Diagnostic and therapeutic approach to children with malformations, dysmorphological phenotype or features suggestive of genetic disease
Diagnostic and therapeutic approach to children with malformations, dysmorphological phenotype or features suggestive of genetic disease  |
| Topics on Continuous Training |
R. Arroyo Ruiz*, P. Prieto Matos**![]()
*Rare Diseases Diagnosis Unit of Castilla y León. **Department of Biomedical and Diagnostic Sciences. University of Salamanca. Rare Diseases Diagnostic Unit of Castilla y León. Salamanca University Hospital. Salamanca Biomedical Research Institute.
| Abstract
The diagnostic and therapeutic approach to patients with rare diseases is challenging, due to the current rapid progress and development in the field of Genetics. The characteristics of the multiple pathologies, both in natural history and in the mechanism of production and development, highlight the importance of a detailed medical history and an exhaustive and systematic clinical examination as essential pillars in this process, as well as relevant background information that can guide us in the selection of the correct diagnostic test. In this regard, a comprehensive approach that combines research, specialized clinical care and long-term follow-up in rare diseases in the pediatric setting is key. To this end, collaboration between primary care pediatricians, hospital specialists and pediatricians trained in Genetics is crucial in order to achieve comprehensive care for affected families. The aim of this review is to describe in a succinct but precise way, the diagnostic approach to these patients, focusing on the relevance and rationale of a proper medical history and examination, and then with this information to perform a correct clinical interpretation and genetic counseling of the variants obtained, along with a review of the current therapeutic options. |
| Resumen
El abordaje diagnóstico y terapéutico de los pacientes con enfermedades raras supone un reto, debido a la rápida evolución y auge existente en la actualidad en el ámbito genético. Las peculiaridades de las múltiples patologías, tanto en historia natural como en mecanismo de producción y desarrollo, hacen que la importancia de una anamnesis detallada y una exploración clínica exhaustiva y sistemática sean pilares fundamentales en este proceso, así como antecedentes relevantes que nos puedan orientar a la elección de la prueba diagnóstica correcta. En este aspecto, es clave un enfoque integral que combine la investigación, la atención clínica especializada y el seguimiento a largo plazo en las enfermedades raras en el ámbito pediátrico. Para ello, es importante la colaboración entre pediatras de Atención Primaria, especialistas de consultas y pediatras con formación en genética, con el fin de conseguir una atención integral a las familias afectadas. El objetivo de esta revisión es describir de forma sucinta pero precisa, el enfoque diagnóstico de estos pacientes, centrándose en la relevancia y justificación de una buena anamnesis y exploración, para posteriormente con esa información realizar una correcta interpretación clínica y asesoramiento genético de las variantes obtenidas, junto a un repaso de las opciones terapéuticas actuales. |
Key words: Malformation; Dysmorphology; Massive sequencing; Diagnosis; Treatment.
Palabras clave: Malformación; Dismorfología; Secuenciación masiva; Diagnóstico; Tratamiento.
Pediatr Integral 2024; XXVIII (5): 289 – 298
AIMS
• To highlight the importance of comprehensive assessment of individuals in whom a rare disease is suspected.
• To establish the steps that must be taken, from suspicion of the rare disease to reaching the diagnosis.
• To aid the pediatrician to correctly interpret the results of genetic studies and communicate this information effectively to the patient.
• To provide information about additional resources that may be useful in the diagnostic process of genetic diseases.
|
|
|


Diagnostic and therapeutic approach to children with malformations, dysmorphological phenotype or features suggestive of genetic disease
Introduction
Relevance of Genetics and definitions
Genetics in medicine advances with technology, facilitating the identification of diseases. Rare diseases, often genetic, require high suspicion to reach a diagnosis and treatment.
The importance of Genetics in medical practice has been increasing in relation to the great advance of molecular and genomic technology, making it easier to identify the genes that cause diseases, allowing the establishment of phenotype-genotype relationships and developing treatments from the genetic view.
Rare disease is defined as one whose frequency is less than 1 in 2,000 people. A large part of these diseases are due to a genetic cause, and knowledge of the particularities of the diagnosis of these diseases is important, both at a clinical and at a molecular study level(1,2). The part of Genetics that is dedicated to the diagnosis and prevention of these pathologies is Clinical Genetics, with health professionals, such as clinical geneticists and dysmorphologists, playing a main role(3).
Dysmorphology, on the other hand, would be the science that deals with the study of human morphological variants and anomalies, with the intention of recognizing certain patterns and particular combinations that guide the discernment of different genetic diseases(4).
Malformations are anomalies that occur throughout fetal development and can affect part or all of an organ or an anatomical structure, and may be major (they have important consequences for the health or life of the individual) or minor (they produce little impact on the health of the individual).
When to suspect a rare disease
Suspecting one of these diseases, usually genetic, is not always easy, but there are certain factors that could help us determine which patients should be considered to have a genetic-based pathology.
Malformations, abnormalities in the morphology of a bodily structure or organ produced by abnormal development, can be indicative(5). Two or more major or organic malformations, or one major and two minor ones, should make us think that the probability of a genetic disorder is very high(6).
Also, there are certain signs and symptoms, which will be commented later, that can make us suspect genetic syndromes, such as different sexual development, loss of already acquired developmental milestones, congenital hypotonia, skeletal dysplasias, marked congenital hypoacusis, chronic syndromes resistant to conventional treatment, consanguinity… All these symptoms should raise suspicion and referral to a rare diseases clinic(7).
Initial patient evaluation
Medical history
Family history, detailed with a family tree, is essential to identify genetic diseases. In addition, the personal history should highlight developmental milestones and review all medical aspects relevant to an accurate diagnosis. This meticulous process should provide a comprehensive view of the patient’s situation.
Family history is one of the most important tools to be used when determining the suspicion of a genetic disease, since it will inform of the type of inheritance pattern that the disease may have.
When collecting this history, it is important to record the moment in which they are obtained, since evolutionary changes can help us make the diagnosis, making it necessary to know the moment in which these have occurred.
Among the most relevant family history in the diagnostic evaluation (Table I) are: the age of the parents at the time of conception, where advanced age, especially in paternity, suggests the possibility of de novo diseases, while in maternity may indicate a chromosomal pathology; consanguinity in the family points towards diseases with a recessive inheritance pattern; Repeated miscarriages in family members or in the same individual may be associated with diseases that are repeated in family members (leading to recessive diseases or the existence of mosaicism) or balanced chromosomal translocations(8) that, although they do not present clear phenotypic manifestations, can cause problems of infertility and recurrent miscarriages; the use of assisted reproduction techniques has been linked to a greater risk of pathologies related to imprinting, such as Beckwith-Wiedemann syndrome(9); in addition, family history of important childhood diseases, such as congenital malformations, early hearing loss, cardiac or neurological defects, are also relevant elements to consider in the diagnostic evaluation.

All this family history must be collected in the most exhaustive and detailed way, and must be accompanied by a family tree that visually collects the family history. This family tree will allow us, graphically, to estimate inheritance patterns, identify relatives who are at risk and facilitate the graphical interpretation of segregation studies. The preparation of the family tree must be carried out using a series of standardized symbols, which are defined in international recommendations(10-12) (Fig. 1).
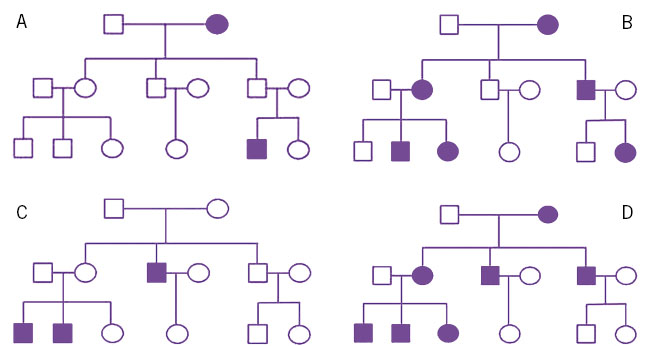
Figure 1. Family trees, showing different inheritance patterns. A. Autosomal recessive. B. Autosomal dominant. C. Linked to X. D. Mitochondrial.
At the current time, the need to move towards “paperless hospitals” presents a challenge for the development of quality family trees, but there are applications and resources (which will be named later) that can be helpful in this fundamental process for interpretation and management of information.
There are some peculiarities that can generate confusion in the interpretation of inheritance, such as incomplete penetrance, variable expressivity, de novo variants and mosaicisms. Incomplete penetrance refers to the fact that some carriers of a specific genotype do not express the associated trait, while others do. On the other hand, variable expressivity implies that the same genetic alteration can manifest itself in different people with varying degrees of severity. De novo variants are those that arise spontaneously in an individual and are not present in any of his/her parents. Mosaicism refers to the presence of two or more genetically distinct cell populations within the same individual, which can influence the phenotypic expression of a genetic disease. These peculiarities must be taken into account in the interpretation of heredity and in genetic counseling.
Personal history, from prenatal and perinatal to postnatal, is crucial to understand rare diseases(13). Relevant aspects include: the length of gestation, both due to excess and deficiency, which can be associated with chromosomopathies; alterations in the amniotic fluid, which may indicate conditions, such as esophageal atresia in polyhydramnios(14) or kidney malformations in oligohydramnios; infections during pregnancy, useful for the differential diagnosis of genetic syndromes and congenital infections; the type of birth, which can trigger neurological sequelae that help distinguish between birth or congenital causes; gestational age; weight-length gain, which may indicate disorders, such as Silver-Russel syndrome or bone dysplasias. Developmental milestones and their progression play a fundamental role due to the high prevalence of global developmental delay and intellectual disability in rare and genetic diseases. Therefore, in any patient with malformations, alterations in phenotype or in whom we suspect a genetic disease, it is essential to pay attention to the warning signs that may indicate a neurodevelopmental anomaly(15).
The complexity of genetic diseases means that the information obtained in the current disease section must be as complete, orderly and precise as possible. It should start with open questions so that parents can point out the most important problems. Once the initial information is obtained, it is important that the clinical history is directed with semi-directed questions, obtaining symptoms and their onset time from all the devices and systems. Obtaining information, chronic or not, on all diseases: neurological, cardiological, ophthalmological, endocrinological, immunological… will allow us to subsequently make a good clinical-genetic relationship of all the patient’s data.
It is equally important to record both positive and negative symptoms. Many times, in subsequent clinical-genetic evaluations we may encounter findings that make us doubt, and the fact that we have also recorded negative data can help us rule out possible diagnoses.
Clinical reassessment may be as important or more important than the initial assessment. Longitudinal follow-up can give us new points of view, demonstrate new symptoms or others that went unnoticed. In this type of consultation, all data is important and should be reflected in the clinical history, as it may be useful to interpret subsequent genetic studies.
Dysmorphological examination
The examination must be complete, systematic, orderly and detailed. Every detail counts and everything must be reflected in the medical history, both positive and negative data. Any detail can be relevant. It is important to collect photographs to be able to consult data that may have escaped in the initial examination.
If the physical examination in a medical consultation is important, in the study of rare diseases it is crucial and needs to be complete, systematic, orderly and detailed. In today’s medicine, so focused on complementary tests and with advances in molecular genetics that allow a genome to be sequenced in less than a week, physical examination in rare diseases is essential. Every detail, no matter how small it may seem, can be essential to reach the diagnosis. In some cases, it will guide the genetic study, while in others, if well performed, it can be diagnostic. Every clinical geneticist, dysmorphologist or general pediatrician must know the gratifying sensation of, performing an exhaustive examination, reaching a diagnosis that seemed impossible minutes before. Details, such as large central incisors, a dimple in the earlobe, a small heterochromia in the iris, ulnar hypertrichosis, Madelung deformity or a prominent heel, can point towards syndromes, such as KBG, Mowat-Wilson, Waardenburg, Wiedemann-Steiner, Leri-Weill or 3M, respectively. This approach of maximum detail and systematicity guarantees a comprehensive analysis of the patient, facilitating the detection of clinical patterns and favoring early diagnosis, especially in dysmorphology consultation.
The examination systematic can be established in any way, we recommend establishing a descending order to help remember all the necessary sections. This examination must be accompanied by taking photographs (with written permission) that allow them to be consulted with hindsight, either by studying the clinical case or receiving candidate variants from a genetic study that allow us to make a correct interpretation of it.
This examination should begin with a general inspection, which constitutes the first evaluation of the individual. In this phase, the following are evaluated: body attitude, somatotype, posture, gait, possible asymmetries and any physical resemblance to other family members.
Subsequently, basic anthropometry is performed, which includes measurements of weight, height and head circumference, and can be extended to other common measurements, such as breaststroke, sitting height and lower segment and even other less frequent measurements, taking into account the existence of normality tables for a wide variety of parameters(16). All anthropometric assessments should be percentilated and, if possible, accompanied by a standard deviation score.
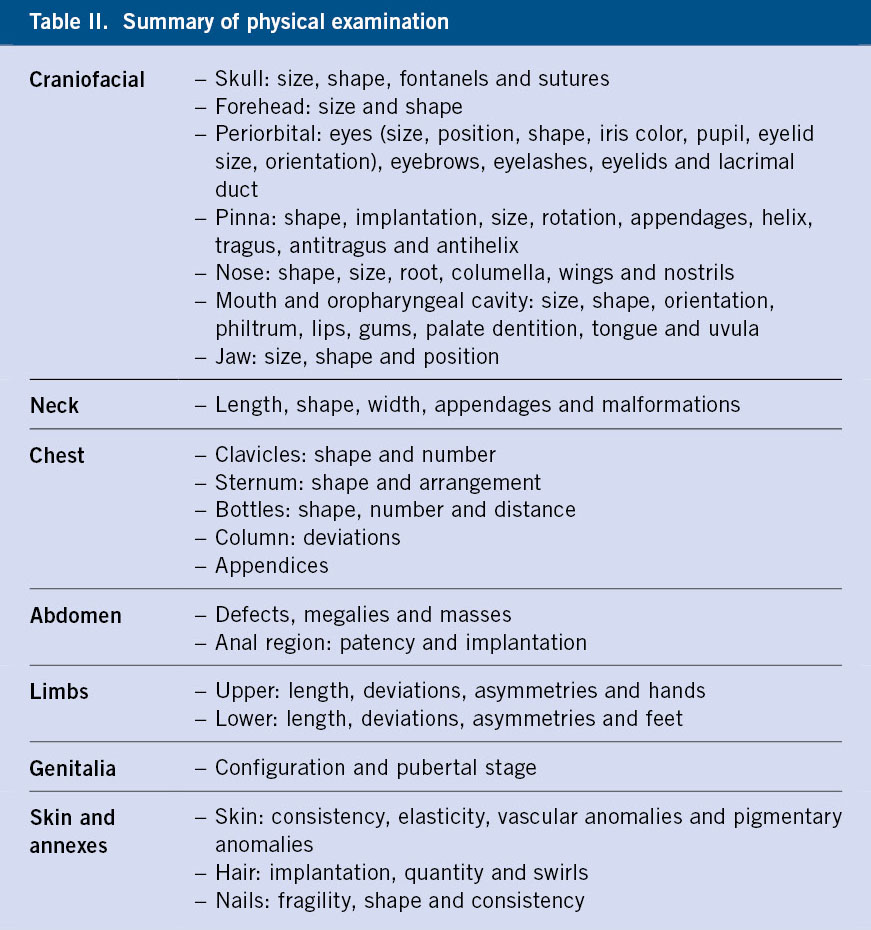
Then we will move on to the examination itself, which will be divided according to each body region (Table II), with the craniofacial region being, without a doubt, the most important part and the one that will provide us with the most information in the analysis of these patients(17-21). We will continue the dysmorphic examination, paying special attention to the neck, thorax, abdomen (including anorectal area)(22-25), limbs(26,27), skin and adnexa, adding to this the Tanner pubertal stage(28,29), with the description of the genitals. It is not the objective of these lines to make a systematic review of each of the sections, but we invite the reader to consult some of the quotes from this article or consult the article “Back to Basics: Dysmorphological semiology of the head and face” of this journal issue or articles from previous issues(5), to delve deeper into what a correct dysmorphological examination should be.
Decision making in the initial evaluation
After the first evaluation in consultation with the patient, the most relevant malformations and/or phenotypic characteristics will have been identified, distinguishing them from those of less importance. Based on these findings, a first approach will be made to determine which alterations could be directly related to the possible disease, which could be secondary to others, which could be interconnected with each other and which could be incidental findings without an apparent relationship with the disease in question.
From here, the extension to other tests may vary depending on the findings obtained so far and specific diagnostic suspicions. It is common to perform imaging tests, such as abdominal ultrasound, left hand x-ray, bone series, and brain imaging studies (CT or MRI). Regarding laboratory tests, general blood and urine tests may be ordered, as well as hormonal tests and more advanced metabolic and enzymatic studies, as necessary. Also, it is common that, given the high incidence of alterations in patients with malformations or dysmorphic phenotype, consultation with other specialists, such as ophthalmologists, otorhinolaryngologists, cardiologists and others, is required to evaluate possible associations with additional problems.
It is necessary to take into account that the diagnosis of these complex diseases usually requires teamwork with the involvement of several pediatric specialties and other specialties, both clinical-surgical (ophthalmology, ENT, etc.), as well as related to the diagnosis (laboratory, radiology, pathological anatomy, etc.).
Diagnostic approach
HPO (Human Phenotype Ontology) codes and differential diagnosis
Technological advances in diagnosis require standardizing clinical information to interpret it efficiently. HPO codes facilitate this task, describing human phenotypes that we must prioritize based on relevance to the diagnosis. Artificial intelligence and databases are transforming the diagnostic process by storing and integrating multiple information. It is important to know what to expect from each genetic test to be able to select the one that best suits our patient’s situation. In the coming years, whole genome sequencing is expected to become the most informative test.
Technological advances in the use of diagnostic systems have made it necessary to standardize clinical information for its correct interpretation by bioinformatic systems. This implies that, once we have our patient’s clinical information, we must take time to transform it from our clinical language to a standardized one. To do this, we will use the HPO (Human Phenotype Ontology)(30,31) codes. HPOs provide a standardized vocabulary of phenotypic abnormalities found in human diseases. Each HPO term, which is accompanied by a unique code, describes an anomaly. At present there are more than 13,000 HPO terms that can be very general (Growth abnormality HP:0001507) or more specific (Infancy onset short-trunk short stature HP:0011406), being interrelated and being able to select the most specific or general ones depending on the circumstances. It is advisable to order the selected terms of our patients by their importance, according to the characteristics of each case. Thus, it is common that a major malformation such as esophageal atresia (Esophageal atresia HP:0002032) or microphthalmia (Microphthalmia HP:0000568) is more helpful in the diagnosis than short stature (Short stature HP:0004322) or motor delay (Motor delay HP:0001270), especially if these are mild.
Taking into account the particularities of each clinical case, as well as our knowledge and experience, in the best of cases, we will be able to reach a clear diagnosis (Noonan syndrome with lentigines), make a differential diagnosis that guides us towards a group of diseases (RASopathies) and, in other cases, we can only guide the diagnosis through HPOs (Hypertelorism, Short stature, Multiple lentigines).
Current advances in artificial intelligence, big data and their integration into a multitude of databases are giving rise to the creation of innovative resources that offer us additional tools (face2gen, Phenomizer, among others) that facilitate the diagnostic process, offering a more accurate and efficient approach in the identification and understanding of these diseases.
Request for genetic testing
Once, after a complete history and examination, we have established a clinical diagnostic suspicion or an orientation for HPOs, it is time to decide which genetic test we are going to perform on our patient. To make this decision, it is essential to understand what type of genetic variant is responsible for the disease or group of diseases that we consider as the possible cause of our patient’s clinical situation.
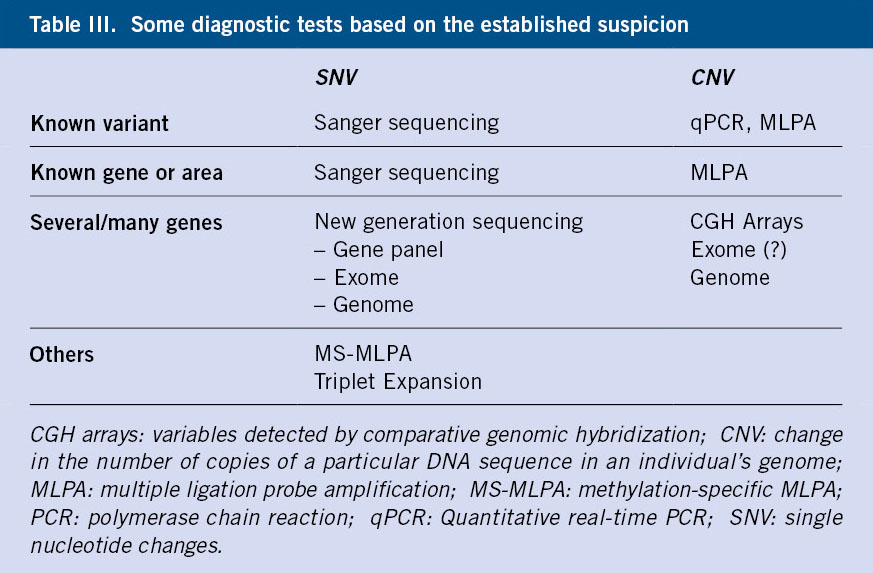
Generally, two large groups of genetic variants are distinguished: SNVs (single nucleotide changes) and CNVs (change in the number of copies of a particular DNA sequence in the genome of an individual, including: nucleotide insertions, duplications or deletions in the chain). From the latter, dynamic variants can be derived that cause an increase in the number of repetitions of a given microsatellite, known as triplet expansions(32,33).
If the suspicion is clear and the variant is known, due to family history or other reasons, Sanger sequencing or qPCR (quantitative real-time PCR) or MLPA (multiple ligation probe amplification) will be requested, depending on whether the variant is an SNV or a CNV. For example, if a parent is affected by a variant in the RET gene (responsible for MEN2A) and we want to determine if the child is a carrier of said variant, we will request a Sanger of the specific variant. We will request an MLPA or qPCR in case the variant we must look for is a CNV, as in the case where we study a child of a woman affected by neurofibromatosis, who has a deletion of the NF1 gene.
In the event that the disease is suspected, but the variant is unknown, MLPA is used to detect CNVs (7q deletion if we suspect Williams syndrome) or sequencing of a specific gene using Sanger (EXT1 gene in multiple enchondromatosis).
In situations where the disease can be caused by variants in multiple genes or regions, or when only HPO targeting of the disease can be performed, tests capable of analyzing SNVs in multiple genes are needed (next-generation sequencing)(34) or search for CNVs in all genomic DNA (CGH arrays)(35). It is common to combine several techniques, due to the possibility that the variant responsible for the disease could be a CNV or a SNV.
There are other tests that are used when the disease we suspect is caused by triplet expansion or methylation alteration, in which case it will be necessary to request a TP-PCR (Triplet Repeat Primed PCR)(36), or an MS-MLPA (specific methylation-specific multiple ligand-dependent probe amplification)(37).
In recent years, thanks to technical and bioinformatic advances, new massive sequencing techniques, which were previously incapable of detecting CNVs, are becoming more capable of identifying them. Currently, we also have at our disposal complete genome sequencing, which is the diagnostic test that can provide us with the most information and which is expected to, in the coming years, become the only test capable of detecting most genetic diseases. Using this technique, we will obtain the variants present, both in the coding and non-coding regions, which will also allow us to find variants at a structural level.
Table III summarizes the diagnostic techniques that can be used to detect the different types of variants.
Clinical interpretation of genetic tests and counseling
The diagnostic process does not end with the results of the genetic test. In inconclusive cases, it must be determined whether to end the process, perform more tests, or reorient with new HPOs. The interpretation of the results, whether conclusive or not, is crucial to offer complete genetic counseling to the family, considering possible phenotypic implications, treatment options and inheritance of the variant.
The diagnostic process does not conclude when the results of the genetic test are received. Regardless of whether the test was positive or inconclusive, the result must be interpreted.
In cases of inconclusive genetic tests, it is necessary to determine whether the diagnostic process is completed, expanded with new tests or redirected with new HPOs. Closure of the diagnostic process occurs if the goal was to rule out a specific genetic variant, disease, or if it is suggested that the suspected disease may not have an identifiable genetic cause. In these cases, the family is informed of the results and the process is concluded.
However, if the result is inconclusive, but there is certainty of a clear genetic suspicion, we must insist on obtaining a diagnosis. We must consider the limitations of the initial genetic test. We may need to look for another type of variant. For example, if an array CGH was ordered due to a possible CNV, but SNVs also need to be ruled out by bulk sequencing. We must be aware of the possibility of errors inherent to the diagnostic technique. In many cases, it will be necessary to reorient the case with new HPOs, or request a reanalysis of the initial test which, due to advances in bioinformatics or new scientific knowledge, may be fruitful. Therefore, when faced with inconclusive tests, it is essential to maintain direct communication with the laboratory that performed the genetic test or with laboratory geneticists, to support the interpretation process and make appropriate decisions.
If genetic testing reveals a conclusive genetic variant, it is essential to interpret it in the context of the patient. Each variant must be evaluated in terms of its pathogenicity according to the criteria of the American College of Medical Genetics (ACMG)(38). Although a pathogenic variant is more likely to be the cause of the patient’s symptoms, this is not always the case. It may be a unique variant in a recessively inherited disease, or a variant that only explains part of the symptoms. On the other hand, a variant classified as “of uncertain significance” could be diagnostic. Additional segregation studies, very compatible clinical features or new knowledge may change the interpretation of pathogenicity. Therefore, it is essential for clinicians to understand that this interpretation depends on multiple factors and may evolve over time (Table IV).

Once the variant is interpreted in the context of the patient, we can determine whether the study is conclusive or not. If we consider that it is not conclusive and that it does not explain our patient’s symptoms, we must behave as we have explained before. If so, comprehensive genetic counseling should be offered to the family, including information about the diagnosis, possible phenotypic implications, treatment options, investigation, psychological support and considerations about the inheritance of the variant. All of this must be communicated with empathy and clarity by a doctor with appropriate training(7).
Current treatments for genetic diseases
Advances in research and knowledge in the pathophysiology of diseases are changing the treatment of genetic diseases. Currently, there are more than 1,300 ongoing studies and 4,000 recruiting studies on ClinicalTrials.gov on orphan treatments. Genetic, replacement and symptomatic therapies are being explored to improve treatment.
New findings in relation to the pathophysiology of rare and genetic diseases have led to a paradigm shift in the treatment and management of these diseases. Currently, in the Clinical trials database, if filtered by genetic disease more than 1,300 active studies and more than 4,000 in the recruitment phase may be found(39).
These advances inform us of a growing interest in specific and targeted therapies; however, currently, treatment that acts directly on the affected gene only occurs in few diseases. Depending on the therapeutic target, we can differentiate the treatment of rare diseases into: genetic treatments, those that try to repair or modulate genetic expression; replacement treatments; and symptomatic treatments(40).
Genetic treatments can act at the level of DNA, RNA or proteins. At the DNA level we can find treatments with viral vectors, antisense oligonucleotides or RNA interference(41). Examples of these treatments would be Onasemnogene Abeparvovec (Zolgensma®), Betibeglogen Autotemcel (Zynteglo®) or Voretigén Neparvovec (Luxturna®), used in spinal cord atrophy type 1(42), in beta-thalassemia(43) or Leber disease(44), all of these with very severe consequences whose prognosis has radically changed with these new treatments.
In the case of antisense oligonucleotides, there is the example of Duchenne muscular dystrophy with Eteplirsén®, which acts at the level of RNA splicing, eliminating exon 51, which means that there is no premature termination codon. This induces a truncated but functional protein, causing a much milder pathology(41).
Another current line of treatment would be one that acts at a molecular level, altering protein folding, such as Trikafta® in cystic fibrosis. This treatment is approved in patients aged 2 years or older who have at least one copy of the F508 variant of the CFTR gene(45).
Finally, regarding enzyme replacement therapies, whose objective is to exogenously provide the deficient enzyme, there is the case of some lysosomal diseases, such as Hunter syndrome.
In the rest of the cases, those pathologies that do not have any precision medicine treatment, they must be provided with adequate monitoring and support, both to the patient and to the families, coordinating in a multidisciplinary manner, ensuring the best possible support treatment for the illness of our patients.
Therefore, currently we are experiencing an explosion of studies that allow us to demonstrate the effectiveness of new treatments that, we are sure that in the near future, with the advance of gene therapy, many of the diseases that are currently even difficult to diagnose will have palliative and even curative treatment that radically changes the prognosis of these patients.
Online resources
There are a series of online resources that can make it easier to search for information with public databases or even websites that can help in the process of diagnostic-therapeutic guidance for children with malformations or a dysmorphological phenotype or one suggestive of a genetic disease. Some of them include:
• Orphanet: A unique resource that brings together and enhances knowledge about rare diseases to improve the diagnosis, care and treatment of patients with rare diseases. It has a database on all aspects related to rare diseases, with information on rare diseases, clinical guides, genes, information sheets for families, etc. Available in: https://www.orpha.net/es.
• OMIM (Online Mendelian Inheritance in Men): catalog that provides complete and referenced descriptions of all known Mendelian diseases and more than 15,000 genes. Available in: https://www.omim.org/.
• GeneReviews: Web resource with multiple reviews and information on genetic diseases. Available in: https://www.ncbi.nlm.nih.gov/books/NBK1116/.
• Face2Gene: partially free of charge group of applications for phenotyping, which use patient photographs and HPO terms, and thanks to artificial intelligence technologies, facilitate the early detection of syndromes. Available in: https://www.face2gene.com/.
• Human Genome Variation Society Nomenclature: web page with information on the standard nomenclature of variants and genes. Available in: https://hgvs-nomenclature.org/stable/.
• HPO (Human Phenotype Ontology): web page with standardized terminology for phenotypic anomalies. Available in: https://hpo.jax.org/app/.
• Clinical Trials: portal that brings together clinical trials being carried out worldwide. Available in: https://clinicaltrials.gov/.
• Phenomizer: application included within the HPO website that helps with diagnosis, offering possible diagnoses in the presence of HPOs. Available in: https://hpo.jax.org/app/tools/phenomizer.
• POSSUMweb: Pay-per-view dysmorphology database that provides tools that can assist in the diagnosis of dysmorphic syndromes. Available in: https://www.possum.net.au/.
• Dx29: software for symptom analysis and management, creation and exchange of medical history to help obtain a diagnosis. Available in: https://dx29.ai/.
• GenoPro and TreeStudio: applications that offer a practical solution to the creation of family trees and genograms, in an open way. Available in: https://genopro.com/es/ and https://treestudio.healthincode.com/.
• MalaCards: disease and gene database that collects information from more than 44 sources, integrating it and creating specific annotations of diseases and connections between them. Available in: https://www.malacards.org/.
• Varsome: platform that provides tools to interpret and filter genetic variants, as well as access to an extensive database of genomic and clinical information. Available in: https://varsome.com/.
• Franklin: advanced platform for the interpretation of genetic variants. Available in: https://franklin.genoox.com/.
• GnomAd: database that integrates exome and genome variant data worldwide, being very useful for knowing the prevalence of variants in the general population. Available in: https://gnomad.broadinstitute.org/.
• ClinVar: resource from the National Library of Medicine that collects genetic variants and relates them to specific phenotypes and classifies them by confidence levels represented by stars. Available in: https://www.ncbi.nlm.nih.gov/clinvar/.
Role of the Primary Care pediatrician
The Primary Care pediatrician plays a fundamental role in the early diagnosis of rare diseases through suspicion. His familiarity with the patient since birth allows him to recognize unusual symptoms and signs, as well as to integrate information from multiple consultations for an early diagnosis. He is the crucial starting point for early identification of these conditions.
Once the diagnosis is made, the Primary Care pediatrician assumes a central role in coordinating the patient’s care, acting as a liaison with specialized medicine to facilitate communication between different specialists. In addition, he plays a crucial role in supporting families, especially in severe and chronic illnesses, being their point of reference throughout the diagnostic and treatment process.
Conflict of interest
There is no conflict of interest in the preparation of the manuscript. Declaration of interests: none.
References
The asterisks show the interest of the article in the authors’ opinion.
1. González-Lamuño D, García Fuentes M. Enfermedades Raras En Pediatría. Rare Diseases in Pediatrics. An Sist Sanit Nava. 2008; 31: 21-9.
2. González-Lamuño D. Una visión general sobre las enfermedades raras. An overview of rare diseases. Pediatr Integral. 2014; XVIII: 550-63. Available in: https://www.pediatriaintegral.es/publicacion-2014-10/una-vision-general-sobre-las-enfermedades-raras/.
3. Guillén Navarro E. Genética clínica y dismorfología: generalidades. Clinical genetics and dysmorphology: generalities. Rev Esp Pediatrics. 2009; 65: 12-4.
4. Aase JM. Diagnostic Dysmorphology. New York and London: Plenum Medical Book Company; 1990. p. 1-4.
5. Ramos Fuentes FJ, Ramons Cáceres M, Ribate Molina MP. Semiología de las malformaciones y deformaciones craneofaciales. Semiology of craniofacial malformations and deformations. Pediatr Integral. 2014; XVIII: 529-38. Available in: https://www.pediatriaintegral.es/publicacion-2014-10/semiologia-de-las-malformaciones-y-deformaciones-craneofaciales/.
6. Jones KL, Adam MP. Evaluation and diagnosis of the dysmorphic infant. Clin Perinatol. 2015; 42: 243-61.
7. Garcia Miñaúr S. Consulta de genética clínica y diagnóstico genético prenatal. Clinical genetics consultation and prenatal genetic diagnosis. Pediatr Integral. 2014; XVIII: 507-14. Available in: https://www.pediatriaintegral.es/publicacion-2014-10/consulta-de-genetica-clinica-y-diagnostico-genetico-prenatal/.
8. Hasanzadeh-NazarAbadi M, Baghbani F, Namazi I, Mirzaee S. Robertsonian translocation between chromosomes (no. 21/14) in relation to the history of spontaneous abortion in a family. Iran J Reprod Med. 2014; 12: 581-5.
9. Mussa A, Molinatto C, Cerrato F, Palumbo O, Carella M, Baldassarre G. Assisted Reproductive Techniques and Risk of Beckwith-Wiedemann Syndrome. Pediatrics. 2017; 140: e20164311.
10.*** Bennett RL, Steinhaus KA, Uhrich SB, O’Sullivan CK, Resta RG, Lochner-Doyle D, et al. Recommendations for standardized human pedigree nomenclature. Pedigree Standardization Task Force of the National Society of Genetic Counselors. Am J Hum Genet. 1995; 56: 745-52.
11. Bennett RL, French KS, Resta RG, Doyle DL. Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J Genet Couns. 2008; 17: 424-33.
12. Sheehan E, Bennett RL, Harris M, Chan-Smutko G. Assessing transgender and gender non-conforming pedigree nomenclature in current genetic counselors’ practice: The case for geometric inclusivity. J Genet Couns. 2020; 29: 1114-25.
13. Gómez EG. Indicaciones del estudio genético. Documento AEPED. Indications for genetic study. AEPED document. Available in: https://www.aeped.es/sites/default/files/documentos/3-estudiogene.pdf.
14. García H, Franco Gutiérrez M. Manejo multidisciplinario de los pacientes con atresia de esófago. Multidisciplinary management of patients with esophageal atresia. Bol. Med. Hosp. Infant. Mex. 2011: 68; 467-75.
15.*** Soto Insuga V, González Alguacil E, García Peñas JJ. Detección y manejo del retraso psicomotor en la infancia. Detection and management of psychomotor delay in childhood. Pediatr Integral. 2020; XXIV: 303-15. Available in: https://www.pediatriaintegral.es/publicacion-2020-09/deteccion-y-manejo-del-retraso-psicomotor-en-la-infancia-2/.
16. Lapunzina P, Aiello H. Manual de antropometría normal y patológica. Fetal, neonatal, niños y adultos. Manual of normal and pathological anthropometry. Fetal, neonatal, children and adults. 1st ed. Barcelona: Masson; 2002.
17.*** Allanson JE, Cunniff C, Hoyme HE, McGaughran J, Muenke M, Neri G. Elements of morphology: standard terminology for the head and face. Am J Med Genet A. 2009; 149A: 6-28.
18. Carey JC, Cohen MMJr, Curry CJ, Devriendt K, Holmes LB, Verloes A. Elements of morphology: standard terminology for the lips, mouth, and oral region. Am J Med Genet A. 2009; 149A: 77-92.
19. Hall BD, Graham JMJr, Cassidy SB, Opitz JM. Elements of morphology: standard terminology for the periorbital region. Am J Med Genet A. 2009; 149A: 29-39.
20. Hunter A, Frias JL, Gillessen-Kaesbach G, Hughes H, Jones KL, Wilson L. Elements of morphology: standard terminology for the ear. Am J Med Genet A. 2009; 149A: 40-60.
21. Hennekam RCM, Cormier-Daire V, Hall JG, Méhes K, Patton M, Stevenson RE. Elements of morphology: standard terminology for the nose and philtrum. Am J Med Genet A. 2009; 149A: 61-76.
22. Díaz C, Copado Y, Muñoz, G, Muñoz H. Malformaciones de la pared abdominal. Malformations of the abdominal wall. Revista Médica Clínica Las Condes. 2016; 27; 499-05.
23. Levitt MA, Peña A. Anorectal malformations. Orphanet J Rare Dis. 2007; 2: 33. Erratum in: Orphanet J Rare Dis. 2012; 7: 98.
24. Moog R. Malformaciones Congénitas Del Pene. Congenital Penile Malformations. EMC – Pediatría. 2008; 43: 1-10.
25. Louis-Sylvestre C. Malformaciones Congénitas de La Vulva. Congenital Malformations of the Vulva. EMC – Gynecol.-Obstet. 2022; 58: 1-9.
26. Espandar R, Mortazavi SM, Baghdadi T. Angular deformities of the lower limb in children. Asian J Sports Med. 2010; 1: 46-53.
27. Sass P, Hassan G. Lower extremity abnormalities in children. Am Fam Physician. 2003; 68: 461-8. Erratum in: Am Fam Physician. 2004; 69: 1049.
28. Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969; 44: 291-303.
29. Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970; 45: 13-23.
30. Gargano MA, Matentzoglu N, Coleman B, Addo-Lartey EB, Anagnostopoulos AV, Anderton J, et al. The Human Phenotype Ontology in 2024: phenotypes around the world. Nucleic Acids Res. 2024; 52: D1333-D46.
31. Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008; 83: 610-5.
32. Corella D, Ordovas JM. Basic Concepts in Molecular Biology Related to Genetics and Epigenetics. Rev Esp Cardiol (Engl Ed). 2017; 70: 744-53.
33. Civeira F, Rodríguez-Rey JC, Pocoví M. Introducción a la genética y su utilidad en el diagnóstico de las enfermedades cardiovasculares: conceptos básicos y el ejemplo de la hipercolesterolemia familiar. Introduction to genetics and its usefulness in the diagnosis of cardiovascular diseases: basic concepts and the example of familial hypercholesterolemia. Rev Esp Cardiol. 2009: 9; 14-23.
34. Santillana-Garzón S, Diego-Álvarez D, Buades C, Romera-López A, Pérez-Cabornero L, Valero-Hervás D, et al. Diagnóstico molecular de enfermedades genéticas: del diagnóstico genético al diagnóstico genómico con la secuenciación masiva. Molecular diagnosis of genetic diseases: from genetic diagnosis to genomic diagnosis with massive sequencing. Rev Méd Clín Condes. 2015; 26: 458-69.
35. Palacios Verdú MG, Pérez-Jurado LA. Nuevas metodologías en el estudio de enfermedades genéticas y sus indicaciones. New methodologies in the study of genetic diseases and their indications. Pediatr Integral. 2014; XVIII: 51528. Available in: https://www.pediatriaintegral.es/publicacion-2014-10/nuevas-metodologias-en-el-estudio-de-enfermedades-geneticas-y-sus-indicaciones/.
36. Benítez J. Implicaciones clínicas y genéticas de las mutaciones dinámicas en clínica neuropediátrica Clinical and genetic implications of dynamic mutations in neuropediatric practice. Rev Neurol. 1999; 28: 60-3.
37. Acosta-Fernández E, Corona-Rivera JR, Ríos-Flores Izabel M, Torres-Anguiano E, Corona-Rivera A, Peña-Padilla C, et al. Utilidad de la técnica de MS-MLPA en el diagnóstico de los síndromes de Beckwith-Wiedemann y Silver-Russell. Usefulness of the MS-MLPA technique in the diagnosis of Beckwith-Wiedemann and Silver-Russell syndromes. Gac Méd Méx. 2022; 158: 210-8.
38. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405-24.
39. ClinicalTrials.gov. Available in: https://clinicaltrials.gov/.
40. Covers Belly HH, Trubnykova M, Castro Mujica MC. Tratamiento de Las Enfermedades Genéticas: Presente y Futuro. Treatment of Genetic Diseases: Present and Future. Rev Fac Med Hum. 2021: 21; 399-416.
41. Rossor AM, Reilly MM, Sleigh JN. Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol. 2018; 18: 126-31.
42. Today SM. Onasemnogene Abeparvovec: First Global Approval. Drugs. 2019; 79: 1255-62.
43. Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. 2018; 378: 1479-93.
44. Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019; 126: 1273-85.
45. Bacalhau M, Camargo M, Magalhães-Ghiotto GAV, Drumond S, Castelletti CHM, Lopes-Pacheco M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals (Basel). 2023; 16: 410.
Recommended bibliography
– Bennett RL, Steinhaus KA, Uhrich SB, O’Sullivan CK, Resta RG, Lochner-Doyle D, et al. Recommendations for standardized human pedigree nomenclature. Pedigree Standardization Task Force of the National Society of Genetic Counselors. Am J Hum Genet. 1995; 56: 745-52.
A must-read article to properly create a family tree.
– Soto Insuga V, González Alguacil E, García Peñas JJ. Detección y manejo del retraso psicomotor en la infancia. Detection and management of psychomotor delay in childhood. Pediatr Integral. 2020; XXIV: 303-15. Available in: https://www.pediatriaintegral.es/publicacion-2020-09/deteccion-y-manejo-del-retraso-psicomotor-en-la-infancia-2/.
It clearly indicates the detection and management of psychomotor delay in childhood, which is important to know given the high frequency of these alterations in rare diseases.
– Allanson JE, Cunniff C, Hoyme HE, McGaughran J, Muenke M, Neri G. Elements of morphology: standard terminology for the head and face. Am J Med Genet A. 2009; 149A: 6-28 et seq. that establish the terminology of the exploration.
Mandatory articles to carry out a correct examination in dysmorphological patients.
– Online resources: it is important to know how to use many of the online resources mentioned in this article, since they will serve as support and reference in the diagnosis of rare diseases.
| Clinical case |
|
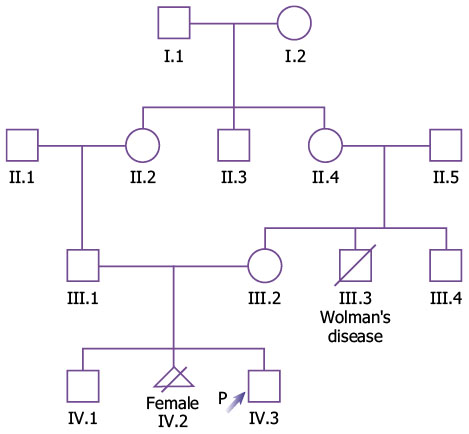
A 3-year-old boy followed up in Primary Care, whose neonatal history highlights the presence of a diaphragmatic hernia and an altered neonatal screening for hearing loss, is referred to the Genetics-rare diseases clinic. Family history: Mother: 25 years old, healthy, Gestations: 3, Miscarriages/losses: 1 (legal interruption due to multiple malformations), live births: 2. Father: 28 years old, healthy. There is consanguinity. Maternal uncle died at 6 months due to Wolman’s disease. No recurrent miscarriages. Family tree (Fig. 2).
Figure 2. Family tree of the clinical case. Personal background: Gestational age 40+1, followed in the High-risk clinic for previous pregnancy with malformations, no infections, no exposure to ionizing radiation. Neonatal anthropometry: weight: 3,650 g; length: 53 cm; cephalic perimeter: 36.5 cm. Neonatal period: he required advanced resuscitation for diaphragmatic hernia (surgery at 48 hours of life). He did not pass the hearing screening. He presents certain delay in developmental milestones. Current illness: Cardiology: he was evaluated in the neonatal stage when muscular IVC and pulmonary hypertension were identified, both resolved and, currently presents a “normal heart”. Neurology: he started ambulation at 2.5 years, but he is not currently able to run and climbs stairs with help. In terms of language, he has the ability to say and express yes/no and say two monosyllables (mom, water). He has no sphincter control. He reports interest in other children, but he does not play with them, he does not have symbolic play. He has not had seizures and has a normal EEG. Ophthalmology: he was referred to Ophthalmology at 18 months due to nystagmus, being diagnosed with retinal dystrophy with poor visual prognosis, and he currently poorly tolerates optical correction. ENT: he has severe sensorineural hearing loss. Treatment with cochlear implant has been started, presenting meningitis in the postoperative period. Traumatology: he has no bone deformities, no scoliosis at the current time. Dermatology: he presented atopic dermatitis as an infant. Nephrology: he has not had any urinary infections. A normal renal US was performed in the neonatal stage. Pulmonology: admission at 6 months of age for RSV bronchiolitis, requiring high flow O2. Other information: no similarities in the family. Physical examination: (not all negative signs are included due to word limit). General: small umbilical hernia. Auxology: weight: -1.9 SD; height: -2.1 SD; Head circumference: 0.8 SD. Skull, neck and face: hypertelorism, ear pinnae present a low implantation and are posteriorly rotated, mild micrognathia, short nose with long and wide philtrum. Oropharyngeal cavity: normal teeth, normal uvula. Limbs: normal, normal hands and feet. Genitals: Tanner stage I, G1, P1, A1 with 2 ml testes, one of them slightly raised. Skin and annexes: normal. Other pending tests: brain MRI. HPOs. Main: Congenital diaphragmatic hernia HP:0000776; Retinal dystrophy HP:0000556. Secondary: Sensorineural hearing impairment HP:0000407; Global developmental delay HP:0001263; Autosomal recessive inheritance HP:0000007; Abnormal heart morphology HP:0001627; Hypertelorism HP:0000316; Abnormal location of ears HP:0000357. Others: Atopic dermatitis HP:0001047; Short stature HP:0004322; Relative macrocephaly HP:0004482; Micrognathia HP:0000347.
|