 |
| Temas de FC |
F. J. Ramos Fuentes*, M. Ramos Cáceres**, M.P. Ribate Molina***
*Unidad de Genética Clínica, Servicio de Pediatría, Hospital Clínico Universitario “Lozano Blesa”, Facultad de Medicina, Universidad de Zaragoza. **Facultad de Medicina, Universidad de Extremadura, Badajoz. ***Departamento de Fisiología, Universidad de San Jorge, Zaragoza
| Resumen
La Semiología es el campo del conocimiento médico que estudia los signos y síntomas de las enfermedades y síndromes humanos, con el fin de obtener un diagnóstico y establecer un tratamiento. En Pediatría, la Dismorfología es un área de especial interés, debido a que las malformaciones congénitas y muchas deformaciones están presentes al nacimiento o en la primera infancia. Debido a las limitaciones de espacio, este trabajo se centrará en las malformaciones y deformaciones del cráneo y la cara, que en muchos casos configuran un fenotipo característico craneofacial reconocible, que proporciona una pista útil para el diagnóstico. Dado que alrededor del 2% de los recién nacidos nacen con una malformación congénita, es necesario identificarlas precozmente y tratar de llegar a un diagnóstico correcto para iniciar el tratamiento lo más rápidamente posible. Por otro lado, es importante informar a los padres sobre el riesgo de recurrencia (consejo genético), en casos de síndromes hereditarios |
| Abstract
Semiology refers to the field of the medical knowledge that studies the signs and symptoms of the human diseases (and syndromes) in order to obtain a diagnosis and establish a treatment for it. In Pediatrics, Dysmorphology is an area of special interest because congenital malformations and many deformations are all virtually present at birth or early childhood. Due to the space limitations, this paper will be focused on the malformations and deformations of the skull and face, that in many cases display a characteristic recognizable craneofacial phenotype, that provides a useful clue for the diagnosis. Since around 2% of neonates are born with a congenital malformation, it is important to identify them early and try to reach a correct diagnosis to establish the adecuate treatment as soon as possible. Moreover, it is important to inform the parents about the recurrence risk (genetic counselling), in cases of inherited conditions |
Palabras clave: Semiología; Malformación; Deformación; Dismorfología.
Key words: Semiology; Malformation; Deformation; Dysmorphology.
Pediatr Integral 2014; XVIII(8): 529-538
Semiología de las malformaciones y deformaciones craneofaciales
Introducción
En Medicina, la Semiología clínica es el cuerpo de conocimientos que se ocupa de la identificación de las diversas manifestaciones patológicas (síntomas y signos) o datos, de cómo buscarlos (semiotecnia), cómo reunirlos en síndromes, y cómo interpretarlos (clínica semiológica).
El método de trabajo o procedimientos desarrollados para la obtención de los datos (fundamentalmente el interrogatorio y el examen físico del paciente) se conoce como método clínico. Parte de observaciones simples y construye conocimientos de complejidad creciente, permitiendo al clínico no solo orientarse en el diagnóstico, sino tener una apreciación pronóstica y plantear las líneas generales del tratamiento. De ahí, la aserción de Laubry: “La Semiología no es solo la gramática de la medicina, sino la Medicina misma”. El contexto de su aplicación es la consulta médica y el instrumento de registro es la historia clínica.
Aunque el valor de esta es fundamental, el examen físico es, si cabe, más importante para un diagnóstico dismorfológico (Tabla I). Los recién nacidos con síndromes polimalformativos presentan una innumerable serie de anomalías individuales que, en muchas ocasiones, representan un difícil reto para el profesional que se enfrenta a ellas.

Digamos que, en primer lugar, es necesario identificar y reconocer dichas anomalías, a veces menores y poco evidentes al ojo no experimentado. En segundo lugar es imprescindible que, tras identificar una primera anomalía malformativa, se complete una buena exploración física para buscar otras.
En el campo de la dismorfología, las anomalías del área craneofacial cobran muchas veces una importancia singular; ya que, en conjunto pueden configurar un fenotipo dismórfico reconocible, que permite realizar un diagnóstico prácticamente inmediato (p. ej.: síndrome de Down) o sospechar algún cuadro clínico que sea susceptible de confirmar o descartar un estudio genético.
Este artículo tiene como principal objetivo describir las malformaciones y deformaciones más prevalentes de las principales estructuras anatómicas externas (visibles en la exploración física), que conforman el área craneofacial y, por lo tanto, ser una herramienta útil para la práctica clínica del Pediatra General cuando se enfrenta a un niño dismórfico. Los aspectos hereditarios y los relacionados con el diagnóstico genético de los síndromes que se mencionan, no están incluidos en el objetivo de este trabajo.
Definiciones
Semiología: área del conocimiento médico que estudia los signos (manifestaciones clínicas objetivas) y síntomas (percepciones subjetivas) presentados y referidos, respectivamente, por el paciente, para, mediante su organización en síndromes, jerarquización y razonamiento.
Malformación: anomalía intrínseca en la morfología de un órgano, parte del mismo, o de una estructura anatómica, producida por un desarrollo anormal del mismo. Suele producirse durante las primeras 8 semanas de vida intrauterina (organogénesis) y la mayoría es de causa genética. Ejemplos: labio leporino, mielomeningocele.
Deformación: anomalía en la forma o posición de un órgano, parte del mismo, o de una estructura anatómica normalmente formada, producida por una causa mecánica (extrínseca) que actúa de forma prolongada tras finalizar el periodo de organogénesis embrionaria. En los casos pertinentes, la mayoría son susceptibles de corrección con medidas ortopédicas. Ejemplo: plagiocefalia.
Síndrome: es el conjunto de anomalías (generalmente malformaciones), que se suele presentar conjuntamente en los pacientes afectados y cuya causa es única y conocida. La mayoría es de origen genético, monogénico o multifactorial, aunque también puede ser de origen ambiental. Ejemplos: síndrome de Down, síndrome de alcohol fetal.
Semiología según el tipo de anomalía en las diferentes estructuras afectadas
Las alteraciones en la forma y tamaño del cráneo y las estructuras de la cara pueden ser debidas a fuerzas mecánicas externas, anomalías en la conformación del cerebro subyacente (deformaciones), o patología intrínseca del crecimiento de los diferentes tejidos que los forman (malformaciones).
Cráneo
Malformaciones
Las malformaciones primarias del cráneo prácticamente se limitan a las asociadas al cierre incompleto del tubo neural. Los encefaloceles y meningoceles son malformaciones aisladas de la línea media del hueso craneal que, en su forma más grave, dan lugar a la anencefalia, debida a la ausencia total del neurocráneo, en la que solo se observan restos desorganizados del cerebro, sin membranas protectoras (meninges) que lo cubran.
Deformaciones
Las anomalías más frecuentes en el cráneo son de tipo deformacional. Ello se debe a que, durante el periodo perinatal, la cabeza del feto sufre transitoriamente importantes presiones de naturaleza mecánica, que configuran su forma al nacimiento. En primer lugar, por su frecuencia, haremos mención al modelamiento craneal, debido al paso de la cabeza fetal por el canal del parto, que da lugar a una cabeza alargada, fundamentalmente por la prominencia del occipucio (presentación). Los recién nacidos que nacen de nalgas (cabeza retroflexionada en el útero) tienen un cráneo con un aplanamiento del occipucio y un hueso occipital prominente.
La modelación de los huesos del cráneo se asocia habitualmente a un acabalgamiento de suturas, debido al solapamiento de los bordes de huesos craneales contiguos. Esta deformación puede modificar a la baja la medida del perímetro craneal y dar la impresión de que las fontanelas están cerradas, por lo que el pediatra debe tenerlo en cuenta cuando explore al recién nacido. La sutura más frecuentemente afectada es la sagital. En general, esta deformidad suele ser transitoria y desaparecer en unas semanas. Si se observa un acabalgamiento de la sutura metópica (en el medio de la frente) es obligado descartar una craneosinostosis (ver más adelante).
La plagiocefalia o asinclitismo craneal es la deformación de la cabeza debida a un aplanamiento en diagonal (extremos opuestos) del cráneo que, visto desde arriba, recuerda a un paralelogramo en lugar de un rectángulo.
Su origen suele estar en la presión que soporta la cabeza fetal en el eje diagonal cuando está retenida demasiado tiempo en el canal del parto. En la mayoría de los casos, a pesar de la alarma inicial que produce en los padres, esta deformación se remodela y desaparece en pocos meses. En las formas más graves, la normalización se retrasa y puede ser necesaria la colocación de un casco terapéutico. En estos pacientes puede asociarse una asimetría facial e incluso anomalías en la visión (Fig. 1).

Figura 1. Plagiocefalia por craneosinostosis coronal unilateral (derecha). La paciente tiene un síndrome de Saëthre-Chotzen. Obsérvese la asimetría facial y las anomalías oculares.
La craneosinostosis (fusión prematura de las suturas craneales) es una de las causas más importantes de deformidad craneal. En dependencia de las suturas afectadas, la cabeza tendrá una configuración diferente y específica.
La dolicocefalia (escafocefalia) es una deformidad producida por la fusión de la sutura sagital que da lugar a un cráneo alargado en el eje antero-posterior. Esta deformidad suele verse en el síndrome de Edwards (trisomía 18). La braquicefalia,cráneo acortado y ancho, se produce por la fusión de ambas suturas coronales; es frecuente en el síndrome de Down. La turricefalia es el alargamiento vertical del cráneo (forma de torre) y es debida a la sinóstosis combinada de las suturas coronales, metópica y lambdoideas. Por último, la trigonocefalia, debida a la desaparición de la sutura metópica, consiste en un cráneo en forma triangular (visto desde arriba) con una frente estrechada, que recuerda a la proa de un barco. Dos deformidades más raras, generalmente asociadas a síndromes polimalformativos son: la acrocefalia (cráneo en pico), debida a la fusión prematura de las suturas de la base, y el cráneo en trébol, cuando se fusionan todas las suturas.
Pelo
Malformaciones
Las anomalías pueden ser localizadas o generalizadas; es decir, afectar a zonas delimitadas o a todo el cuero cabelludo. En el primer grupo estarían las patillas alargadas, que se extienden desde la sien hacia abajo y adelante (mejilla) y que son un signo muy sugerente de síndrome de Treacher-Collins.
La implantación baja del pelo en la nuca puede ser el resultado de la existencia de edema en esa zona durante la vida fetal. En algunos pacientes, la línea del pelo adquiere una forma de “M”.
La alopecia areata es la ausencia de cabello en una zona delimitada del cuero cabelludo. Sus causas pueden ser diferentes, siendo la más frecuente la forma espontánea, que aparece de forma repentina y a cualquier edad. Estas zonas son redondeadas u ovaladas, están bien delimitadas y la piel subyacente (visible) es normal. La evolución puede ser hacia la desaparición progresiva o hacia la alopecia total, cuando las zonas sin pelo se van fusionando hasta cubrir toda la cabeza. Se sospecha un origen genético en algunos casos familiares, aunque se conoce su aparición en trastornos endocrinos o síndromes dismórficos.
La poliosis es la aparición de mechas de cabello hipopigmentado. Su variante más conocida es el mechón blanco del síndrome de Waardenburg (sordera, anomalías del iris y cantos de los ojos), aunque también puede observarse en formas parciales de albinismo (piebaldismo)e incluso en individuos normales y siguiendo un patrón de herencia autosómico dominante.
El pelo rizado, principalmente cuando no es un rasgo familiar, puede ser un signo asociado a un síndrome dismórfico, generalmente afectando también a los dientes y a otros tejidos de origen ectodérmico.
El pelo frágil consiste en un cabello corto, fino y escaso. Puede acompañarse de roturas a lo largo del tallo, teniendo una apariencia de pelo ensortijado, el cual es muy característico del síndrome de Menkes, una enfermedad ligada al X debida a un déficit de cobre (Fig. 2).
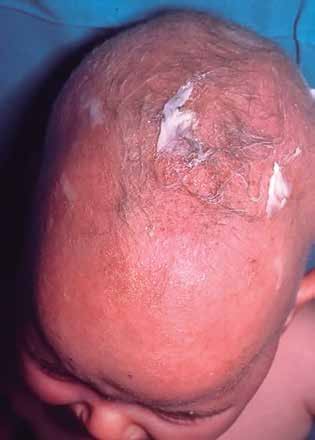
Figura 2. Cuero cabelludo con pelo escaso y ensortijado de paciente con síndrome de Menkes.
En el pelo no hay prácticamente deformaciones, salvo las que se observan transitoriamente tras algunos procedimientos cosméticos.
Cara
Malformaciones
La facies triangular se debe, a menudo, a la desproporción entre el crecimiento normal de los huesos del cráneo y el crecimiento reducido de los de la cara. Esta malformación es típica del síndrome de Silver-Russel (baja talla, asimetría de extremidades inferiores, braquiclinodactilia bilateral del quinto dedo de las manos).
La facies aplanada es debida al fallo de crecimiento de los maxilares superiores, dando a veces la cara una falsa impresión de prognatismo. Esta malformación es un hallazgo inespecífico asociada a numerosos síndromes dismórficos.
La mayoría de los pacientes afectados de déficit de hormona de crecimiento presentan una facies de aspecto inmaduro, que les da una apariencia más infantil que la que les correspondería a su edad cronológica. Suele ser debida a un fallo en la maduración del esqueleto.
Deformaciones
La asimetría facial es debida, al igual que la plagiocefalia con la que a menudo se asocia, a la acción prolongada de fuerzas externas que comprimen las estructuras de uno de los lados de la cara del feto dentro del útero. Suele desaparecer a los 6-9 meses de vida.
Esta deformidad debe ser distinguida de la asimetría facial secundaria a una craneosinostosis de una de las suturas coronales. Esta, si no es tratada, acaba produciendo una ambliopía que obliga a que el niño, inconscientemente, incline su cabeza para mantener ambos ojos en el mismo plano horizontal, provocando primero una tortícolis y, con el paso del tiempo, una escoliosis compensadora. Esta situación puede verse en el síndrome de Müenke y en el síndrome de Saethre-Chotzen.
La facies miopática se observa en pacientes con enfermedades neuromusculares graves (p. ej.: distrofia miotónica de Steinert) (Fig. 3) o síndromes asociados a hipotonía grave. El crecimiento óseo suele ser normal.

Figura 3. Facies miopática de paciente con distrofia miotónica de Steinert (forma congénita).
La compresión prolongada de toda la cara, feto contra la pared del útero, asociada habitualmente a oligohidramnios, da lugar a la facies de Potter, que incluye una nariz aplanada, crestas verticales subpalpebrales y orejas grandes y blandas.
Ojos y región periocular
Malformaciones
El hipertelorismo ocular (Fig. 4) es la distancia aumentada entre las pupilas (superior a 3 de la media). Es una malformación que se observa en numerosos síndromes dismórficos, confiriendo a la cara una apariencia característica. Es habitual que los pacientes afectados desarrollen estrabismo y/o ambliopía.
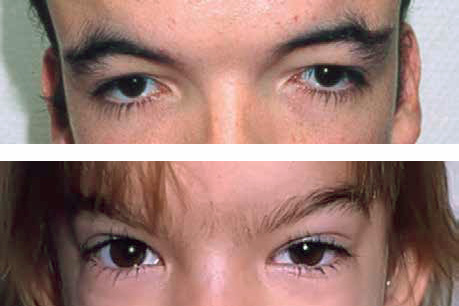
Figura 4. A. Hipertelorismo ocular verdadero; B. Telecanto con ptosis palpebral izquierda.
Por el contrario, la presencia de una distancia disminuida entre los ojos se denomina hipotelorismo ocular, el cual puede ser secundario a un desarrollo anómalo del bulbo olfatorio y los lóbulos frontales del cerebro. Su grado extremo da lugar a la ciclopía o fusión de ambos ojos en uno.
Se denomina telecanto, al desplazamiento lateral de los cantos internos de ambos ojos, cuyas órbitas y globos oculares están normalmente situados. La ausencia o disminución de esclera del lado medial al iris, permite distinguirlo del hipertelorismo ocular verdadero.
Las indentaciones palpebrales pueden formar parte de una fisura labial (labio leporino) que se extiende hasta la órbita. En otras ocasiones, forman parte de síndromes dismórficos como el síndrome de Treacher-Collins (párpado inferior) o el síndrome de Goldenhar (párpado superior).
El ectropión es una malformación del párpado inferior, en la que existe una eversión del mismo y lo separa de su posición normal respecto al globo ocular en la parte inferior del iris. Suele dar lugar a una conjuntivitis crónica.
La reducción del espacio entre el párpado superior e inferior, con globos oculares y órbitas normales se denomina blefarofimosis. Dicha reducción se observa, tanto en altura como en anchura, debido a la asociación de telecanto con ptosis palpebral. Las fisuras palpebrales cortas, características del síndrome de alcohol fetal, se deben a una disminución de la distancia entre los cantos externo e interno de los ojos, siendo normal la distancia entre los bordes de ambos párpados. En algunos casos, puede asociarse a una microftalmia leve.
Las malformaciones del globo ocular se pueden producir en cualquiera de los periodos de desarrollo del mismo. En el periodo de organogénesis (hasta la 5ª semana de gestación), se produce la anoftalmía o ausencia completa de globo ocular, por fallo del desarrollo de la vesícula óptica. Se suele asociar a una falta de desarrollo de las órbitas y de otras estructuras perioculares. Más frecuente es la microftalmia, que consiste en la disminución del tamaño del globo ocular y que suele acompañarse de hipoplasia del párpado, del sistema lacrimal y de las órbitas.
El coloboma ocular es un fallo completo del cierre de la fisura coroidea en el botón óptico embrionario, pudiendo afectar al nervio óptico, la retina, el iris y el cuerpo ciliar.
Durante la segunda fase del desarrollo ocular (de la 6ª a la 12º semana de gestación), puede aparecer una hipoplasia del nervio óptico, debida a un fallo de proliferación de sus células nerviosas, causante de ambliopía, nistagmo y disminución de la agudeza visual.
La tercera fase del desarrollo ocular (de la 13ª a la 26ª semana de gestación) se caracteriza por el desarrollo final de las células de la retina (conos y bastones), del iris y el cuerpo ciliar. La malformación más importante de este periodo es la aniridia o ausencia total o parcial del desarrollo del iris. En los pacientes afectados por esta malformación, puede aparecer: nistagmo, ptosis, glaucoma o ceguera. Es importante recordar la asociación entre aniridia y tumor de Wilms (síndrome WARG), en pacientes portadores de una deleción intersticial del cromosoma 11 (11p13) que afecta a varios genes.
La debilidad del colágeno, que forma los ligamentos suspensorios que lo mantienen en su posición normal, da lugar a una luxación del cristalino, un hallazgo característico del síndrome de Marfan y que puede asociarse a estrabismo y disminución de la agudeza visual. En este la luxación es superior, a diferencia de la homocistinuria, en la que el cristalino se desplaza hacia abajo.
La criptoftalmia es el fallo de la apertura definitiva de los párpados, que normalmente se produce durante la 26ª semana de vida intrauterina. Una de las pocas entidades que incluye esta anomalía es el síndrome de Fraser.
Deformaciones
La asimetría de las órbitas puede producirse por compresión externa del feto o ser secundaria a una sinóstosis prematura de sus estructuras óseas. Suele acompañarse de estrabismo y ambliopía.
La horizontalización o caída de las pestañas es una deformación que suele asociarse a ptosis palpebral o ser un signo sutil de hipotonía generalizada (p. ej.: síndrome de Prader-Willi).
El exoftalmos (globos oculares protruyentes), a veces, se produce por edema o hiperplasia del tejido blando infraorbitario, como ocurre en los casos de hipertiroidismo. También es un hallazgo habitual en síndromes con craneosinostosis, como el síndrome de Crouzon o el síndrome de Apert.
Nariz
Malformaciones
La atresia de coanas es debida a la persistencia de la membrana oronasal, dando lugar a una obstrucción total de la apertura posterior de la cavidad nasal.
La obstrucción puede ser parcial en los casos de estenosis de coanas. La atresia de coanas es una malformación grave que requiere de una intervención terapéutica rápida y eficaz, generalmente quirúrgica.
La displasia frontonasal se produce por un desarrollo o migración excesivos de tejido por encima de las mejillas. Estos pacientes tienen un puente nasal muy ancho y un importante hipertelorismo ocular.
La presencia de una foseta nasal media debe obligarnos a descartar la existencia de malformaciones ocultas de la línea media, concretamente del sistema nervioso central. En contraposición, si existe una prominencia sobre el puente nasal que se abomba durante el llanto del niño, hay que sospechar la existencia de un cefalocele anterior, que requiere una consulta con el neurocirujano.
Deformaciones
Son generalmente debidas a la compresión de la cara del feto dentro del útero materno e incluyen: la desviación del tabique nasal, la asimetría de los orificios nasales y la desviación de la punta de la nariz. La facies de Potter secundaria a oligohidramnios, también puede asociar deformaciones en la nariz.
Orejas
Malformaciones
Incluyen las derivadas de un desarrollo anormal del esqueleto cartilaginoso del pabellón auricular. En ocasiones, se presentan de forma aislada, pero en muchos pacientes son un hallazgo más de un síndrome polimalformativo.
Para la mejor compresión de este apartado, es necesario recordar la anatomía del pabellón auricular.
La microtia se produce por un desarrollo incompleto del pabellón auricular (pinna), derivado del primer y segundo arcos branquiales. Es la malformación más frecuente de la oreja y puede ser de grado variable y, en los casos más graves, el pabellón se reduce a un pequeño resto de cartílago cubierto de piel que delimita el canal auditivo externo.
Puede ser unilateral (afecta con mayor frecuencia al lado derecho) o bilateral. En ambos casos, es obligado realizar un estudio de audición en el paciente; dado que, es frecuente la co-existencia de hipoacusia, incluso en el lado aparentemente no afectado. Una forma sutil de microtia es la ausencia de la crus superior del antihélix, que habitualmente pasa desapercibida, pero que, en un 15% de casos, se asocia a hipoacusia del mismo lado. Por último, es importante recordar que la microtia forma parte de numerosos síndromes dismórficos, especialmente los pertenecientes al grupo “oto-renal”.
Como parte del cuadro clínico de numerosos síndromes, las malformaciones óticas no suelen ser muy específicas, aunque en algún caso, pueden dar una pista al clínico en el diagnóstico diferencial. Algunos ejemplos son: la oreja (hélix) arrugada en el síndrome de Beals, la oreja triangular del síndrome de Turner y del síndrome de Noonan, la cresta diagonal en el lóbulo del síndrome de Beckwith-Wiedemann o el hélix en raíles de tren,en el síndrome alcohol fetal.
La presencia de fosetas o pedículos preauriculares (Fig. 5), considerados variantes de la normalidad, es debida a la persistencia de restos embrionarios del arco o hendidura branquial. Están presentes en el 0,5-1% de la población general y existe una gran variabilidad entre diferentes grupos étnicos.

Figura 5. A. Pedículo preauricular en pabellón auricular anormal; B. Foseta preauricular a nivel de la inserción del hélix.
Deformaciones
La distorsión del pabellón auricular es una deformación habitual y sin significado clínico, ya que desaparece cuando cesan las fuerzas que lo producen (compresión intraútero del hombro homolateral).
Las orejas grandes y aplanadas se suelen ver en recién nacidos que han sufrido oligohidramnios en la vida fetal y se deben a la compresión permanente de los pabellones. Es uno de los hallazgos presentes en la llamada facies de Potter.
En ocasiones, las deformaciones de la oreja no se deben a anomalías de su estructura cartilaginosa, sino a problemas con los músculos (posterior y superior) que la mantienen unida al hueso subyacente. La oreja (hélix) colgantees debida a la ausencia o disfunción del músculo superior, la oreja saliente (de soplillo)se ve cuando es el músculo posterior el afectado, y, por último, la oreja ahuecada (en taza), cuando son anormales ambos músculos.
Boca y región perioral
Malformaciones
La malformación mayor, más frecuente, que afecta a la boca y región perioral es el labio leporino, resultado de un fallo en la proliferación y posterior fusión del tejido embrionario en la zona de unión de las protuberancias nasales, medial y lateral, y la maxilar.
Puede ser parcial o completo y afectar uno o ambos lados (Fig. 6a). La forma más leve puede incluso pasar desapercibida (pequeña indentación del labio superior paralela al philtrum) y la más grave se asocia a fisura palatina (Fig. 6b)(v. más adelante). Es importante recordar que, en los casos de labio leporino bilateral, suele existir un mamelón de tejido rudimentario en la línea media y que si este no existe, es un signo de mal pronóstico, ya que indicaría un desarrollo anormal del cerebro anterior, generalmente presente en la holoprosencefalia.

Figura 6. A. Labio leporino bilateral con fisura palatina (obsérvese el mamelón de tejido que ocupa el hueco de la nariz); B. Paladar ojival.
La macrostomía (boca grande) se produce por la fusión incompleta entre los segmentos maxilares y mandibulares del primer arco branquial. Es una anomalía poco frecuente, pero que frecuentemente se asocia a hipoacusia. En estos casos, las comisuras bucales están inclinadas hacia arriba, extendiéndose más allá de los bordes labiales y es habitual la presencia de fosetas o pedículos a lo largo de la línea imaginaria que une la comisura bucal y el canal auditivo externo homolateral.
La presencia de fosetas en el labio inferior es una rara malformación presente en algunos síndromes genéticos (p.ej.: síndrome de Wan der Woude). Generalmente son bilaterales, situadas en el borde inferior y pueden acompañarse de fístulas ocultas que, a veces, expulsan una sustancia mucoide. Salvo que se produzca esta circunstancia, suelen pasar desapercibidas si no se explora la zona cuidadosamente.
La micrognatia (mandíbula pequeña) puede ser debida a un fallo del crecimiento del hueso maxilar y se observa en numerosos síndromes dismórficos. En su forma más grave, puede comprometer la capacidad de la cavidad oral para contener la lengua y da lugar al llamado complejo Pierre-Robin.
La malformación más frecuente e importante de la cavidad oral es la fisura palatina, debida a un fallo en el cierre medial de ambas prominencias palatinas, que da lugar a una comunicación entre la boca y la cavidad nasal superior y que, en su forma más grave, se asocia a labio leporino (Fig. 6a).
Existen formas intermedias, como la fisura palatina unilateral o la fisura palatina incompleta o posterior, esta última puede pasar inadvertida en su variante más leve (fisura palatina submucosa). La úvula bífida puede considerarse la forma más leve de esta malformación.
El crecimiento excesivo de zonas de la mucosa oral puede dar lugar a frénulas aberrantes entre las encías y los labios.
La microglosia es una lengua de tamaño inferior al normal y puede asociarse a micrognatia o a persistencia de los bordes alveolares secundarios.
En contrapunto, la macroglosia es la presencia de una lengua grande y puede causar dificultades en la respiración del niño y deformidades de las arcadas dentarias. Esta malformación es habitual en pacientes con síndrome de Down y con síndrome de Beckwith-Wiedemann.
La lengua asimétrica o la lengua lobulada son malformaciones muy raras que pueden ser la pista para identificar entidades o anomalías asociadas (hemihipertrofia o hamartoma respectivamente).
Deformaciones
La micrognatia también puede ser una deformación, cuando es secundaria a fuerzas de compresión intraútero sobre la mandíbula el feto. Suele observarse en neonatos nacidos de cara o de nalgas y tiene carácter leve o moderado.
Cuando la presión del feto sobre esta región ha sido asimétrica se produce una hipoplasia mandibular asimétrica. En ambos casos, dada la integridad del potencial de crecimiento del hueso maxilar inferior, la recuperación es rápida y en pocos meses su tamaño será proporcional al resto de las estructuras faciales del niño.
La presencia de una cresta en el mentón (horizontal o en forma de “H”) es un hallazgo poco habitual, que es debido a una función anormal de los músculos subyacentes y suele hacerse más evidente con la edad.
En la cavidad oral, la mayoría de las deformaciones son secundarias a alteraciones de la masticación o de la movilidad de la lengua. Así, la persistencia de un borde alveolar secundario es debida a una inadecuada presión sobre el paladar por parte de la lengua, dando lugar a un paladar muy estrecho y elevado (paladar ojival) (Fig. 6b).
Por último, la presencia de una distorsión del arco dentario también puede ser debida a una excesiva y repetida presión de la lengua sobre uno de los maxilares, como puede ocurrir en pacientes con macroglosia.
Bibliografía recomendada
Los asteriscos reflejan el interés del artículo a juicio del autor.
*** Aase JM. Diagnostic Dysmorphology. New York: Plenum Publishing Corp. 1990.
Libro de referencia para la realización de una buena exploración dismorfológica. Es muy completo y en él se da una orientación diagnóstica ante numerosos hallazgos dismórficos. En él está basado este trabajo.
* American Academy of Pediatrics. The role of the primary care pediatrician in the management of high risk newborn infants. Pediatrics. 1996; 98: 786-788.
Documento-guía de la Academia Americana de Pediatría sobre del papel del Pediatra de cabecera en el cuidado y seguimiento de recién nacidos de alto riesgo, incluyendo los que presentan malformaciones congénitas.
* American Academy of Pediatrics. General principles in the care of children and adolescents with genetic disorders and other chronic health conditions. Pediatrics. 1997; 99: 643-644.
Documento-guía de la Academia Americana de Pediatría sobre los cuidados y seguimiento de niños y adolescentes con problemas genéticos (entre otros), en los que se incluyen los síndromes polimalformativos.
*** Reardon W. Ed. The bedside dysmorphologist: Classic clinical signs in human malformation syndromes and their diagnostic significance. Oxford. Oxford University Press; 2008.
Libro de referencia (de bolsillo) obligado para todo genetista clínico y/o dismorfólogo. El autor se basa en el reconocimiento y descripción de los signos (dismórficos) más relevantes (signos guía), en las distintas regiones anatómicas del organismo como herramienta de ayuda para el clínico, en el diagnóstico diferencial de los síndromes polimalformativos más importantes.
* Hunter AG. Medical Genetics 2. The diagnostic approach to the child with dysmorphic signs. Can Med Assoc J. 2002; 167: 367-372.
Artículo sobre la forma de proceder para llegar al diagnóstico de un niño con malformaciones. Está ilustrado con 2 casos reales, en los que se aplica de forma práctica lo que se ha explicado de forma teórica.
** Jones KL, Jones MC. A clinical approach to the dysmorphic child. En: Rimoin DL, Connor JM, Pyeritz RE, Eds. Emery and Rimoin´s Principles and Practice of Medical Genetics. 5th ed. Vol. I. New York: Churchill Livingstone. 2007; pp. 889-899.
Capítulo sobre la actuación clínica frente al niño dismórfico, con una primera parte dedicada a conceptos embriológicos. Está escrito por 2 expertos y pertenece a un tratado imprescindible en Genética Médica.
*** Jones KL. Smith´s Recognizable Pattern of Human Malformation. 6th ed. Philadelphia: W.B. Saunders. 2006.
Puede considerarse “la Biblia” de la sindromología. Incluye más de 500 síndromes polimalformativos, la mayoría de origen genético, de los que ofrece un resumen de los principales hallazgos clínicos por sistemas, acompañado por fotografías representativas de cada patología. También se incluye un apartado sobre la etiología.
** Pérez-Aytés A. Actitud ante el recién nacido con malformaciones congénitas. En Protocolos diagnósticos y terapéuticos en Pediatría. Tomo 1. Barcelona: Ergon. 2000, pp 19-22.
Se realiza un repaso de los aspectos más relevantes para la obtención de una buena historia clínica, ante un recién nacido con malformaciones congénitas. Termina con una recopilación de los principales términos dismorfológicos.
* Smith DW. An approach to clinical dysmorphology. J Pediatr. 1977; 91: 690-692.
Uno de los trabajos pioneros en Dismorfología, escrito por una de las grandes autoridades mundiales en la materia, en el que se definen las bases conceptuales de la sindromología moderna.
* Ramos FJ. Seguimiento y cuidados del recién nacido con malformaciones. Pediatr Integral. 2010; XIV: 461-468.
Artículo reciente de uno de los autores del presente trabajo, que incluye la exploración del recién nacido dismórfico que servirá de base para la aproximación diagnóstica (diagnóstico diferencial) y seguimiento del paciente. Contiene las definiciones de los conceptos principales en Dismorfología.
Nota: La bibliografía recomendada que se incluye al final de este trabajo y que debe servir principalmente para consultar y ampliar lo que en este trabajo se ha expuesto, no está citada en el texto del artículo, ya que son principalmente libros de consulta o recomendaciones generales publicadas en revistas científicas.
| Caso clínico |
|
Varón de 3 años y medio que acude a la consulta de Pediatría General por asimetría craneofacial. Los antecedentes personales indican un embarazo a término sin complicaciones, con serologías negativas y sin exposición conocida a teratógenos. Parto por cesárea por desproporción pélvico-cefálica. Puntuación de Apgar 8/9. Somatometría neonatal normal, con todos los parámetros entre los percentiles 75 y 90. En la exploración física al nacer, se observó una asimetría craneofacial con braquicefalia y, aparentemente, sin otras anomalías externas asociadas. Dicha asimetría se ha ido haciendo más evidente con la edad. Los estudios (bioquímicos) de laboratorio fueron normales, al igual que un estudio radiológico del esqueleto, que incluía el cráneo. En la ecografía transfontanelar, se informó de la existencia de una asimetría de los lóbulos frontales, con ligero desplazamiento de las estructuras de la línea media y de los ventrículos laterales. El desarrollo psicomotor ha sido aparentemente normal, aunque refieren ciertas dificultades con el (inicio) lenguaje. Los antecedentes familiares incluyen: madre y abuela materna con braquicefalia y frente alta, y aplanada. Ambas tienen ptosis palpebral unilateral leve-moderada y pabellones auriculares dismórficos. La exploración física del paciente en el momento de la primera visita (3a y 9m): peso y talla en percentiles 50-75; perímetro cefálico >97%. Proporción normal tronco/extremidades. Asimetría craneal y facial con hipoplasia del lado izquierdo. Tortícolis izquierda, secundaria a problema en la visión (Fig. 1). En la cara se observa: hipertelorismo ocular, ptosis palpebral izquierda, pabellones auriculares malformados, maloclusión dentaria y paladar ojival. Tórax normal, salvo un leve pectus excavatum. No soplo cardíaco. Genitales normales. En extremidades: braquidactilia bilateral del 5º dedo en manos y sindactilia leve entre los dedos 2 y 3. Lenguaje escaso. Tono y reflejos normales. Resto de exploración, sin datos relevantes para el caso.
|

* Modificado de Wilson, 2000.
