 |
| Temas de FC |
J. Lirio Casero, J. García Pérez
Unidad de Pediatría Social. Hospital Infantil Universitario Niño Jesús. Madrid
| Resumen
El síndrome de Down (SD) es la primera causa congénita de retraso mental en nuestro mundo. El principal problema médico al nacimiento es la existencia de cardiopatía congénita que, hasta hace unos años, resultaba un factor pronóstico clave. Sin embargo, hay una lista importante de enfermedades que pueden desarrollar los individuos con SD a lo largo de su vida y que justifica protocolos de seguimiento específicos. Hoy en día, los cuidados médicos de Atención Primaria deben incluir aspectos educativos y del desarrollo, y no centrarse solo en problemas estrictamente médicos. |
| Abstract
Down syndrome (DS) is the first cause of congenital mental retardation in our world. The main medical problem at birth is congenital heart disease, which until recently, was a key prognostic factor. However, there is a substantial list of diseases that can develop DS children along their life, and that justifies specific protocols. Nowadays, health primary care should include educational and developmental aspects, not only medical problems. |
Palabras clave: Síndrome de Down; Diagnóstico prenatal; Trisomía 21; Utilización de los servicios de salud.
Key words: Down syndrome; Prenatal diagnosis; Trisomy 21; Health care utilisation.
Pediatr Integral 2014; XVIII(8): 539-549
Protocolo de seguimiento del síndrome de Down
El SD es la primera causa genética de retraso mental. Aunque su incidencia ha ido disminuyendo progresivamente, su esperanza y calidad de vida han mejorado gracias a los programas específicos de salud.
En nuestro país nacen alrededor de 600 niños con SD cada año. El 95% de los casos son causados por una trisomía 21 (forma no familiar por la no-disyunción de los cromosomas del óvulo o espermatozoide); mientras que, en el 3-4% aparece como resultado de una translocación del cromosoma 21 con otro cromosoma acrocéntrico, generalmente el 14 (siendo ¼ parte de origen familiar) y, en el 1-3% restante debido a mosaicismo. El fenotipo de estos últimos es variable, desde la casi normalidad de rasgos, a formas indistinguibles de las trisomías, en función del porcentaje de células alteradas.
El síndrome de Down (SD) es el defecto congénito cuya frecuencia al nacimiento ha experimentado un descenso más acusado; ya que, ha disminuido a razón de una media de 4 nacidos menos con SD por cada 100.000 nacimientos anualmente, hasta situarse en el 2007 (último año del que se disponen datos en nuestro país), en una incidencia de 8,09 nacidos con SD por cada 100.000/año. El descenso es mucho más intenso en el grupo de madres con más de 34 años, entre las cuales la disminución media anual es de casi 34 (33,6) niños con SD por cada 100.000 nacimientos (Fig. 1). Ello es debido a que existen planes de diagnóstico prenatal específicamente dirigidos a la detección del SD, y a que están especialmente enfocados a los grupos de mayor riesgo, es decir, a las madres de mayor edad. Los motivos del éxito del cribado en madres jóvenes hay que buscarlos en la existencia de buenos indicadores ecográficos, que pueden hacer sospechar el diagnóstico prenatal de SD antes de pasar al empleo de técnicas invasoras(1).
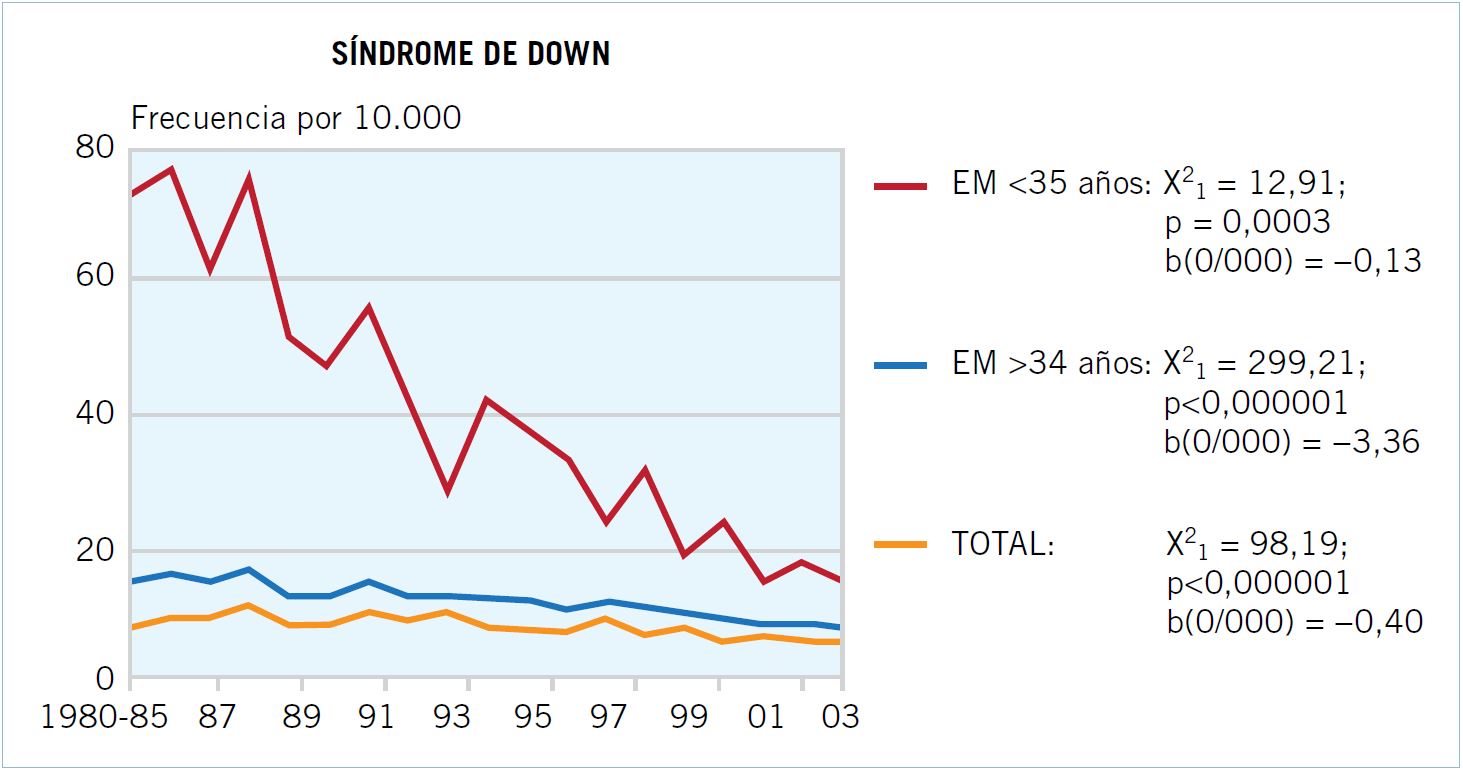
Figura 1. Evolución de nacimientos con SD.
También, la calidad y la esperanza de vida de estos niños han cambiado radicalmente en las dos últimas décadas, alcanzándose mejor estado de salud, mayor grado de autonomía personal e integración en la comunidad. En los Estados Unidos de América, la supervivencia al año de vida de los nacidos entre 1942 a 1952 era inferior al 50%; mientras que, en los nacidos entre 1980 a 1996 llegó al 91%. Paralelamente, la edad media de fallecimiento fue de 25 años en 1983 y de 49 años en 1997(2). En Suecia, la mortalidad entre los nacidos en 1970 a 1980 fue del 44,1% en los primeros 10 años, cuando presentaban cardiopatía, frente al 4,5% si no la padecían(3).
El mayor conocimiento de los riesgos y problemas asociados al SD permite conocer qué alteraciones pueden aparecer y en qué momentos de la vida del individuo (Tabla I), siendo posible añadir a las recomendaciones generales de control de salud para la población infantil en general, un grupo de actividades preventivas y exploraciones que permitan corregir, aliviar o evitar los problemas de salud de niños con SD.

Criterios diagnósticos
Fenotipo Down
Los rasgos más característicos del SD son los siguientes: hipotonía, cara aplanada, hendiduras palpebrales oblicuas hacia arriba (inclinación mongoloide), epicantus, iris moteado (manchas de Brushfield), puente nasal aplanado, orejas pequeñas, paladar ojival, exceso de piel en nuca, surco palmar transverso único (pliegue simiesco), manos cortas y anchas, hipoplasia de falange media del quinto dedo, separación entre primer y segundo dedo del pie (signo de la sandalia)… En casi todos los casos, existe retraso mental, de grado variable. A pesar de los intentos en diversos estudios, no se ha encontrado una correlación fenotipo/genotipo.
Confirmación mediante estudios genéticos
La confirmación se realiza mediante estudios genéticos: cariotipo o técnicas de hibridación in situ (HIS) ya sea prenatal (amniocentesis/biopsia de corion) o postnatalmente. El estudio HIS permite hacer un diagnóstico de urgencia, pudiendo obtener el resultado en unas pocas horas(4).
Información a los padres
Constituye la primera actividad preventiva en estos niños. Aunque el problema suele detectarse en la etapa neonatal, cualquier pediatra puede verse en la necesidad de dar la noticia del diagnóstico y, en todo caso, conviene revisar la información recibida previamente. La forma de plantear una noticia inesperada que desestructura el presente de la familia y cambia sus perspectivas de futuro, puede determinar la actitud de los padres. Es recomendable informar conjuntamente a ambos progenitores, mientras sujetan al niño entre sus brazos, de forma clara y comprensible, dosificando el exceso de información, lo antes posible y en un ambiente tranquilo que facilite la intimidad(5). Tras esa primera conversación, puede ser útil que los padres acudan a una Fundación o Asociación de Síndrome de Down, donde otros padres en su misma situación podrán hablarles de forma más directa, y convocarles para otra reunión informativa posterior. Algunas familias tardan en aceptar la noticia, debemos ofrecerles el apoyo de los Servicios Sociales (ayudas económicas, recursos administrativos…) y ayuda psicológica. El consejo genético conviene que sea realizado por un especialista en el tema.
Justificación de las intervenciones preventivas
El médico de Atención Primaria puede desarrollar toda la actividad preventiva de los niños con SD para lo que es fundamental conocer los problemas de salud asociados.
Desarrollo psicomotor, incluido el lenguaje
La función cognitiva varía ampliamente de un niño a otro y no se puede predecir al nacimiento. Tampoco existe relación entre el fenotipo concreto de un niño con SD y el nivel de función cognitiva. El cociente intelectual va de rango bajo a retraso moderado o profundo, siendo este último raro, tal como puede observarse en la figura 2, según datos extraídos de la Base de Datos Estatal de Personas con Discapacidad (IMSERSO), en España, en enero del 2001.

Figura 2. Distribución de las personas con síndrome de Down según el grado de retraso mental.
En general, los bebés con SD se desarrollan y progresan de modo muy parecido a como lo hacen los demás niños en la mayoría de las áreas de desarrollo, aunque lo hacen a una velocidad más lenta(6). En realidad, su desarrollo avanza más deprisa en unas áreas (el desarrollo social es uno de sus puntos fuertes); mientras que, el progreso motor y el aprendizaje del lenguaje se retrasan más (Tabla II). En lo que respecta a la comunicación, funcionan bien en la utilización de gestos para comunicarse, pero muestran mayor dificultad para el habla; de modo que, entienden más de lo que pueden decir. Respecto a la cognición, tienen mayor capacidad de procesamiento y recuerdo de la información visual, que de la información verbal.

Diversos estudios con casos y controles han observado que los programas de intervención temprana mejoran el desarrollo global, los trastornos del comportamiento alimentario, el lenguaje, la integración social y la adaptación entre padres e hijos(7). También pueden mejorar el pronóstico académico de estos niños. Los programas de intervención temprana conviene que sean atendidos por profesionales con experiencia, habitualmente las asociaciones o fundaciones para personas con SD ofrecen este servicio con personal y recursos adecuados.
Desarrollo físico (póndero-estatural)
El desarrollo físico es más lento que los grupos poblacionales por edad y sexo de niños no afectados de SD. Por ello, las medidas póndero-estaturales deben ser referidas a estándares específicos para estos niños. Las primeras publicadas y de uso extendido son las tablas de Cronk. Recientemente, se ha publicado la actualización de tablas de crecimiento de niños españoles con síndrome de Down(8). Se pueden consultar otras tablas de crecimiento editadas en Internet: www.growthcharts.com y en www.growthcharts.com/charts/DS/hccharts.htm. También se ha observado, que estos niños tardan un mes en recuperar el peso del nacimiento, probablemente por todas las dificultades que plantean con su alimentación durante los primeros días de vida.
El crecimiento más lento no es atribuible, de forma generalizada, a déficit de hormona de crecimiento (GH) y, actualmente, se especula sobre el papel del IGF-1. Si observamos una disminución del crecimiento en referencia a las tablas estándares del SD, debemos investigar causas como: cardiopatía congénita, hipotiroidismo, enfermedad celíaca, déficit de hormona del crecimiento o ausencia de soporte nutricional, entre otras.
El estirón puberal es menos vigoroso que en la población general y suele ocurrir antes. También la prevalencia de obesidad en este grupo es mayor que en la población general, y debe ser considerada un problema de salud, en el que deben involucrarse médicos, enfermeras, miembros de la familia e individuos con SD. Se sugiere que la intervención debe combinar una dieta equilibrada, sin restricción energética y aumento de la actividad física.
Alteraciones de la función tiroidea
Sabemos que las alteraciones más frecuentes en la función tiroidea en niños con SD están incrementadas significativamente en todas las edades, aproximadamente el 45% de las personas con SD presentan disfunción de la glándula tiroidea, la mayor incidencia corresponde a elevaciones aisladas de la TSH (20-60%).
Los casos de hipotiroidismo adquirido a partir de la segunda década de la vida representan el 12-17% de los casos en pacientes con SD, de los que el 33% son de causa autoinmune, en cambio los casos de hipotiroidismo primario persistente, se observan solo en el 0,7% de los nacidos con SD, por lo que podemos decir que la disfunción tiroidea se incrementa con la edad, particularmente por encima de los 15 años.
La prevalencia de anticuerpos antitiroideos aumenta por encima de los 8 años de vida y, en ocasiones, antecede al estado hipotiroideo en 12 a 18 meses. En el 35% de los adolescentes con anticuerpos antitiroideos y estado de hipotiroidismo subclínico (elevación aislada de TSH con T4 normal), se desarrollará un hipotiroidismo franco.
Gibson et al.(9) hicieron el seguimiento de una cohorte de 103 niños con SD de 6 a 13 años de edad, y control a la edad de 10 a 20 años. Según estos autores, la probabilidad de presentar hipotiroidismo y precisar levotiroxina antes de los diez años de vida fue del 2%, si presentaban anticuerpos antitiroideos positivos en la segunda década de la vida, se elevaba esta probabilidad al 28% y llegaba al 34%, cuando se asociaba aumento aislado de la TSH. A la luz de estos resultados, los autores proponían el cribado anual de TSH solo cuando los resultados iniciales estén alterados (elevación aislada de TSH) y cada cinco años si la TSH es normal y no aparecen signos clínicos de sospecha de hipotiroidismo. En la adolescencia es conveniente, además, determinar la presencia de anticuerpos antitiroideos.
Dadas las limitaciones de este estudio (pequeño tamaño muestral, pérdida de pacientes), a falta de más pruebas, el grupo PrevInfad/PAPPS Infancia y Adolescencia(10) aconseja el cribado sistemático de hipotiroidismo mediante la determinación de TSH en los controles habituales de salud. En casos de elevaciones aisladas de TSH, se aconseja control anual para confirmar un estado de hipotiroidismo franco, añadiéndose en la evaluación la determinación de rT3. En los controles de salud de la edad escolar, se determinarán los anticuerpos antitiroideos si se detecta aumento de la TSH. Por otra parte, no existe evidencia sobre el beneficio del tratamiento hormonal sustitutivo en elevaciones aisladas de la TSH(11).
Problemas cardíacos
La frecuencia de cardiopatías congénitas es mayor en estos niños que en la población general. Cerca de la mitad de los sujetos con SD padecen algún tipo de cardiopatía susceptible de control por un cardiólogo pediátrico y/o de cirugía correctora (Tabla III).

Un examen clínico normal no excluye la presencia de cardiopatía. En la etapa neonatal, la mitad de los niños con cardiopatía no presenta síntomas y quedan sin diagnosticar, y a las 6 semanas, un tercio de los casos pueden seguir sin diagnóstico.
La ecografía cardíaca es la prueba diagnóstica más adecuada para detectar las anomalías del corazón y debe realizarse en todo niño con SD, siempre en la etapa neonatal, en niños mayores a los que nunca se haya realizado exploración (aunque no muestren signos de cardiopatía) y en la etapa de adolescente o adulto joven(12), ya que en estos, es frecuente la presencia de enfermedades cardíacas no congénitas, tales como: prolapso de válvula mitral (46%), en menor proporción prolapso de la válvula tricúspide, regurgitación aórtica, disfunción valvular y aumento del septo membranoso.
La mortalidad para cada tipo de malformación cardíaca es similar a la de niños sin SD, excepto en presencia de defecto atrioventricular completo, asociado a hipertensión pulmonar (13% vs 5%).
Trastornos odontológicos
Cabe destacar las siguientes características(13) en los pacientes con SD:
• Notable retraso en la erupción dentaria, tanto temporal como permanente. Erupción irregular de dientes, hipodontias (presente en el 60% de los niños), anodontias y agenesias en la dentición (frecuencia 4-5 veces mayor que en la población general) y dientes supernumerarios en el 6%. Aparición de manchas blanquecinas de hipocalcificación en el 18% de casos.
• Grave y acusado compromiso periodontal, que afecta sobre todo al sector anteroinferior. La severidad de la enfermedad periodontal aumenta con la edad, pudiendo afectar al 39% de la población adulta. En la población pediátrica con SD, se encuentra inflamación gingival hasta en un 67% de casos. La causa puede atribuirse a mala higiene bucal y alimentación inadecuada, junto a factores locales como: maloclusión, bruxismo y malposición dentaria.
• Alta tendencia a maloclusiones dentarias, debido en parte a la macroglosia y la hipoplasia del maxilar. La más frecuente es la mordida cruzada (78%) y la mordida abierta.
• Menor incidencia de caries, relacionada con el retraso de la erupción dentaria y la función tamponante de la saliva, entre otros.
• El bruxismo se observa hasta en el 70% de los niños, afectando a las superficies triturantes de los dientes.
Por todo ello, deben revisarse e indicarse todas las medidas de higiene bucodental tendentes a mejorar la técnica del cepillado, uso de pastas dentífricas fluoradas, enjuague con colutorios con flúor, utilización de la seda dental, control de la dieta y de hábitos perniciosos (chupete, biberón de noche) en el domicilio, y control de la placa bacteriana, junto a los sellados de fisuras. En los casos en los que sea preciso, debe instaurarse el tratamiento con ortodoncia a pesar de las dificultades debidas a la gravedad de las maloclusiones y a la deficiente colaboración de estos muchachos para el mantenimiento de la aparatología.
Enfermedad celíaca
La enfermedad celíaca (EC), como ocurre con otras patologías autoinmunes, es más frecuente que en la población general (4 al 7%), habitualmente de forma silente, asintomática o atípica, pasando desapercibida en niños con SD.
Por esta razón, en ausencia de síntomas sugestivos de EC, se recomienda el cribado sistemático mediante la determinación de marcadores serológicos a los 2-3 años de edad, siempre que haya estado tomando alimentación que contenga gluten, al menos durante un año. La determinación inicial será de anticuerpos antitransglutaminasa (ATGt-IgA) o ATGt tipo IgG, en los casos en que se asocia con déficit de inmunoglobulina tipo A (IgA).
Un resultado inicial negativo de los test serológicos de EC no excluye la posibilidad de que desarrolle la enfermedad a lo largo de la vida. La estrategia, para algunos autores, sería repetir los marcadores celíacos a los 6-7 años, o bien determinar la presencia de marcadores genéticos HLA DQ2 o DQ8, y si son positivos, continuar con los controles serológicos cada 2-3 años. No obstante, según un estudio que analizaba el coste-eficacia del cribado en niños con SD, no parece adecuado insistir en aquellos niños con un primer test negativo y sin ninguna sintomatología(14).
Trastornos de la audición
La prevalencia de hipoacusia es elevada, a veces manifestada en forma de conductas desajustadas pseudopsiquiátricas. Algunos estudios demuestran, en niños con SD con edades comprendidas entre 2 meses a 3 años de edad, que el 34% presentan normoaudición, el 28% tiene sordera unilateral y un 38% padecen sordera bilateral. Solo el 4% de los niños presentó sordera neurosensorial y la mayoría de ellos presentaba hipoacusia conductiva(15).
La correlación entre la audición, los problemas de adquisición y elaboración del lenguaje en niños con SD, obliga a manejar este problema de forma enérgica, especialmente en lo relativo a la hipoacusia de conducción secundaria a: otitis media serosa, colesteatoma, estenosis del conducto auditivo externo o impactaciones de cerumen en dicho conducto.
Los actuales protocolos proponen realizar cribado universal de hipoacusia sensorial en el primer semestre de vida (potenciales auditivos automatizados, test de otoemisiones acústicas, o bien evaluación de potenciales evocados auditivos del tronco cerebral (PET)). Después de los 6 meses, el cribado se realizará con pruebas de valoración basadas en reflejos conductuales audiológicos, impedanciometría o prueba de otoemisiones acústicas (OEA), dependiendo de la edad, nivel intelectual y estado de la audición.
Inestabilidad atlantoaxoidea
La inestabilidad atlantoaxoidea o subluxación atlantoaxoidea, definida por la existencia de un espacio de 5 mm o más entre el atlas y la apófisis odontoides del axis, está presente en el 10-20% de los menores de 21 años con SD, y es debida a la laxitud ligamentosa. Aunque la mayoría carece de síntomas, las formas sintomáticas pueden alcanzar el 1-2% de todos los niños con SD.
El diagnóstico se realiza mediante radiografía lateral de la columna cervical, en posición de flexión, neutra y en extensión, obtenida entre los tres y cinco años de edad.
Todos los niños con espacios superiores a 5 mm deben ser examinados en busca de síntomas de compresión medular (cansancio precoz, marcha anormal, parestesias en miembros, pérdida de fuerza, dolor o contracturas cervicales de repetición), estando indicada la realización de una resonancia magnética del área antes de decidir la restricción de la actividad deportiva o cualquier procedimiento que precise anestesia (maniobras que precisen de la hiperextensión del cuello).
La indicación de cribado en fase asintomática y, por ello, la incorporación de la técnica diagnóstica en las recomendaciones de actividades preventivas, es controvertida. Mientras que, el Medical Advisory Committee of the Special Olympics recomienda la realización de la radiografía previa a la participación en los Juegos Olímpicos desde 1983, el Committee on Sports Medicine and Fitness de la AAP indica que, no hay evidencia científica que permita concluir, que las radiografías laterales de columna cervical tengan en los pacientes con SD un valor detector del riesgo de desarrollar lesión del cordón espinal y consideran de mayor utilidad el seguimiento clínico para el reconocimiento de los pacientes con síntomas de compresión medular(16). Algunos autores de gran prestigio como Pueschel, resaltan que la inestabilidad atlantoaxial asintomática es una alteración, tan grave, como para justificar el trabajo y el gasto del cribado mediante radiología lateral(17).
En nuestro servicio, preconizamos realizar cribado universal a los 3 años de edad y seguimiento clínico en todas las revisiones. Para evitar la irradiación innecesaria, solo realizamos radiografía cervical dinámica pasados los 3 años cuando no tengan realizada la prueba previamente o antes de aquellos procesos quirúrgicos o anestésicos que precisen de la manipulación del cuello. Algunos protocolos repiten el cribado a los 6 años, nunca después de los 10, ya que no se ha demostrado su utilidad después de esa edad, en ausencia de signos o síntomas relacionados(18).
Inmunizaciones
Las vacunas recomendadas en la población con SD son las establecidas en los calendarios de vacunación para la población. Dada la “inmunosupresión relativa” de estos niños (déficit de subclases IgG2 e IgG4), las malformaciones anatómicas y la comorbilidad con cardiopatías y enfermedad respiratoria crónica, se recomienda vacunación antigripal anual, junto con otras vacunas como: varicela y neumocócica (tipo conjugada en menores de 5 años + vacuna polisacárida 23 valente, en mayores de 36 meses).
Otros problemas médicos
Los pacientes con SD pueden tener además otros problemas, algunos de ellos son observables mediante la aplicación del cribado recomendado en la población infantil en general, por ejemplo la criptorquidia; mientras que, en otros no existe evidencia científica de que necesiten intervenciones preventivas, pero que deben ser conocidos para diagnosticarlos en fases tempranas y permitir intervenciones precoces (Tabla IV).

Guía de actividades preventivas por grupos de edad, en niños con síndrome de Down
Teniendo en cuenta todas las patologías que se pueden asociar al síndrome, se han desarrollado una serie de guías de atención de la salud, en niños con SD. Entre ellas destaca la editada en 1999 por la Down Syndrome Quarterly(19) o la Medical Care and Monitoring for the Adolescent with Down Syndrome(20). Respecto al control de salud en adultos con síndrome Down, cabe destacar Health Care Management of Adults with Down Syndrome.
En nuestro servicio, recomendamos una serie de actividades que quedan reflejadas en la Tabla V:
• Iniciar programas de intervención temprana del desarrollo psicomotor, lenguaje y conducta alimentaria. Valorar el desarrollo psicomotor con especial referencia al área del lenguaje.
• Controlar el crecimiento físico con tablas estándar para niños con SD (preferiblemente de población autóctona), en cada visita (semestrales durante los 2 primeros años, anuales entre los 2 y 6 años, bianuales a partir de esa edad).
• Determinar en todas las visitas la TSH (al nacer se incluiría dentro del cribado de metabolopatías universal a los recién nacidos). En caso de disfunción tiroidea compensada (elevación aislada de TSH), repetir cada seis meses TSH, T4 y rT3 hasta que se normalice la función o se diagnostique de hipotiroidismo franco. En la edad escolar, determinar anticuerpos antitiroideos al menos en una ocasión (entre los 9 a 12 años).
• En la etapa neonatal, debe realizarse una ecografía cardíaca a los niños con SD. En niños mayores en los que nunca se haya realizado exploración y no muestren signos de cardiopatía, además de la exploración clínica, es recomendable realizarla también. En la etapa de adolescente y adulto joven, se repetirá ecografía cardíaca para descartar la disfunción de alguna de las válvulas del corazón.
• Realizar control, limpieza de la placa bacteriana y sellado de fisuras, a partir de los seis años y cada 6-12 meses por un odontólogo. A partir de los 8 años, debe realizarse estudio de maloclusión dentaria al menos bienalmente.
• En ausencia de clínica sugestiva de enfermedad celíaca, se determinarán a los 2-4 años los anticuerpos antitransglutaminasa (ATGtIgA) junto con cuantificación de IgA. Después de esa edad, repetir periódicamente la determinación de ATGtIgA cada 2-3 años o, al menos, hacer seguimiento clínico.
• Realizar cribado de hipoacusia en los primeros seis meses de vida, mediante test de otoemisiones acústicas, evaluación de potenciales evocados auditivos del tronco cerebral o de potenciales auditivos automatizados. Realizar cribado de hipoacusia con pruebas basadas en reflejos auditivos conductuales, impedanciometría u otoemisiones acústicas, cada año hasta los 6 años y después cada dos años.
• Realizar exploración oftalmológica al nacer, 6 y 12 meses y al menos cada 2 años.
• Realizar radiografía lateral cervical en posición neutra, flexión y extensión, entre los 3 y 5 años de edad.
• Inmunizar a los niños con SD, según el calendario vacunal vigente en cada comunidad autónoma. Inmunizar frente a neumococo, varicela y gripe, según pautas vacunales recomendadas para grupos de riesgo.

Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Bermejo E, Cuevas L, Mendioroz J, Martínez-Frías ML Vigilancia epidemiológica de anomalías congénitas en España en los últimos 24 años. Rev. de Dismorfología y Epidemiología. 2004; Serie V(3): 68-81.
2. Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002; 359 (9311): 1019-25.
3. Frid C, Drott P, Lundell B, Rasmussen F, Anneren G. Mortality in Down’s syndrome in relation to congenital malformations. J Intellect Disabil Res. 1999; 43 (Pt 3): 234-41.
4.*** Pueschel SM, Anneren G, Durlach R, Flores J, Sustrova M, Verman IC. Guidelines for optimal medical care of persons with Down syndrome. International League of Societies for persons with mental handicap (ILSMH). Acta Paediatr. 1995; 84 (7): 823-7.
5.*** VV. AA. Programa Español de Salud para personas con Síndrome de Down. Ed. FEISD (Federación Española del Síndrome de Down) Madrid 2011; 89 Págs. [disponible en Internet http://www.sindromedown.net/adjuntos/cPublicaciones/90L_downsalud.pdf con acceso 27-04-2014].
6. Riquelme I, Manzanal B. Factores que influyen en el desarrollo motor de los niños con síndrome de Down. Rev. Med Inter. Síndrome Down. 2006; 10 (2): 18-24.
7. Hines S, Bennett F. Effectiveness of early intervention for children with Down syndrome. Ment Retard Dev Disabil Res Rev. 1996; 2: 96-101.
8. Pastor X, Quintó L, Corretger M, Gassió R, Hernández M, Serés A. Tablas de crecimiento actualizadas de los niños españoles con síndrome de Down. Rev Med Intern síndrome de Down. 2004; 8 (3): 34-36
9. Gibson PA, Newton RW, Selby K, Price DA, Leyland K, Addison GM. Longitudinal study of thyroid function in Down’s syndrome in the first two decades. Arch Dis Child 2005; 90: 574-8.
10.*** Soriano J. Actividades Preventivas en síndrome de Down. Ed. PrevInfad (AEPap)/PAPPS Infancia y adolescencia. Madrid 2007: 19 Págs. [disponible en Internet https://www.aepap.org/previnfad/pdfs/previnfad_down.pdf con acceso 27-04-2014].
11. Tirosh E, Taub Y, Scher A, Jaffe M, Hochberg Z. Short-term efficacy of thyroid hormone supplementation for patients with Down syndrome and low-bordeline thyroid function. Am J Ment Retard. 1989; 93 (6): 652-6.
12. Geggel R, O`Brien J, Feingold M. Development of valve dysfunction in adolescents and young adults with Down syndrome and no known congenital heart disease. J Pediatr. 1993; 122: 821-3.
13. Pipa A, Álvarez J, Ruiz J. Síndrome de Down: alteraciones estomatológicas. Aspectos preventivos. Rev Esp Pediatr. 1999; 55 (4): 353-60.
14. Swigonski N, Kuhlenschmidt H, Bull M, Corkins M, Downs S. Screening for celiac disease in asymptomatic children with Down syndrome: Cost-effectiveness of preventing lymphoma. Pediatrics. 2006; 118: 594-02.
15. Shott SR, Joseph A, Heithaus D. Hearing loss in children with Down syndrome. Int J Pediatr Otorhinolaryngol. 2001; 61 (3): 199-205.
16. Committee on Sports Medicine and Fitness. AAP. Atlantoaxial instability in Down syndrome: Subject review (RE9528). Pediatrics. 1995; 96 (1): 151-4.
17. Pueschel SM. Should children with Down syndrome be screened for atlantoaxial instability? Arch Pediatr Adolesc Med. 1998; 152 (2): 123-5.
18. Brockmeyer D. Down syndrome and craniovertebral instability: Topic review and treatment recommendations. Pediatr Neurosurg. 1999; 31 (2): 71-7.
19. Cohen W. Health care guidelines for individuals with Down syndrome: 1999 revision. Down Syndrome Quarterly. 1999; 4 (3).
20. Roizen NJ. Medical care and monitoring for the adolescent with Down syndrome. Adolesc Med. 2002; 13 (2): 345-58, vii.
Bibliografía recomendada
- Pueschel SM, Anneren G, Durlach R, Flores J, Sustrova M, Verman IC. Guidelines for optimal medical care of persons with Down syndrome. International League of Societies for persons with mental handicap (ILSMH). Acta Paediatr. 1995; 84 (7): 823-7.
Una de las guías clásicas de cuidados médicos para personas con SD, la mayor parte de sus consejos todavía siguen vigentes. Por otra parte, su primer autor es una de las referencias, en cuanto a publicaciones relacionadas con SD.
- VV. AA. Programa Español de Salud para personas con Síndrome de Down. Ed. FEISD (Federación Española del Síndrome de Down) Madrid 2011; 89 Págs. [disponible en Internet http://www.sindromedown.net/adjuntos/cPublicaciones/90L_downsalud.pdf con acceso 27-04-2014].
Una revisión en español muy actualizada (la última revisión es del 2011) y ampliada. Da una visión pormenorizada del SD válida, tanto para profesionales como para familiares y tiene la ventaja de poder ser consultada por internet.
- Soriano J. Actividades Preventivas en síndrome de Down. Ed. PrevInfad (AEPap)/PAPPS Infancia y adolescencia. Madrid 2007: 19 Págs. [disponible en Internet https://www.aepap.org/previnfad/pdfs/previnfad_down.pdf con acceso 27-04-2014].
Protocolo de la AEPap para el seguimiento del síndrome de Down. Incluye valoración de los diferentes estudios en función de la evidencia científica.
| Caso clínico |
|
Varón de 15 días de vida diagnosticado postnatalmente de síndrome de Down, acude a nuestra consulta de Pediatría para iniciar controles de salud. Antecedentes familiares: padres jóvenes de 23 años, sin hábitos tóxicos ni antecedentes de consanguinidad. Un hermano sano de 2 años de edad. Historia familiar: un primo materno con SD (cariotipo 47 XY, + 21: trisomía primaria). Antecedentes personales: embarazo controlado de curso normal. Control ecográfico del embarazo: no se evidencian malformaciones cardíacas ni pliegue nucal. Parto hospitalario, a término (39 + 2 semanas), eutócico, presentación cefálica. Peso al nacimiento: 2.600 gramos. A la exploración en paritorio, destaca hipotonía generalizada, fenotipo Down, signo de la sandalia, mano en tridente, clinodactilia de 5º dedo de ambas manos y pliegue simiesco. Pruebas complementarias: Screening de metabolopatías pendiente de resultados. Test de hibridación in situ (HIS), compatible con trisomía 21 libre.
|
