|
| Temas de FC |
F. Santos Simarro*,**, E. Vallespín García*,**, M. Palomares Bralo*,**
*Instituto de Genética Médica y Molecular (INGEMM); Hospital Universitario La Paz, Madrid. **Unidad 753, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Instituto de Salud Carlos III, Madrid
| Resumen
En los últimos años se han producido grandes avances en genética, con un impacto global en la práctica de la medicina. Entre estos avances se encuentran las técnicas que permiten el análisis genómico de forma global o masiva, mediante los microarrays o la secuenciación masiva. Los microarrays más utilizados en la clínica son los de hibridación genómica comparada (array-CGH), que permiten detectar cambios en la dosis genómica (deleciones o duplicaciones), con un nivel de resolución muy superior al cariotipo. Actualmente, es la técnica inicial de elección para el estudio de pacientes con retraso del desarrollo psicomotor/discapacidad intelectual, trastornos del espectro autista o en pacientes dismórficos o con anomalías congénitas. La secuenciación masiva o NGS de sus siglas en inglés, permite secuenciar de forma paralela millones de fragmentos de ADN, ofreciendo la posibilidad de realizar estudios dirigidos a: un conjunto de genes (panel de genes), estudios de exoma completo o la secuenciación del genoma completo. |
| Abstract
Great advances in genetics with global impact on the practice of medicine have been made in recent years. Among these advances, microarrays and next generation sequencing offer the possibility to analyze great amount of ge-netic material simultaneously or the entire genome. Currently, the most commonly used microarrays are those of comparative genomic hybridization (array-CGH), which allow the detection of changes in genomic dose (deletions or duplications) with a much higher resolution than the karyotype. This is nowadays the technique of choice for the investigations of patients with delayed psychomotor development / intellectual disability, autism spectrum disorders or in patients with dysmorphic features or congenital anomalies. Next generation sequencing or NGS has made sequencing of millions of DNA fragments in parallel possible. Different NGS approaches include the study of targeted gene panels, whole exome or whole genome sequencing. |
Palabras clave: ADN; Gen; Microarray; Array-CGH; Secuenciación masiva; NGS.
Key words: DNA; Gene; Microarray; Array-CGH; Next generation sequencing; NGS.
Pediatr Integral 2019; XXIII (5): 241 – 248
Nuevas metodologías en el estudio de enfermedades genéticas y sus indicaciones
Introducción
Los avances en genética están modificando la práctica médica asistencial, por lo que cualquier profesional de la medicina ha de estar al corriente de dichos avances.
En los últimos años, se han producido grandes avances en genética, lo que ha supuesto un gran impacto, involucrando y modificando la práctica asistencial en todos los ámbitos de la medicina. Esto ocurre de forma particular en la Pediatría, debido a la presentación mayoritaria en edad infantil de las enfermedades genéticas. Entre dichos avances, se encuentran los avances en el diagnóstico molecular, que gracias a la aparición de nuevas tecnologías como son los arrays o la secuenciación masiva (también conocida como NGS – del término inglés Next Generation Sequencing), permiten realizar análisis genómicos globales. En particular, la NGS ha tenido un gran impacto en el descubrimiento e identificación de nuevos genes, lo que ha permitido conocer la causa de trastornos ya conocidos, cuya base molecular no había sido, hasta el momento dilucidada, así como la descripción de gran cantidad de entidades genéticas nuevas. Además, se han producido grandes avances en el diagnóstico prenatal, fundamentalmente con la llegada del diagnóstico prenatal no invasivo, que permite el diagnóstico de enfermedades genéticas del feto en una muestra de sangre materna, o el diagnóstico genético preimplantación, que posibilita la selección de embriones libres de un trastorno cromosómico o monogénico previamente conocido en la familia. Estos avances en el diagnóstico prenatal no se detallan en este artículo y referimos al lector al artículo correspondiente de este mismo número. Por tanto, cualquier profesional de la Pediatría debe estar familiarizado, tanto con estas nuevas técnicas como con conceptos básicos de genética molecular, que le permitan entender y aplicar los nuevos avances genéticos y genómicos en su práctica clínica habitual(1).
Principios de genética molecular
Para poder conocer e interpretar los avances en genética, es necesario estar familiarizado con los principios básicos de genética molecular.
La información genética se almacena en el núcleo de las células en forma de ADN, una doble hélice complementaria formada por la combinación de cuatro nucleótidos (adenina, guanina, citosina y timina). El ADN se empaqueta en el núcleo, alrededor de unas proteínas llamadas histonas, para formar nucleosomas que son la estructura básica de la cromatina. En el momento de la división celular, la información genética se transmite en forma de cromosomas. El ADN permite, mediante su replicación, la transmisión de la información genética de una célula a sus células hijas y, por tanto, de generación en generación. De esta forma, las células somáticas mediante la mitosis se dividen para dar lugar a células idénticas, mientras que la meiosis es el proceso que permite la formación de gametos que tienen un solo juego cromosómico.
El ADN está formado por elementos funcionales o genes que contienen fragmentos codificantes, denominados exones, que dan lugar a las proteínas, separados por regiones intermedias no codificantes, denominadas intrones. Los genes se transcriben a ARN mensajero y este, mediante la traducción, va a dar lugar a las proteínas. Aunque uno siempre se plantea este proceso como unidireccional, realmente la interacción entre el ADN, ARN y las proteínas, para regular la expresión de la información genética, es mucho más compleja y excede el objetivo de este artículo(2-4).
Estudios moleculares tradicionales
Las técnicas tradicionales siguen siendo de utilidad en estudios dirigidos. La secuenciación Sanger permite la detección de mutaciones puntuales, mientras que el MLPA permite detectar cambios de dosis (deleciones o duplicaciones) en regiones concretas del genoma.
La mayoría de los estudios moleculares utilizados en los laboratorios diagnósticos de genética molecular, se basan en la PCR (Polymerase Chain Reaction o reacción en cadena de la polimerasa), que permite seleccionar y amplificar miles de veces un fragmento de ADN de interés. La PCR se basa en la selección de un fragmento de ADN concreto a partir de unos oligonucleóticos o primers específicos, que se unirán a nuestra secuencia diana, la cual se replicará de forma exponencial en diferentes ciclos de desnaturalización, hibridación y elongación del ADN. Entre las técnicas clásicas se incluyen:
• Secuenciación Sanger: utiliza como base un producto de PCR y, en este caso, se leen (secuencian) de forma independiente ambas hebras de ADN con nucleótidos marcados con fluorescencia. El producto de la reacción de secuenciación se lee en un secuenciador automático, mediante una electroforesis capilar. Esta técnica permite detectar mutaciones puntuales y deleciones o inserciones de unos pares de bases en un gen de interés o un fragmento del mismo, pero no es una técnica útil para evaluar la dosis génica (grandes deleciones o duplicaciones) ni detectar alteraciones en mosaico por debajo del 30%.
• MLPA (Multiple Ligation-dependant Probe Amplification): es un método cuantitativo muy fiable que se basa en la hibridación de sondas específicas a una región de interés del ADN y su posterior ligación y amplificación (para más información sobre la técnica, visitar la web MRC-Holland: www.mlpa.com/). Esta técnica permite evaluar simultáneamente hasta 45 secuencias concretas de ADN en un mismo ensayo, detectando cambios en la dosis (deleciones y/o duplicaciones) de uno o varios exones de un gen o de regiones específicas. No permite, sin embargo, la identificación de mutaciones puntuales o reordenamientos en equilibrio. Ha reemplazado al FISH (Hibridación In Situ Fluorescente) en la mayoría de las ocasiones, aunque este sigue siendo útil para el estudio de reordenamientos en equilibrio.
Nuevas técnicas genómicas
Las nuevas técnicas genómicas permiten realizar estudios simultáneos de la totalidad o gran parte del genoma.
Las nuevas técnicas genómicas (microarrays y NGS) han reemplazado, en muchas ocasiones, a las técnicas clásicas como técnica de elección en el estudio de pacientes con sospecha de una enfermedad genética, siendo las técnicas clásicas todavía útiles, para estudios dirigidos o para confirmar hallazgos detectados con las nuevas técnicas genómicas.
Array-CGH
Los arrays-CGH permiten detectar cambios en la dosis genómica (deleciones o duplicaciones) con un nivel de resolución muy superior al cariotipo. Es la técnica inicial de elección para el estudio de pacientes con: retraso del desarrollo psicomotor/discapacidad intelectual, trastornos del espectro autista o en pacientes dismórficos o con anomalías congénitas.
Los microarrays permiten la detección de pérdidas y/o ganancias de material genético. Tradicionalmente, este tipo de alteraciones se han identificado mediante el estudio de los cromosomas en el cariotipo que permitía identificar alteraciones “microscópicas” con un tamaño comprendido entre las 5-10 Mb. Los arrays de hibridación genómica comparada, arrays de CGH (Comparative Genomic Hybridization) o microarrays de CGH permiten analizar simultáneamente cientos o miles de regiones del genoma e identificar pérdidas y/o ganancias genómicas con un nivel de resolución muy superior al cariotipo hasta un tamaño de Kb. El nivel de resolución se determina considerando, tanto el tamaño de la sonda empleada como la distancia genómica entre ellas o el número de sondas que tiene el array (p. ej., un array de 60 k tiene 60.000 sondas u oligonucleótidos distribuidos a lo largo del genoma). Los microarrays pueden diseñarse para cubrir cualquier región de interés y alcanzar distintas resoluciones.
Los arrays de CGH se basan en comparar el ADN del paciente en estudio con un ADN control. Para ello, una cantidad determinada de ADN extraído del paciente a estudiar, se marca con un fluoróforo de un color específico, mientras que la misma cantidad de ADN de la muestra control o ADN de referencia, se marca con un fluoróforo diferente. Los fluoróforos empleados habitualmente son: rojo y verde. Los dos ADN genómicos marcados, del paciente y la referencia, se mezclan, se desnaturalizan para que pasen de cadena doble a cadena sencilla y se hibridan sobre el microarray que contiene las sondas. Los ADN del paciente y la referencia compiten por hibridar con secuencias complementarias en las sondas del microarray. Posteriormente, se emplea un escáner láser de alta resolución para capturar y cuantificar la intensidad de señal fluorescente de uno y otro color que ha hibridado en cada sonda. A continuación, se calcula la relación de la intensidad de señal fluorescente en el ADN del paciente y el ADN de referencia para cada sonda del microarray, obteniéndose información sobre el número de copias relativo de secuencias en el genoma en el paciente en comparación con el genoma de referencia (Fig. 1A y 1B). Esta información es interpretada por un software de análisis que permite representar los datos en forma de “cariotipo molecular” (Fig. 1C).
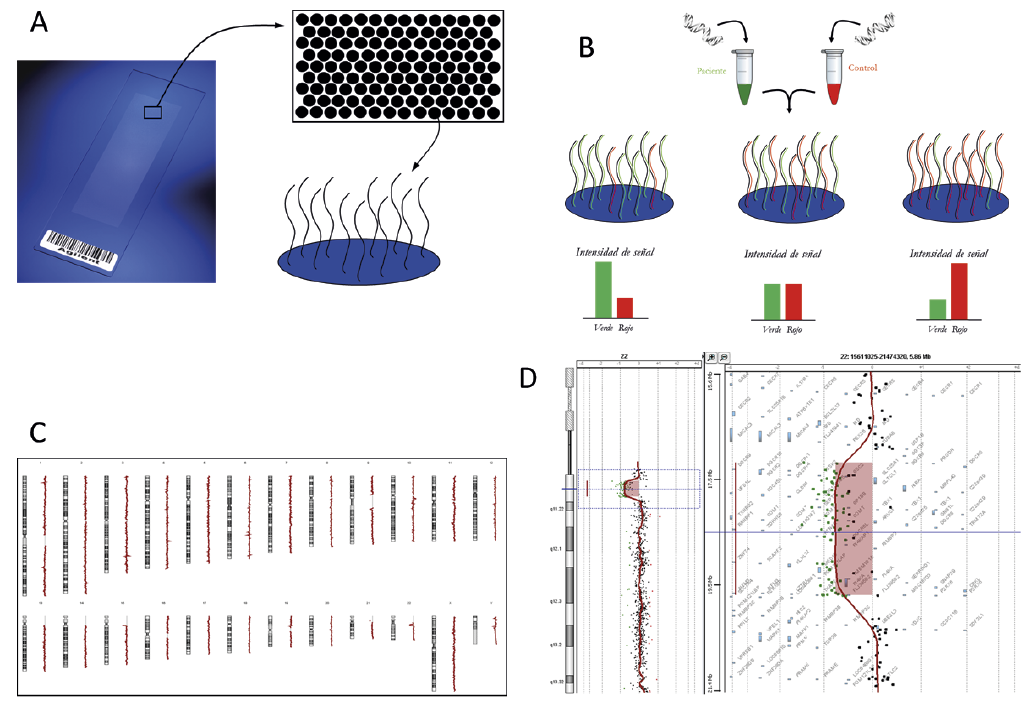
Figura 1. A. Portaobjetos de cristal en cuya superficie se fijan las sondas que componen el microarray. Cada uno de los puntos contiene una sonda, representando el conjunto de puntos el genoma completo. B. La técnica de array CGH consiste en marcar cantidades semejantes de ADN de un paciente y un control con fluoróforos diferentes. Una vez marcados se juntan, y la mezcla compite por hibridar con las sondas del microarray. A continuación, se mide la relación de la intensidad de señal, si es la misma, el paciente no presenta ganancias ni pérdidas genómicas, resultado normal; si la intensidad de señal verde es mayor, indica una ganancia de material genético en el paciente en esa región; una intensidad de señal roja mayor, indica una pérdida de material genético de la región representada por la sonda en el paciente. C. Representación del “cariotipo molecular”. D.Ejemplo de una deleción 22q11 detectada por array-CGH; a la izquierda de la imagen, visión global del cromosoma 22 y, a la derecha, visión ampliada de la región 22q11.
La principal ventaja del array CGH es la capacidad de detectar simultáneamente aneuploidías, deleciones, duplicaciones y/o amplificaciones de cualquier locus del genoma representado en el array. La utilidad de esta tecnología para la detección de ganancias y pérdidas de material genético ha sido bien documentada y, por tanto, los microarrays han de ser considerados como técnica de estudio inicial en pacientes con(5,6):
• Retraso del desarrollo psicomotor o discapacidad intelectual.
• Trastornos del espectro autista.
• Rasgos dismórficos y/o anomalías congénitas.
La tasa diagnóstica del array de forma global, en estas situaciones, se estima en un 10-20%.
Las limitaciones de los arrays de CGH incluyen:
• No identifican reordenamientos cromosómicos equilibrados (como translocaciones e inversiones), en los cuales no se producen pérdidas o ganancias de material genómico.
• No detectan algunos tipos de poliploidía (más de los 2 conjuntos habituales de cromosomas), como la triploidía.
• No detectan cambios (mutaciones) puntuales en el ADN.
• No detectan cambios de dosis en mosaico por debajo del 10-20%.
Además de las plataformas de arrays de CGH, existen otros tipos de microarrays como los arrays de polimorfismos de un solo nucleótido (SNP) que permiten detectar: deleciones y duplicaciones submicroscópicas, amplificaciones, pérdida de heterocigosidad y disomías uniparentales. Además, pueden usarse para realizar estudios de asociación de genoma. Actualmente, estos tipos de arrays se utilizan de forma mayoritaria en el ámbito de la investigación.
Secuenciación masiva (NGS)
La NGS permite secuenciar, de forma paralela, millones de fragmentos de ADN, pudiendo realizar diferentes abordajes, estudios dirigidos a un conjunto de genes mediante: paneles personalizados, estudios de exoma completo (WES-Whole Exome Sequencing) o la secuenciación del genoma completo (WGS-Whole Genome Sequencing).
La técnica de secuenciación masiva (NGS, de sus siglas en inglés Next Generation Sequencing) es una nueva tecnología en el campo de la genética, que está suponiendo una revolución en todos los ámbitos de la biología y de la medicina. De hecho, la secuenciación masiva es una novedosa técnica que tiene muchas ventajas en relación con los microarrays, por lo que es más que posible que con la rápida disminución de los costes(7), esta robusta tecnología acabe desplazando a los microarrays en los próximos años.
La NGS, al contrario que la secuenciación Sanger (también denominada clásica), permite secuenciar, de forma paralela, millones de fragmentos de ADN. Esto hace posible detectar diferentes tipos de cambios en un único experimento, incluyendo: variantes de nucleótido único o mutaciones puntuales, pequeñas inserciones y deleciones y, según el diseño, también variantes estructurales equilibradas y desequilibradas. La secuenciación masiva, debido a su elevada sensibilidad, también permite identificar mutaciones presentes en un porcentaje muy pequeño de células (mosaicismos y/o contaminaciones).
Hasta el momento, la estrategia para el estudio de una patología genética era el estudio secuencial de los genes candidatos conocidos hasta la fecha. Se hacía un barrido, uno por uno, de los genes hasta que se detectaba, o no, la mutación responsable del fenotipo, siendo este abordaje lento y costoso y, especialmente, en patologías con mucha heterogeneidad genética, muchas veces infructuoso. Ahora, con la NGS, el abordaje cambia completamente, pudiendo hacerse, por ejemplo, el estudio de todos los genes responsables de esa patología en un solo experimento. Todo esto ha llevado a una reducción enorme de los costes de secuenciación y a una mejora en los tiempos de repuesta; pero el auge de la NGS ha llegado acompañado de una gran generación de información y datos que, debido a su magnitud, han hecho necesario el desarrollo de plataformas específicas para almacenar y gestionar el volumen que se genera. Esto ha dado lugar a la incorporación de un nuevo perfil profesional dentro del ámbito biosanitario: el bioinformático, que se encarga del análisis, gestión, almacenaje, control de calidad, etc., de los datos derivados de la secuenciación masiva. Tras los análisis llevados a cabo por el bioinformático, los resultados pueden ser evaluados por un especialista en genética humana en el contexto clínico del paciente, quien emitirá un informe de secuenciación masiva (Fig. 2).

Figura 2. Ejemplo del flujo de trabajo en el diagnóstico de una paciente con discapacidad intelectual sindrómica. A.Decisión inicial de estudio de genoma completo, exoma o panel de genes candidatos. B. Flujo de trabajo en el laboratorio (wet-lab) y a nivel bioinformático (dry-lab). C. En la parte superior, visor genómico de análisis de los resultados; en la parte inferior, confirmación por secuenciación Sanger de la mutación detectada en el gen MAGEL2.
Indicaciones y estrategias de estudio con NGS
Con la secuenciación masiva, se pueden plantear diferentes estrategias a la hora del diagnóstico del paciente. Es posible realizar estudios dirigidos a un conjunto de genes mediante paneles personalizados, se pueden hacer estudios de exoma completo (parte codificante del genoma) y que se denominan WES (Whole Exome Sequencing) o incluso la secuenciación del genoma completo, WGS (Whole Genome Sequencing).
• Estrategia dirigida, mediante paneles personalizados y diseñados específicamente para un grupo de patologías. Es la estrategia más adecuada en enfermedades que están muy bien definidas clínicamente, para las cuales además se conocen la mayoría de los genes implicados y muestran heterogeneidad genética baja. Por ejemplo, en el estudio de rasopatías o trastornos del ritmo cardiaco.
• Secuenciación del exoma completo (WES). Es más apropiada para enfermedades que tienen mayor heterogeneidad fenotípica y genética. El exoma constituye aproximadamente el 2-3% del genoma completo. Este abordaje permite identificar mutaciones en las regiones del genoma más susceptibles a producir enfermedades genéticas, sin necesidad de secuenciar todo el material genético. Hay que tener en cuenta que, el 85% de las mutaciones causantes de enfermedad se encuentran en las regiones codificantes o sitios de splicing (procesamiento que permite eliminar las regiones no codificantes de los genes o intrones). La secuenciación del exoma presenta una alternativa eficaz, sin sesgo y coste-efectiva a la secuenciación del genoma para el estudio de las bases genéticas de la enfermedad. Esta técnica ha demostrado, por ejemplo, su utilidad en el estudio de pacientes con discapacidad intelectual sindrómica de causa no aclarada, teniendo un rendimiento diagnóstico dependiendo de las series de entre un 25-40%(8-10).
• Secuenciación del genoma completo (WGS). Consiste en la secuenciación completa de todo el material genético(11). Este tipo de análisis aún no se ha incorporado al diagnóstico clínico de forma rutinaria, porque tanto la cantidad de datos generada como el coste, lo convierten en un estudio poco coste-efectivo, aunque lo más probable es que en un futuro, con el descenso en los precios de la secuenciación, solo se hagan estudios de genoma completo, aplicando filtros bioinformáticos para analizar regiones concretas, pero teniendo toda la información genética del individuo disponible.
Hay que tener en cuenta, que al ser un campo tan novedoso, las técnicas son muy dinámicas y están en constante evolución, por lo que cada pocos años o meses aparecen nuevas aproximaciones y protocolos.
Particularidades de los estudios genéticos
Las nuevas técnicas genómicas han incrementado la posibilidad de detectar hallazgos de significado clínico incierto o incidentales que pueden tener implicación, tanto para el paciente como para su familia, por lo que es fundamental realizar un asesoramiento pre y pos test y obtener el consentimiento informado correspondiente.
Las técnicas que permiten el análisis genómico global como son los microarrays y, sobre todo, la secuenciación masiva, han multiplicado la posibilidad de detección de las bases moleculares de las enfermedades genéticas, pero, a la vez, han incrementado notablemente la detección de hallazgos de significado clínico incierto o de hallazgos incidentales (aquellos que pueden tener consecuencias médicas, pero que no están relacionados con el problema médico que motivó el estudio, como por ejemplo, variantes patogénicas o probablemente patogénicas en genes causantes de enfermedades cardiovasculares que predisponen a muerte súbita o en genes que predisponen al desarrollo de cáncer hereditario), que pueden ser relevantes tanto para el paciente como para su familia(12). Por ello, es necesario un asesoramiento genético pre y pos test con su consentimiento informado correspondiente, en el que se informe a los pacientes y sus familias de estos posibles hallazgos. Para minimizar estos hallazgos, sigue siendo de vital importancia que el estudio genético esté guiado por una sospecha clínica basada en una caracterización clínica exhaustiva del paciente y, en la medida de lo posible, realizar un estudio dirigido a los potenciales genes casuales(13,14).
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Manolio TA, Chisholm RL, Ozenberger B, Roden DM, Williams MS, Wilson R, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013; 15: 258-67.
2. García Nieto V, Exeni R, Medeiros M, Santos F. Nefrología Pediátrica. Universidad de Oviedo, Oviedo, 2019 (en prensa).
3.*** Nussbaum RL, McInnes RR, Willard HF. Thompson and Thompson Genetics in Medicine. 7th edition. Saunders Elsevier. 2007.
4.*** Read A, Donnai D. New Clinical Genetics. 3rd edition. Scion Publishing Limited, Oxfordshire, UK, 2007.
5.*** Moeschler JB, Shevell M, Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014; 134: e903-18.
6. Moeschler JB. Medical genetics diagnostic evaluation of the child with global developmental delay or intellectual disability. Curr Opin Neurol. 2008; 21: 117-22.
7. Van Nimwegen KJM, van Soest RA, Veltman JA, Nelen MR, van der Wilt GJ, Vissers LELM, et al. Is the $1000 Genome as Near as We Think? A Cost Analysis of Next-Generation Sequencing. Clinical Chemistry. 2016; 62: 1458-64.
8. Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, et al. Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. 2017; JAMA; 171: 855-62.
9. LaDuca H, Farwell KD, Vuong H, Lu HM, Mu W, Shahmirzadi L, et al. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PloS One. 2017; doi:10.137.1.
10. Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016; 89: 275-84.
11.*** Van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care. Recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013; 21: 580-4.
12. Blackburn HL, Schroeder B, Turner C, Shriver CD, Ellsworth DL and Ellsworth RE. Management of Incidental Findings in the Era of Next-generation Sequencing. Current Genomics. 2015; 16: 159-74.
13. Hennekam RC, Biesecker LG. Next-generation sequencing demands next-generation phenotyping. Hum Mutat. 2012; 33: 884-6.
14. Guttmacher AE, Collins FS, Carmona RH. The Family History – More Important Than Ever. NEJM. 2004; 351:2333-6.
15. Palacios-Verdú MG, Pérez-Jurado LA. Nuevas metodologías en el estudio de enfermedades genéticas y sus indicaciones. Pediatr Integral. 2014; XVIII(8): 515-28.
Bibliografía recomendada
– Nussbaum RL, McInnes RR, Willard HF. Thompson and Thompson Genetics in Medicine. 7th edition. Saunders Elsevier. 2007.
Texto que explica los principios básicos de la genética, así como las novedades en genética molecular, con una orientación práctica.
– Read A, Donnai D. New Clinical Genetics. 3rd edition. Scion Publishing Limited, Oxfordshire, UK, 2007.
Texto que aborda los diferentes aspectos de la genética con un abordaje diferente, basado en casos prácticos.
– Moeschler JB, Shevell M, Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014; 134(3): e903-18.
Artículo que recoge las indicaciones de estudio en pacientes con retraso del desarrollo psicomotor/discapacidad intelectual.
– Van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care. Recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013; 21: 580-4.
Recomendaciones de la sociedad europea de genética para la implementación de estudios de secuenciación masiva en la práctica asistencial.
| Caso clínico |
|
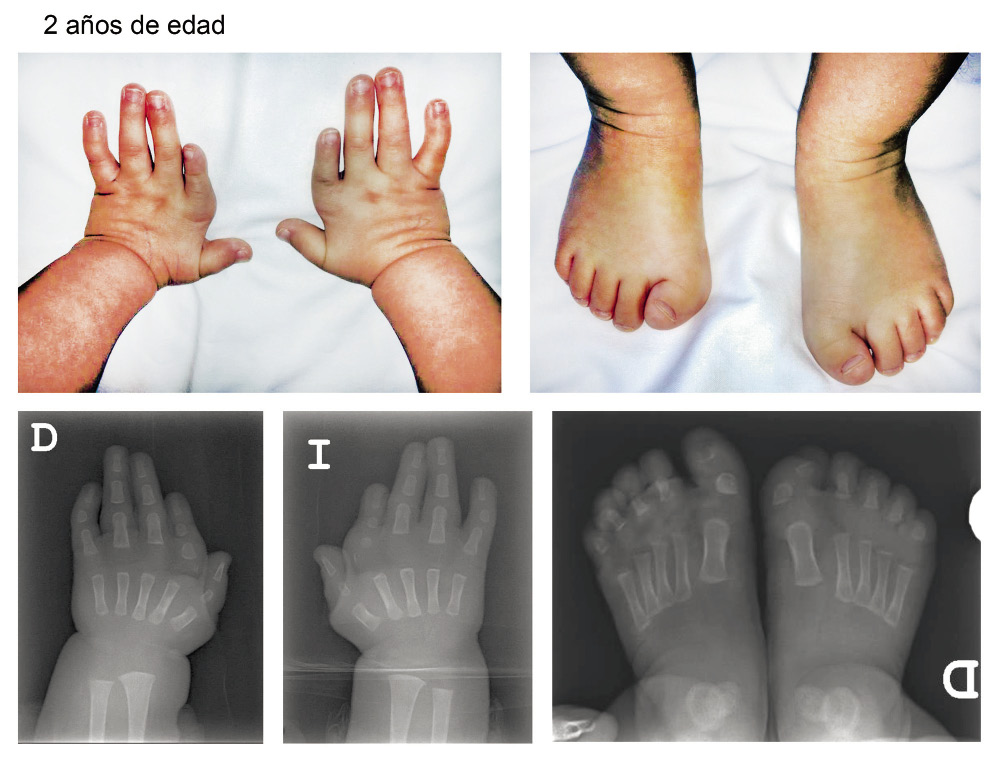
Niña con braquidactilia y hallux valgus remitida a los 2 años de edad para valoración y estudio. Antecedentes familiares y personales Primera y única hija de padres no consanguíneos. Madre con neuropatía idiopática. Sin otros antecedentes familiares de interés. Embarazo controlado, de curso normal. Parto normal, a término, peso al nacimiento 3.200 g. Periodo neonatal: diagnóstico de la malformación de manos y pies, consistente en: desviación cubital de los dedos y acortamiento del 2º dedo, pulgar adducto, falange delta en primeros dedos de los pies. Hasta el momento, ha recibido tratamiento ortopédico. No otros problemas médicos de relevancia. Crecimiento y desarrollo psicomotor dentro de la normalidad. Ha sido valorada en Neurología y Cardiología en su hospital de origen. Exploración física Talla: 85 cm (p10), perímetro cefálico 48,5 cm (p50). Proporciones corporales normales, piel normal. Craneofacial: sin rasgos faciales particulares, paladar un poco alto y estrecho. Tórax y abdomen normales. Extremidades: pulgares adductos, acortamiento del 2º dedo. 5º con clinodactilia. Halluces con desviación externa. Estudios complementarios solicitados - Serie ósea: falange proximal del primer dedo de las manos en forma de delta, así como la del segundo y tercero. Braquidactilia de 2º dedo de manos. Clinodactilia del quinto dedo de ambas manos. Hallux valgus bilateral. - Estudio de hibridación genómica comparada con array de 60.000 oligonucleótidos (KaryoArray v2.0, Agilent): resultado normal, no se detectan pérdidas ni ganancias significativas de material genómico. - Estudio molecular mediante secuenciación Sanger de los genes GDF5, BMPR1B, BMP2 asociados a braquidactilia: resultado normal, no se detectan variantes de relevancia clínica. Evolución Se mantiene el seguimiento y, en la evolución a los 5 años de edad, tiene un desarrollo psicomotor dentro de la normalidad. Refieren bastantes episodios de infecciones de vías respiratorias superiores. Seguimiento en endocrinología por talla baja, estudio hormonal normal. Usa gafas por astigmatismo e hipermetropía, no problemas de audición. Seguimiento en traumatología, utiliza plantillas.
Exploración física a los 5 años de edad Talla: 108 cm (p10), perímetro cefálico: 49,5 cm (p15). Proporciones corporales normales, piel atópica. Craneofacial: cejas perfiladas, sinofridia, ojos un poco hundidos. Tórax ligeramente asimétrico, abdomen normal. Extremidades: pulgares adductos, acortamiento del 2º dedo. 5º con clinodactilia. Halluces con desviación externa. Resultado de nuevos estudios genéticos realizados Estudio molecular mediante secuenciación masiva de un panel de 327 genes implicados en displasias esqueléticas (SkeletalSeq V4): resultado normal, no se detectan variantes de relevancia clínica. Posteriormente, se revisan las manifestaciones clínicas y radiológicas y la literatura científica en la que ha habido novedades, ya que en el año 2017 (Balasubramanian, et al.) se ha identificado la variante c.266A>G; p.Tyr89Cys en heterocigosis en el gen ERF, en pacientes con diagnóstico de síndrome Chitayat (OMIM 617180), cuyas manifestaciones solapan con las de nuestra paciente. Se revisa el estudio de secuenciación masiva del panel de displasias esqueléticas realizado, en el que se confirma la presencia de la variante en heterocigosis c.266A>G; p.Tyr89Cys en el gen ERF (NM_006494.2). Dicha variante se había filtrado en el análisis inicial, al no estar relacionada en ese momento con patología. Se confirma este hallazgo mediante secuenciación Sanger, así como su ausencia en la muestra de sus padres. Impresión diagnóstica y asesoramiento genético El estudio de secuenciación masiva ha confirmado la presencia de la variante en heterocigosis c.266A>G; p.Tyr89Cys en el gen ERF (NM_006494.2) en la paciente, así como la ausencia de la misma en sus padres, lo que confirma el diagnóstico de síndrome de Chitayat. Hasta el momento, se han descrito solamente una media docena de casos en la literatura con este diagnóstico, siendo sus manifestaciones ortopédicas similares a la de la paciente. De forma adicional, se describen problemas respiratorios, incluyendo: distrés respiratorio neonatal, broncomalacia o enfermedad intersticial pulmonar. El diagnóstico permite realizar un seguimiento personalizado de la paciente. Las mutaciones en este gen, se transmiten siguiendo un patrón de herencia autosómica dominante, lo que significa que una persona afectada tiene una probabilidad de dos (50%) de trasmitirla a cada uno de sus hijos. En el caso de esta paciente, la mutación habría aparecido por primera vez (“de novo”) en ella. Por tanto, la probabilidad de que se repita el problema en una próxima gestación de padres sanos se considera mínima y se estima en un 1%, debido a la hipotética y remota posibilidad de “mosaicismo germinal o gonadal”. No estaría, por tanto, indicado estudiar a su hermana sana. Plan Valoración y seguimiento por parte de Neumología; mantener seguimiento por parte de Traumatología.
|