurofibromas son los más frecuentes. Los tumores son probablemente la complicación más temida, porque incluso tumores de estirpe benigna, según el tamaño y la localización, pueden causar grandes complicaciones.
Los neurofibromas son tumores benignos que derivan de la vaina neural de los nervios periféricos. Se calcula que aproximadamente el 38% de los pacientes presenta uno o varios tipos de neurofibroma antes de los 17 años(10). La clasificación de los neurofibromas varía según los autores y, a menudo, resulta confusa, porque mezcla términos clínicos e histológicos, y porque algunos neurofibromas presentan un componente mixto. Los podemos clasificar en dos grandes grupos(10):
1. Neurofibromas superficiales (palpables): cutáneos y subcutáneos.
2. Neurofibromas profundos (internos): neurofibromatosis visceral, neurofibromatosis espinal o neurofibromatosis orbitaria.
Tanto los neurofibromas subcutáneos como los profundos, suelen corresponder histológicamente a neurofibromas plexiformes(10). En algunos casos, la presencia de los neurofibromas es la característica que predomina en el cuadro clínico, siendo referida como una entidad por su agresividad. Es el caso de la neurofibromatosis espinal, en la que existen neurofibromas profundos bilaterales en todas las raíces espinales con o sin otros estigmas de NF1, y la neurofibromatosis orbitaria terriblemente desfigurante, en la que el neurofibroma ocupa toda la órbita, invade los músculos orbitarios y se asocia a: exoftalmos, asimetría ocular, deformidad temporal, displasia del esfenoides y herniación del lóbulo temporal. La transformación maligna de un neurofibroma en un tumor maligno de la vaina nerviosa periférica o neurofibrosarcoma es rara en la infancia. Los síntomas de alarma incluyen: aumento brusco de tamaño, cambio de textura (aumento de la consistencia), dolor intenso o incoercible y sintomatología neurológica distinta de la habitual(14).
Gliomas de la vía óptica (GVO). Se trata del tumor más frecuente del SNC, suelen ser astrocitomas pilociticos de bajo grado y aparecen en cualquier zona a lo largo de la vía visual afectando hasta un 30% de los niños con NF1(3) (Fig. 4).
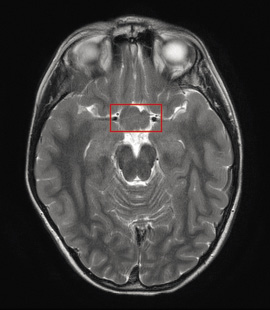
Figura 4. Resonancia craneal. Imagen axial FLAIR. Lesión en el quiasma óptico, compatible con glioma de vía óptica.
La mayoría cursa de forma asintomática con una evolución estable e incluso, en algunos casos, se ha descrito regresión espontánea(18). Sin embargo, la evolución natural del GVO en la NF1 es impredecible y aproximadamente un 7-17% de los casos serán sintomáticos. Los casos sintomáticos generalmente, se diagnostican antes de los 6 años de edad(18). La sintomatología incluye: disminución de agudeza visual uni o bilateral, proptosis, estrabismo, nistagmo y precocidad sexual o hidrocefalia (cefalea y vómitos) si el tumor se origina en el quiasma o comprime estructuras vecinas(3,19). Aquellos GVO de localización posterior (quiasma, tractos y radiaciones ópticas), o con presentación antes de los 2 años o después de los 10 años, o con una proptosis rápidamente progresiva se consideran de peor pronóstico, con un peor resultado visual(20).
Otros gliomas. Aunque el GVO es el más frecuente, también hay una mayor incidencia (entre un 3-8%) de gliomas en otra localización, cerebral o espinal (principalmente en mesencéfalo)(3,19). Suelen presentarse a mayor edad que los GVO, alrededor de los 7 años(19). Pueden cursar de forma asintomática, presentándose como un hallazgo en la neuroimagen o presentar: cefalea, náuseas, vómitos, alteración de pares craneales (disfagia, disartria, desviación de la úvula…) o ataxia, precisando tratamiento(19).
Otras neoplasias frecuentes incluyen: neurofibrosarcomas y tumores neuroepiteliales disembrioplásicos, rabdomiosarcoma, el neuroblastoma, el tumor de Wilms, el feocromocitoma y las leucemias mieloides(12). También se han descrito segundas neoplasias tras tratamientos con radioterapia y quimioterapia.
Otras manifestaciones
Alteraciones en el crecimiento, pubertad, osteoporosis o alteraciones en el sistema cardiovascular (preferentemente son: displasias arteriales fibromusculares de aorta, arterias mesentéricas, arterias renales o cerebrales y estenosis de la válvula pulmonar). La hipertensión arterial que puede producirse por causa idiopática, puede ser debida a la presencia de un feocromocitoma, o secundaria a una vasculopatía renal o aórtica. Obviamente, la posibilidad de que los neurofibromas se presenten en cualquier localización y de que se hayan descrito casos de tumores de afectación endocrinológica, pulmonar o del aparato urinario, e incluso la presencia de metástasis, amplía el potencial de afectación multisistémica de esta enfermedad.
Diagnóstico
El diagnóstico de NF1 generalmente se basa en criterios clínicos; sin embargo, en la infancia no siempre es fácil, dado que muchas de las manifestaciones clínicas van apareciendo con la edad. También puede realizarse el diagnóstico molecular, aunque por su complejidad suele requerir diferentes técnicas(3,12).
El Instituto Nacional de Salud de EE.UU. definió en 1988, un consenso de criterios clínicos para el diagnóstico de NF1 (Tabla I).

Estos criterios están aún vigentes. Tras valorar el número y el tamaño de las MCCL, hay que buscar otros hallazgos clínicos que nos permitan diagnosticar la NF1 con certeza, algo que puede ser difícil en los niños más pequeños, ya que si bien los criterios clínicos son muy específicos y sensibles a partir de los 8 años de vida, apenas el 45% de los niños menores de 1 año los cumplen(7,10). Durante estos años, algunos autores han considerado que otros criterios diagnósticos deberían admitirse, debido a la frecuencia con la que se presentan algunos hallazgos en estos pacientes. Es el caso de la presencia de hiperseñales en secuencias T2 en la RM craneal, que se presentan hasta en un 80-90% según estudios(3), o de las anormalidades coroideas, cuya frecuencia es cercana al 100% en adultos. Por ahora, se ha considerado que no es necesario introducir ni cambiar los criterios diagnósticos(12).
Los lactantes suelen acudir por MCCL, pero habitualmente si no presentan antecedentes familiares, no cumplen ningún otro criterio para su diagnóstico. Por eso, consideramos de especial relevancia detectar la presencia de otras manifestaciones clínicas que pueden ya estar presentes a esa edad, en que aún no cumplen criterios clínicos y no disponemos del estudio genético. De este modo, ocasionalmente, en algunos pacientes con NF1 podemos observar extensas máculas hiperpigmentadas de bordes irregulares con o sin hipertricosis que se confunden con MCCL gigantes, y que pueden corresponder con los denominados neurofibromas plexiformes superficiales congénitos, cuya confirmación nos lleva al diagnóstico en edades tempranas(7). Otros hallazgos clínicos frecuentes en menores de dos años, aún sin ser criterios diagnósticos, pueden aumentar la sospecha diagnóstica como: los nevus anémicos(13), el xantogranuloma juvenil o la presencia de un fenotipo de rasopatía (macrocefalia, hipertelorismo)(7).
La confirmación molecular del diagnóstico clínico puede llegar al diagnóstico hasta en un 95%. Sin embargo, es complicado por el tamaño del gen NF1 y la existencia de miles de mutaciones. Por este motivo, los protocolos para el diagnóstico genético suelen combinar varias técnicas. El análisis de secuencia de ADN genómico NF1 (ADNg) suele combinarse con el análisis de ADNc (ADN complementario, copiado de ARNm), ya que existe un 30% de variantes patogénicas que afectan al splicing. Además, se asocia un análisis de deleción/duplicación dirigido a genes por diferentes métodos de PCR o MLPA o microarray dirigidos con el fin de detectar deleciones o duplicaciones intragénicas(12). No en todos los centros la confirmación molecular resulta accesible y, en ocasiones, el resultado se demora más de un año. Además, hay que añadir el hecho de que aún no se ha establecido una correlación fenotipo-genotipo clara, salvo escasas excepciones como en las deleciones que se relacionan con un fenotipo más grave(12,4). No se puede conocer el alcance clínico en un individuo que porte determinada mutación.
Diagnóstico diferencial
Además de considerar otras enfermedades que presenten MCCL o causen tumores, en la infancia debemos establecer el diagnóstico diferencial especialmente con otras rasopatías por su superposición geno-fenotípica(3) (Tabla II).

Queremos destacar especialmente el síndrome de Legius, previamente conocido como NF1-Like que se debe a mutaciones en el gen SPRED1. Clínicamente, pueden presentar: MCCL, pecas axilares y/o inguinales, fenotipo de rasopatía y problemas de aprendizaje. Todo ello puede observarse indistintamente en ambas enfermedades. A pesar de no estar descritos nódulos de Lisch, neurofibromas u otros estigmas de NF1, el hecho de cumplir los criterios diagnósticos de MCCL y efélides, puede llevar al diagnóstico erróneo de una NF1(4).
Tratamiento
La NF1 es una enfermedad multisistémica que precisa de una atención multidisciplinar. Una supervisión periódica ayuda a una detección precoz de las complicaciones y a un tratamiento sintomático de las mismas, si fuera preciso. Recientemente, la FDA (Food and Drug Administration) ha aprobado el uso de Selumetinib en pacientes mayores de 2 años con neurofibromas plexiformes sintomáticos inoperables.
Los pacientes pueden requerir tratamientos farmacológicos, como metilfenidato por TDAH, antihipertensivos, así como cirugías por diversos motivos como: escoliosis, neurofibromas, hidrocefalia o tumores. En los casos de neurofibromas en las raíces espinales, la indicación de cirugía se basa en la progresión de los síntomas y déficits neurológicos.
Basado en el conocimiento creciente en la última década de las rasopatías, se están diseñando nuevas estrategias terapéuticas antineoplásicas con mecanismos de acción selectivos(9), los resultados más alentadores son los obtenidos con inhibidores de MEK, como el selumetinib en neurofibromas plexiformes. También se han realizado estudios en tumores malignos de la vaina del nervio periférico, en la restauración ósea de la pseudoartrosis y en el GVO con buenos resultados(6,9). La activación incontrolada de las vías RAS lleva asociada una disfunción GABAérgica implicada en los déficits cognitivos en la NF1(5), para los cuales también se están intentando encontrar fármacos eficaces. Actualmente, existen estudios con lamotrigina, tras no demostrarse la eficacia de la lovastatina(6).
El tratamiento del GVO en los pacientes con NF1 en general, es conservador, pues no hay que olvidar que en la mayoría de pacientes el GVO cursará de forma asintomática. La combinación de una disminución en la agudeza y campo visual junto con la presencia de cambios progresivos en la neuroimagen, son los principales factores decisivos para iniciar tratamiento(20). La quimioterapia (vincristina y carboplatino) es el tratamiento de primera línea, produce una regresión del tumor y, aunque mejora la visión en muchos niños, en otros casos no se produce una recuperación de la agudeza visual. El tratamiento con radioterapia no se recomienda en niños con NF1, debido al alto riesgo de tumores secundarios. El tratamiento quirúrgico está únicamente indicado para tumores orbitales de gran tamaño sin visión útil, con proptosis desfigurante o exposición corneal, o para tratar la hidrocefalia obstructiva o compresiones(20).
Prevención
Aunque la mitad de los individuos con NF1 presentan una forma leve, la severidad resulta muy variable y, por tanto, casi imposible de predecir. La gran variabilidad clínica, los riesgos tumorales y la evolución impredecible imponen el seguimiento anual de los niños asintomáticos(12).
En la evaluación médica, además del fenotipo y examen de la piel, se valora: el crecimiento, la tensión arterial, se realiza examen óseo y neurológico, valoración de la maduración sexual y cribado visual. Es importante supervisar los hitos del desarrollo y alteraciones del neurodesarrollo. Dada la frecuencia del GVO, la edad de presentación y de progresión, deberían realizar revisiones oftalmológicas hasta la pubertad, anuales hasta los 8 años y, posteriormente, bienales(20). La tomografía de coherencia óptica resulta útil para la detección de GVO. Para la supervisión periódica de los pacientes, hay que recordar que las manifestaciones y complicaciones de la NF1 son edad dependiente(4,12).
La detección de los tumores más frecuentes por técnicas de imagen en pacientes con NF1 asintomáticos es controvertida. La realización de una RM craneal programada para la detección del GVO en pacientes asintomáticos, no está aconsejada por la mayoría de autores, pues la identificación incidental de estos tumores rara vez cambian el manejo clínico(19) y conlleva dudas en la repetición posterior de pruebas de control(12). Sin embargo, en la práctica clínica es habitual su realización, pues alrededor de los 2 años de edad, la colaboración para realizar una exploración oftalmológica minuciosa que descarte sintomatología es limitada(3). De forma adicional a esa edad, la detección de un GVO en RM craneal resulta útil como herramienta de diagnóstico clínico, así como para apoyar la sospecha clínica con la presencia de las hiperseñales en las secuencias T2, a pesar de no ser diagnósticas ni patognomónicas. La RM craneal también permite el diagnóstico de complicaciones relacionadas con esta enfermedad, gliomas de otra localización, vasculopatía o malformaciones(3). Al igual que ocurre con la RM craneal, tampoco está establecida una RM corporal en pacientes asintomáticos para la detección de neurofibromas internos. Sin embargo, son muchos los clínicos que la plantean en la adolescencia o al inicio de la edad adulta(12). Tampoco los intervalos para realizar controles de imagen y vigilancia oftalmológica están bien establecidos cuando existe un GVO. En general, se considera inicialmente trimestral, posteriormente semestral y se espacia a anual a los dos años(20). Podríamos considerar también factores pronósticos por edad o localización del GVO.
Los pacientes con NF1 deben recibir consejo genético, en que se explique y asesore sobre la herencia que determina la enfermedad y el riesgo de recurrencia en cada familiar, siendo del 50% en el individuo afecto. Las personas con NF1 en mosaico tienen un riesgo mucho menor (2,5%)(14) de transmitir una NF1 generalizada a su descendencia. En ausencia de hallazgos clínicos de NF1 en los padres, el porcentaje de tener otro hijo con NF1 es muy bajo, de <1%. Para poder ofrecer el diagnóstico preimplantacional o prenatal se precisa conocer la mutación(12).
Funciones del pediatra de Atención Primaria
• Derivación precoz a atención especializada de pacientes sugestivos de NF1.
• Muchos de los niños con NF1 presentarán síntomas cutáneos sin otra sintomatología llamativa y su seguimiento en la atención especializada será anual. El pediatra debe ofrecer un buen manejo clínico, con conocimiento de las manifestaciones clínicas que pueden surgir y aconsejar adelantar la revisión cuando lo considere oportuno.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1.*** Rosser T. Neurocutaneous Disorders. Continuum (Minneap Minn). 2018; 24: 96-129.
2. Fernández-Fernández MA, Morillo Rojas MD. Los síndromes neuro-cutáneos. Pediatr Integral. 2015; XIX: 565-71.
3.*** Duat Rodríguez A, Martos Moreno GA, Martín Santo-Domingo Y, Hernández Martín A, Espejo-Saavedra Roca JM, Ruiz-Falco Rojas ML, et al. Phenotypic and genetic features in neurofibromatosis type 1 in children. An Pediatr (Barc). 2015; 83: 173-82.
4.*** Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019; 143:e20190660. doi: 10.1542/peds.2019-0660.
5. Anderson JL, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol. 2015; 132: 75-86.
6. Ferner RE, Bakker A, Elgersma Y, Evans DGR, Giovannini M, Legius E, et al. From process to progress-2017 International Conference on Neurofibromatosis 1, Neurofibromatosis 2 and Schwannomatosis. Am J Med Genet A. 2019; 179: 1098-106.
7. Bergqvist C, Servy A, Valeyrie-Allanore L, Ferkal S, Combemale P, Wolkenstein P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020; 15: 37.
8.** Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017; 3: 17004.
9. Duat-Rodríguez A, Hernández-Martín A. Update on the treatment of RASopathies. Rev Neurol. 2017; 64: S13-S7.
10.** Hernández-Martín A, Duat-Rodríguez A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots, Freckling, and Neurofibromas. An Update. Part I. Dermatological Clinical Criteria Diagnostic of the Disease. Actas Dermosifiliogr. 2016; 107: 454-64.
11. Vázquez-Osorio I, Duat-Rodríguez A, García-Martínez FJ, Torrelo A, Noguera-Morel L, Hernández-Martín A. Cutaneous and Systemic Findings in Mosaic Neurofibromatosis Type 1. Pediatr Dermatol. 2017; 34: 271-6.
12. Friedman D. Neurofibromatosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews. Updated 2019.
13. Hernández-Martín A, García-Martínez FJ, Duat A, López-Martín I, Noguera-Morel L, Torrelo A. Nevus Anemicus: A Distinctive Cutaneous Finding in Neurofibromatosis Type 1. Pediatr Dermatol. 2015; 32: 342-7.
14.** Hernández-Martín A, Duat-Rodríguez A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots and Freckling. Part II. Other Skin Manifestations Characteristic of NF1. NF1 and Cancer. Actas Dermosifiliogr. 2016; 107: 465-73.
15. Duat-Rodríguez A, Carceller Lechon F, López Pino MA, Rodríguez Fernández C, González-Gutiérrez-Solana L. Neurofibromatosis type 1 associated with moyamoya syndrome in children. Pediatr Neurol. 2014; 50: 96-8.
16. Serdaroglu E, Konuskan B, Karli Oguz K, Gurler G, Yalnizoglu D, Anlar B. Epilepsy in neurofibromatosis type 1: Diffuse cerebral dysfunction? Epilepsy Behav. 2019; 98: 6-9.
17. Marana Pérez AI, Duat Rodríguez A, Soto Insuga V, Domínguez Carral J, Puertas Martín V, González Gutiérrez Solana L. Prevalence of sleep disorders in patients with neurofibromatosis type 1. Neurologia. 2014. 30: 561-5.
18. Oh KS, Hung J, Robertson PL, Garton HJ, Muraszko KM, Sandler HM, et al. Outcomes of multidisciplinary management in pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys. 2011; 81: e481-8.
19. Mahdi J, Shah AC, Sato A, Morris SM, McKinstry RC, Listernick R, et al. A multi-institutional study of brainstem gliomas in children with neurofibromatosis type 1. Neurology. 2017; 88: 1584-9. PMCID: 5395076.
20.** de Blank PMK, Fisher MJ, Liu GT, Gutmann DH, Listernick R, Ferner RE, et al. Optic Pathway Gliomas in Neurofibromatosis Type 1: An Update: Surveillance, Treatment Indications, and Biomarkers of Vision. J Neuroophthalmol. 2017; 37: S23-S32.
Bibliografía recomendada
– Rosser T. Neurocutaneous Disorders. Continuum (Minneap Minn). 2018; 24: 96-129.
Completo artículo de revisión de las enfermedades neurocutáneas.
– Duat Rodríguez A, Martos Moreno GA, Martín Santo-Domingo Y, Hernández Martín A, Espejo-Saavedra Roca JM, Ruiz-Falco Rojas ML, et al. Phenotypic and genetic features in neurofibromatosis type 1 in children. An Pediatr (Barc). 2015; 83: 173-82.
Se recogen los datos clínicos, epidemiológicos, radiológicos y genéticos de 239 niños con NF1.
– Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019; 143:e20190660. doi: 10.1542/peds.2019-0660.
Detalla las recomendaciones para el seguimiento de los pacientes con Neurofibromatosis tipo 1.
– Hernández-Martín A, Duat-Rodríguez A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots, Freckling, and Neurofibromas. An Update. Part I. Dermatological Clinical Criteria Diagnostic of the Disease. Actas Dermosifiliogr. 2016; 107: 454-64.
Recomiendo la completa iconografía.
| Caso clínico |
|
Motivo de consulta Niña de 3 meses con aparición de manchas café con leche de forma progresiva desde el nacimiento. No refieren otra sintomatología. Antecedentes familiares Madre y padre sanos no consanguíneos. Tras preguntar de forma dirigida, el padre refiere presentar varias manchas café con leche. Sin otros antecedentes familiares de interés. Antecedentes personales Embarazo parto y periodo neonatal normales. Sin otros datos de interés. Exploración Peso: 6 kg (percentil 59); talla: 58 cm (percentil 28); perímetro cefálico: 42 cm (percentil 95); y tensión arterial: 89/40 mm Hg (percentil 66/68). Buen estado general, bien hidratada nutrida y perfundida. Auscultación cardiopulmonar normal. Abdomen blando sin palpación de masas ni visceromegalias. Presenta 8 manchas café con leche, de color homogéneo y contorno liso, regular y bien delimitado, que predominan en tronco y piernas de más de 0,5 cm de diámetro. Pectus carinatum. En la región preesternal, tras frotar ligeramente la zona, se observan máculas pálidas de contorno polilobulado de varios centímetros de tamaño. No presenta pecas axilares ni inguinales. No presenta bultos. Buen contacto, fija y sigue con la mirada. Frente prominente y ligero hipertelorismo. Sin asimetrías faciales. Sonrisa social. En decúbito supino, mantiene una postura estable con la cabeza centrada en línea media, realizando coordinación mano a mano y mano boca, llevando las piernas flexionadas hacia el abdomen. En decúbito prono, se apoya en antebrazos con codos en ángulo recto y en la parte baja del abdomen, elevando la cabeza hasta 45º. Sedestación inestable. Buen sostén cefálico. Tono y fuerza normal. Reflejos osteotendinosos normales. No deformidades óseas con columna alineada.
|

