|
| Temas de FC |
M. Pérez Pascual*, P. Sánchez Mascaraque**
*Pediatra. Unidad de Medicina de la Adolescencia. Hospital Universitario La Paz. Madrid. **Psiquiatra Infanto-Juvenil. Centro de Salud Mental Jaime Vera. Coslada. Hospital Universitario del Henares. Madrid
| Resumen
Uno de los trastornos mentales más prevalente en la infancia y la adolescencia son los trastornos de ansiedad. En muchas ocasiones no se presentan solos, sino que se asocian a comorbilidades como depresión, dificultades académicas y abuso de sustancias, entre otras. Las manifestaciones clínicas de la ansiedad en estas etapas de la vida puede ser una tarea complicada para el pediatra. La edad es un factor determinante en la expresión clínica, siendo el miedo, la tristeza, la irritabilidad y las quejas somáticas, síntomas diana que nos deben de hacer sospechar su existencia. Los trastornos de ansiedad en los niños incluyen: trastorno de ansiedad por separación, mutismo selectivo, fobia específica, agorafobia, trastorno de pánico, trastorno de ansiedad social y trastorno de ansiedad generalizada. El pediatra de Atención Primaria es clave para detectar tempranamente estos síntomas e iniciar su abordaje y correcto tratamiento. En este capítulo se va a revisar la etiología, epidemiología, clínica, diagnóstico, prevención y tratamiento de los trastornos de ansiedad. |
| Abstract
One of the most prevalent mental disorders in childhood and adolescence are anxiety disorders. On many occasions they do not appear alone, but are associated with comorbidities such as depression, academic difficulties and substance abuse, among others. The clinical manifestations of anxiety in these stages of life can be a complicated task for the pediatrician to tackle. Age is a determining factor in clinical expression, with fear, sadness, irritability and somatic complaints being target symptoms that should make us suspect its existence. Anxiety disorders in children include: separation anxiety disorder, selective mutism, specific phobia, agoraphobia, panic disorder, social anxiety disorder, and generalized anxiety disorder. The Primary Care pediatrician is key to detecting these symptoms at an early stage and initiating their adequate management and treatment. This article will review the etiology, epidemiology, clinical manifestations, diagnosis, prevention, and treatment of anxiety disorders. |
Palabras clave: Trastornos de ansiedad; Infancia; Adolescencia; Síntomas somáticos; Miedo.
Key words: Anxiety disorders; Childhood; Adolescence; Somatic symptoms; Fear.
Pediatr Integral 2022; XXVI (1): 40 – 47
OBJETIVOS
- Aprender que la etiología de los trastornos de ansiedad es multifactorial, estando implicados en su desarrollo factores biológicos, psicológicos y socioambientales.
- Entender que la ansiedad en la infancia se equipara al miedo. Los miedos cambian según la edad del niño o adolescente.
- Conocer que el diagnóstico es clínico. Existen cuestionarios que pueden apoyar el diagnóstico.
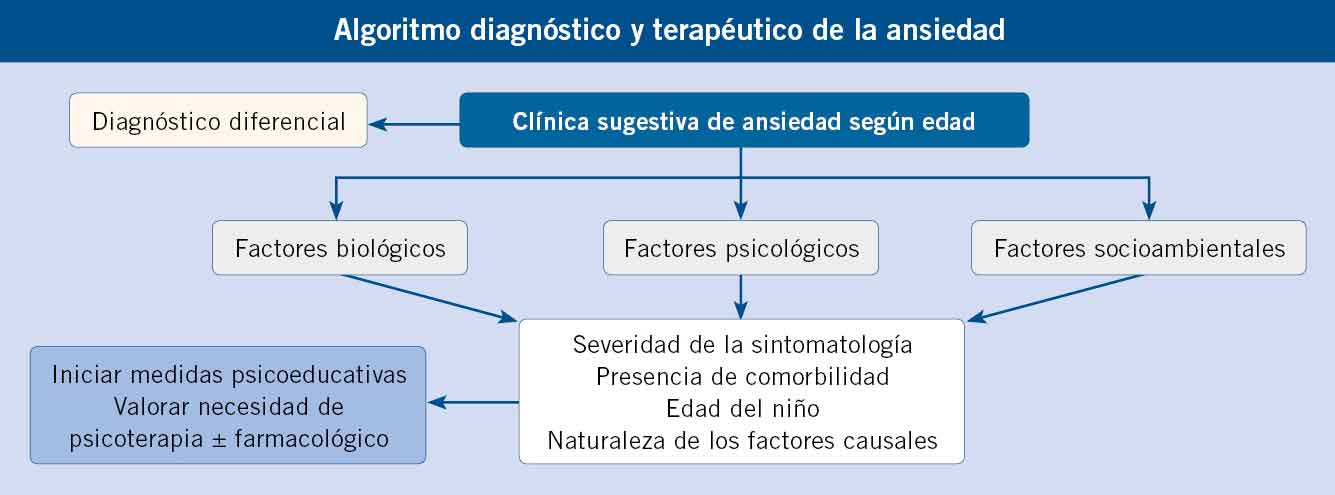
- Saber la importancia que tiene la elección del tratamiento, este debe basarse en: la severidad de la sintomatología, la presencia de comorbilidad, la edad del niño y la naturaleza de los factores causales.
- Comprender que la primera intervención es la psicoeducación del niño y sus padres sobre la ansiedad.
|
|
|


Ansiedad en la infancia y adolescencia
Introducción
La ansiedad, en situaciones de estrés o peligro puede ser normal y adaptativa, incluso necesaria, ya que hace que la persona se proteja ante dicho estímulo potencialmente dañino. Cuando la reacción es excesiva, se considera ansiedad patológica.
Los trastornos de ansiedad, globalmente, son los trastornos psiquiátricos más frecuentes en la infancia y adolescencia, presentando tasas de prevalencia entre un 10 y un 20%, por encima de la depresión y de los trastornos de la conducta(1). Los trastornos de ansiedad a menudo se inician en estas etapas y son progresivos, persistentes y crónicos o de curso recurrente. El diagnóstico precoz y su tratamiento puede reducir el impacto en la vida del niño y del adolescente, en todos sus aspectos, académico, social y familiar, y prevenir la persistencia de un trastorno de ansiedad en la vida adulta(2-4). La ansiedad se puede equiparar al miedo. Los miedos y las preocupaciones son normales en la infancia, tienen un carácter evolutivo, preparan al niño para afrontar situaciones que pueden entrañar un peligro, así como enfrentarse a los cambios. Estos miedos van cambiando con la edad. En edades tempranas, se asustan por estar solos o ante ruidos fuertes. Según crecen aparece el miedo a separarse de sus padres, a la oscuridad, a los extraños. En la edad escolar aparecen miedos a fenómenos naturales, monstruos o enfermedades, y en la adolescencia predomina el miedo a hacer el ridículo ante compañeros, al fracaso académico, la competencia escolar y las cuestiones de salud. La ansiedad aparece cuando existe un peligro inmediato real o imaginario. Tiene carácter adaptativo y es necesaria para la supervivencia. Se hace patológica cuando es excesiva en intensidad, duración (en general, si dura más de 6 meses), o causa un desproporcionado malestar o sufrimiento. También se considera patológica, cuando el desencadenante es un estímulo objetivamente neutro o inofensivo.
La ansiedad se puede desencadenar por factores externos o internos (recuerdos, imágenes, ideas, deseos). Se manifiesta con síntomas neurovegetativos (inquietud psicomotriz, taquicardia, piloerección, sudoración…), cognitivos (miedo, preocupación) y conductuales, siendo la inhibición la respuesta más típica en los trastornos de ansiedad. La adolescencia, en concreto, es una etapa de cambios y retos evolutivos, cambios físicos, elección de estudios, realización de la selectividad como acontecimiento especialmente estresante, inicio de la vida laboral, necesidad de la aceptación e integración al grupo de sus iguales, inicio de relaciones afectivas de pareja, etc(1). A veces, estos cambios tan importantes en la vida precipitan ansiedad en adolescentes vulnerables, pudiendo desencadenar patología. Siguiendo la clasificación del manual diagnóstico y estadístico de los trastornos mentales en su quinta edición (DSM-5)(5), los trastornos de ansiedad más habituales en la infancia son: trastorno por ansiedad de separación (TAS), trastorno por ansiedad generalizada (TAG), fobia social y fobias específicas. En los trastornos de ansiedad es frecuente la comorbilidad, en especial con otro trastorno de ansiedad y con depresión. Es relevante en Pediatría el hecho de que los trastornos somatomorfos, el dolor abdominal, las cefaleas y los dolores crónicos sin patología física identificable, se asocian hasta en un 20% con un trastorno de ansiedad comórbido. Su diagnóstico puede evitar realizar pruebas complementarias innecesarias y tratamientos yatrogénicos. En esta revisión se abordará: epidemiología, etiopatogenia, clínica, tratamiento farmacológico y psicoterapéutico, el pronóstico y la prevención de los trastornos de ansiedad.
Epidemiología
La prevalencia de los trastornos de ansiedad en la edad pediátrica oscila entre el 10 y el 20%, dependiendo del diseño epidemiológico del estudio, los criterios diagnósticos empleados, los trastornos de ansiedad incluidos y la edad de los pacientes.
Los trastornos de ansiedad en la infancia y adolescencia están asociados a dificultades académicas y sociales, a la depresión, a la tentativa autolítica y al abuso de sustancias en la edad adulta. Por otra parte, es frecuente la coocurrencia de varios trastornos de ansiedad en un mismo paciente. Un 33% de los niños y adolescentes con trastornos de ansiedad cumplen criterios para dos o más trastornos de ansiedad. Además, se encuentra comorbilidad con otros trastornos psiquiátricos, fundamentalmente con depresión, con rangos que varían entre el 28 y el 68%.
Además, en los próximos años tendremos que contestar la siguiente pregunta: ¿cuál ha sido el impacto de la pandemia COVID-19 en la prevalencia de ansiedad en la infancia? Las pandemias son crisis poco frecuentes, pero potencialmente devastadoras, que afectan a la vida física, social y psicológica de muchos niño/as y sus familias. Aunque tienen mucho en común con otros desastres naturales: impacto en la comunidad, imprevisibilidad, víctimas y efectos persistentes, la respuesta a las pandemias difiere respecto a la de otros desastres en la necesidad de medidas para evitar su propagación, entre las que destacan el distanciamiento social y la cuarentena(6,7).
La mayoría de los estudios realizados en relación con trastornos de ansiedad secundarios a la pandemia por COVID, están hechos en China. En ellos se muestran altas tasas de ansiedad durante el confinamiento, que llegaban hasta el 37,4%, y se mantenían en la fase de estabilización(8).
Existen también estudios del impacto emocional secundario a COVID-19 en población infantil española, donde se incluyeron a 1.143 padres de niños/as españoles e italianos entre 3 y 18 años. En él se encontró que un alto porcentaje (85,7%) de los padres señalaron cambios en el estado emocional de sus hijos durante la cuarentena. Este estudio tiene la limitación de que no se preguntó directamente a los niños, sino que la encuesta fue referida por los padres(9).
Según lo sucedido en otras pandemias, lo previsible es que durante estos años de estabilización de la pandemia por SARS-Cov2, tengamos un repunte de trastornos por ansiedad, sobre todo, en los niños y adolescentes susceptibles.
Etiopatogenia
La etiología de los trastornos de ansiedad es multifactorial, estando implicados en su desarrollo factores biológicos, psicológicos y socioambientales.
Los distintos factores que intervienen en la etiología de los trastornos de ansiedad se pueden incluir en las siguientes categorías.
Factores del desarrollo
Se debe valorar la progresión de la ansiedad a lo largo de la vida, una perspectiva del desarrollo contribuye a comprender la patogenia de los trastornos de ansiedad. Los bebés que muestran reacciones de aprensión, vacilación o angustia a la novedad tienen más probabilidades de evitar los estímulos nuevos, cuando empiezan a caminar(10). Estos niños pequeños han sido descritos como “conductualmente inhibidos”, teniendo un mayor riesgo de desarrollar trastornos de ansiedad en la infancia(11,12), trastorno de ansiedad social en la adolescencia y, finalmente, más probabilidades de tener trastornos de ansiedad persistentes en la edad adulta.
Factores cognitivos y de aprendizaje
Las personas con trastornos de ansiedad tienen un sesgo de atención hacia los estímulos relacionados con la amenaza, se encuentran hipervigilantes hacia los mismos, incluso interpretan estímulos neutros como potencialmente dañinos.
Otro posible determinante cognitivo es la negatividad relacionada con el error, la baja tolerancia hacia el error a lo largo del desarrollo (de 6 a 18 años) puede predecir ansiedad en diferentes periodos de la vida(13).
Factores neurobiológicos
Las regiones cerebrales que actualmente sabemos que están implicadas en la sensación de ansiedad son la corteza prefrontal, que integra la información exterior y la amígdala, que es la responsable de la respuesta inicial del miedo.
Además de ellas, estructuras posteriores, la corteza cingulada anterior, la ínsula y el cerebelo han sido implicados en los trastornos de ansiedad en estudios funcionales realizados en niños y adolescentes. Respecto a los neurotransmisores, en la ansiedad se produce un aumento de la liberación de noradrenalina, que eleva el glutamato y disminuye el GABA. En cambio, la serotonina produce el efecto contrario. De ahí, la acción terapéutica de los inhibidores selectivos de la recaptación de la serotonina (ISRS) en los trastornos de ansiedad. Las neuronas serotoninérgicas también ejercen una acción inhibidora de las neuronas noradrenérgicas, que tienen un papel esencial en el desencadenamiento de la ansiedad y en su mantenimiento(2).
Factores genéticos
La mayoría de las estimaciones de la heredabilidad del rasgo de ansiedad en los niños son de alrededor del 30%, aunque en algunos estudios llegan del 50 al 60%. Estos hallazgos sugieren que los factores genéticos juegan un papel importante en el desarrollo de estos trastornos en relación con los factores ambientales(14).
Factores ambientales y sociales(3,15)
Los factores ambientales y sociales son muy importantes a la hora de desarrollar y mantener un trastorno de ansiedad. Sobre una base genética y un temperamento susceptible, el contexto en el que se encuentra el niño o el adolescente es determinante a la hora de enfermar. Se estima que el ambiente contribuye en un 60% en los trastornos de ansiedad.
• Dentro de los estilos de crianza parental, la sobreprotección excesiva, estilos educativos excesivamente punitivos y la transmisión de miedos específicos por parte de los padres, pueden contribuir a la génesis de dichos trastornos.
• Los acontecimientos vitales estresantes (conflictividad familiar, escolar o social, situaciones traumáticas, pérdidas o duelo de un ser querido, cambio de colegio o domicilio) pueden actuar como factores desencadenantes o mantenedores.
• Familias disfuncionales con condiciones desfavorables de salud (trastornos neuróticos, enfermedades crónicas no compensadas), niveles altos de violencia y escasa capacidad para resolver problemas.
• Situación social desfavorable (nivel socioeconómico bajo, adversidad económica, condiciones de vida desfavorables) pueden generar una sensación de inseguridad crónica que colabore en la génesis de un trastorno de ansiedad.
Clínica
Las manifestaciones clínicas de los trastornos de ansiedad están marcadas por la edad y el desarrollo cognitivo de cada niño y adolescente, así como el tipo de miedos o preocupaciones.
Identificar el miedo o la motivación central detrás de síntomas o comportamientos particulares es fundamental para un diagnóstico preciso:
• Trastorno de ansiedad por separación: estar lejos de sus padres o cuidadores.
• Trastorno de ansiedad social: avergonzarse de sí mismo frente a sus compañeros.
• Trastorno de ansiedad generalizada: tener una sensación constante de temor o preocupación.
• Trastorno de pánico con agorafobia: miedo a tener un ataque de pánico y no poder escapar.
Por ejemplo, un niño puede sentir ansiedad por ir a la escuela por varias razones. La evaluación de la preocupación específica es fundamental para el diagnóstico y el plan de tratamiento.
Dentro de la clínica se puede destacar(1):
• En los bebés: llanto, irritabilidad, hipertonía muscular, vómitos, hiperventilación, espasmos de sollozo.
• En la edad escolar: miedos, síntomas somáticos (dolor abdominal, cefaleas), irritabilidad, alteración de conducta (inquietud, desobediencia, rabietas), problemas de memoria, atención y concentración, problemas relacionados con el sueño (insomnio, pesadillas), rituales.
• En la adolescencia: irritabilidad, mareo, dolor torácico, insomnio, fatiga y miedos sociales. Es la etapa del desarrollo en que aparecen los síntomas de despersonalización y desrealización. La despersonalización es un sentimiento de extrañeza hacia el propio yo, como si el adolescente se sintiera vacío. En la desrealización, el mundo circundante se percibe como si no existiera, como si no fuera real ni tuviera vida.
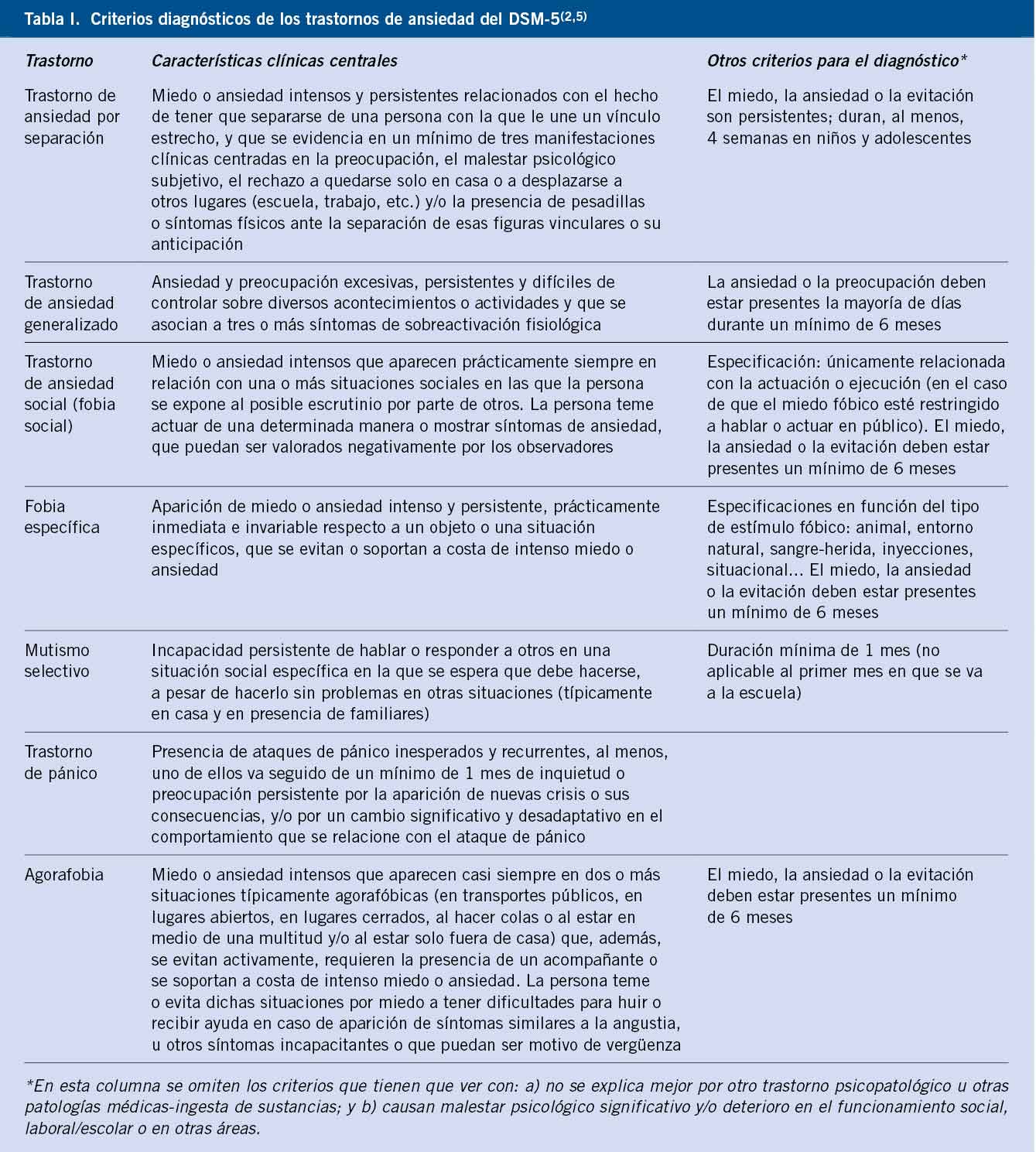
En la tabla I, se muestran los criterios diagnósticos de los trastornos de ansiedad del DSM-5(5).
Comorbilidad
En los trastornos de ansiedad, la comorbilidad es muy frecuente, a pesar de ello suele pasar desapercibida y es infradiagnosticada.
Cuando se diagnostica un trastorno de ansiedad hay que explorar las comorbilidades. La existencia de estas agrava la sintomatología, incrementa el deterioro académico y laboral, y condiciona una mala respuesta al tratamiento, por lo que son importantes, tanto su prevención como su diagnóstico precoz, a veces, difícil por el solapamiento de los síntomas. Los trastornos comórbidos se benefician de un tratamiento específico concomitante. Pueden presentarse de manera transversal, si ocurren varios trastornos en un periodo corto de tiempo, o bien longitudinal, cuando estos trastornos se van desarrollando en un periodo de tiempo más prolongado. Las comorbilidades más frecuentes son:
• Con otro trastorno de ansiedad, 50%.
• Depresión, 33%. La depresión mayor se asocia a trastornos de ansiedad severos, los síntomas de ansiedad suelen preceder a la aparición de los síntomas depresivos.
• Trastorno por síntomas somáticos.
• Trastorno por déficit de atención con hiperactividad (TDAH), 20-40%.
• Trastorno por abuso de sustancias. El tratamiento de la ansiedad ha demostrado acompañarse de una disminución en el consumo de sustancias, y de los problemas derivados de ello a largo plazo.
• Trastorno del sueño, hasta en un 90%.
Diagnóstico
La experiencia del profesional a la hora de reconocer la clínica de ansiedad en la infancia y la adolescencia es determinante para poder detectar a tiempo los trastornos de ansiedad, puesto que el diagnóstico es clínico.
El diagnóstico de los trastornos de ansiedad es clínico y se realiza siguiendo los criterios diagnósticos de las clasificaciones internacionales: CIE-11 y DSM-5.
Existen algunos cuestionarios y escalas de apoyo al diagnóstico para detectar ansiedad en los niños. Entre ellas se encuentran: la SCAS (Escala de Ansiedad para Niños de Spence), para ansiedad prescolar e infantil; y la CMAS-R2 (Escala de Ansiedad Manifiesta en Niños revisada) para niños y adolescentes de 6 a 19 años.
Otro de los cuestionarios es STAIC (State-Trait Anxiety Inventory for Children) o cuestionario de ansiedad estado-rasgo para niños, que evalúa la ansiedad en el momento actual y la predisposición del sujeto a la ansiedad respectivamente(16,17). También es útil el cuestionario CBCL (Child Behavior Checklist), que discrimina síntomas internalizantes.
En la tabla II se muestra un ejemplo de valoración diagnóstica de los trastornos de ansiedad en la infancia y adolescencia(3,15).

Diagnóstico diferencial
El diagnóstico diferencial de los trastornos de ansiedad en la infancia es complejo, debido a las altas tasas de superposición de varios trastornos ansiosos en el mismo paciente y a su com.orbilidad con otros procesos psicológicos-psiquiátricos.
En líneas generales, es necesario hacer el diagnóstico diferencial con(16,2,3):
• Síntomas somáticos y trastornos relacionados.
• Trastorno obsesivo compulsivo (TOC).
• Trastorno de estrés postraumático.
• Trastorno por déficit de atención e hiperactividad (TDAH).
• Trastorno del comportamiento.
• Esquizofrenia.
• Patología orgánica que pueda dar síntomas parecidos a los de la ansiedad: hipotiroidismo e hipertiroidismo, feocromocitoma, arritmias cardiacas (taquicardia supraventricular paroxística), enfermedad de Wilson, epilepsia, hipoglucemia, enfermedad vestibular, asma, esclerosis múltiple, corea de Huntington, tumores del sistema nervioso central, etc.
• Consumo de tóxicos (alcohol, opiáceos, cocaína, cafeína, anfetaminas, etc.) o medicamentos responsables de los síntomas como, por ejemplo: síntomas del síndrome de abstinencia o efectos secundarios de psicoestimulantes, beta-agonistas, esteroides, teofilina, estimulantes alfa-adrenérgicos o bloqueadores de los canales del calcio.
Prevención
El pediatra está en una situación privilegiada para la prevención de estos trastornos, que se basa en la identificación de los factores de riesgo ya descritos para poder hacer un diagnóstico precoz.
Saber detectar los miedos exagerados e inadecuados a la edad del niño, las preocupaciones exageradas y las conductas evitativas, deben hacer sospechar patología y valorar si precisa tratamiento. Son factores de riesgo: antecedentes personales o familiares de ansiedad, presencia de acontecimientos vitales estresantes, sexo femenino, tener una enfermedad médica crónica y timidez e inhibición del comportamiento. Una vez diagnosticados, su correcto tratamiento evitará su cronicidad e impacto negativo en la vida del niño.
Tratamiento
El abordaje de los trastornos de ansiedad es multimodal. La elección del tratamiento debe basarse en: la severidad de la sintomatología, la presencia de comorbilidad, la edad del niño y la naturaleza de los factores causales.
El tratamiento de los trastornos de ansiedad requiere de una aproximación multimodal y de un enfoque terapéutico global, con el objetivo de disminuir la sintomatología, evitar complicaciones a largo plazo, prevenir la aparición de comorbilidad psiquiátrica y el desarrollo de trastornos ansiosos en la edad adulta(1).
La primera intervención es la psicoeducación del niño y sus padres sobre la ansiedad. Los tratamientos eficaces son la psicoterapia (cognitivo-conductual) y el tratamiento farmacológico (inhibidores selectivos de la recaptación de serotonina, ISRS).
El objetivo del tratamiento es reducir la angustia y el estrés del niño o adolescente. Para diseñar un plan de tratamiento habrá que tener en cuenta lo siguiente:
• La gravedad del trastorno.
• El diagnóstico específico del trastorno de ansiedad.
• Tiempo de evolución del trastorno (si es de inicio reciente, con síntomas de larga evolución con empeoramiento progresivo o fluctuante).
• Situaciones en las que presenta síntomas (al ir a colegio, al irse a dormir, etc.).
• Comorbilidades (TDAH, trastorno del espectro autista, trastorno de conducta…).
• Edad y grado de desarrollo del niño. Cuanto más pequeño sea el niño, más debemos centrar la intervención en el entrenamiento de los padres.
• Situación social, familiar y escolar del niño o adolescente.
• Características familiares y psicopatología en la familia.
• Recursos disponibles del sistema de salud y familiares.
• Tratamientos anteriores que hayan sido efectivos o hayan fracasado.
El papel del pediatra de Atención Primaria es clave. En primer lugar, porque es el profesional que conoce el entorno de cada niño y desde su posición puede realizar prevención primaria para evitar la ansiedad, por ejemplo, detectando niños con inhibición conductual y enseñándoles estrategias para que se expongan a situaciones novedosas; enseñando técnicas de afrontamiento enfocadas al problema, como: búsqueda activa de información, autoinstrucciones positivas, distracción, relajación, parar pensamientos negativos, etc.
Por otro lado, una vez que el pediatra detecta un problema de ansiedad en un niño o adolescente, deberá formar en lo posible a las figuras de referencia del menor, sobre los principios básicos del manejo conductual en niños con ansiedad (Tabla III).

Un buen manejo conductual puede frenar o retrasar la evolución de los síntomas, enlentecer la evolución e incluso revertir el trastorno.
Cuando los síntomas provocan problemas, generalmente el manejo solo no es suficiente, y es preciso iniciar tratamiento. El tratamiento de primera elección es la psicoterapia y, en segundo lugar, el tratamiento farmacológico en niños a partir de los 6 años, con síntomas moderados o graves y si ha fracasado la psicoterapia. La terapia cognitivo-conductual (TCC) es el método de psicoterapia con mayor evidencia científica en ensayos controlados aleatorizados para el tratamiento de trastornos de ansiedad.
El objetivo de estas intervenciones es entrenar al niño para que adquiera habilidades de afrontamiento de problemas, mejore su autoconfianza, reestructure sus cogniciones erróneas y modifique sus conductas con la práctica de nuevos comportamientos (técnicas de relajación y respiración, técnicas de estudio, entrenamiento en habilidades sociales, ejercicios de dramatización o “role-play” y exposición gradual a situaciones que provocan ansiedad)(3,18).
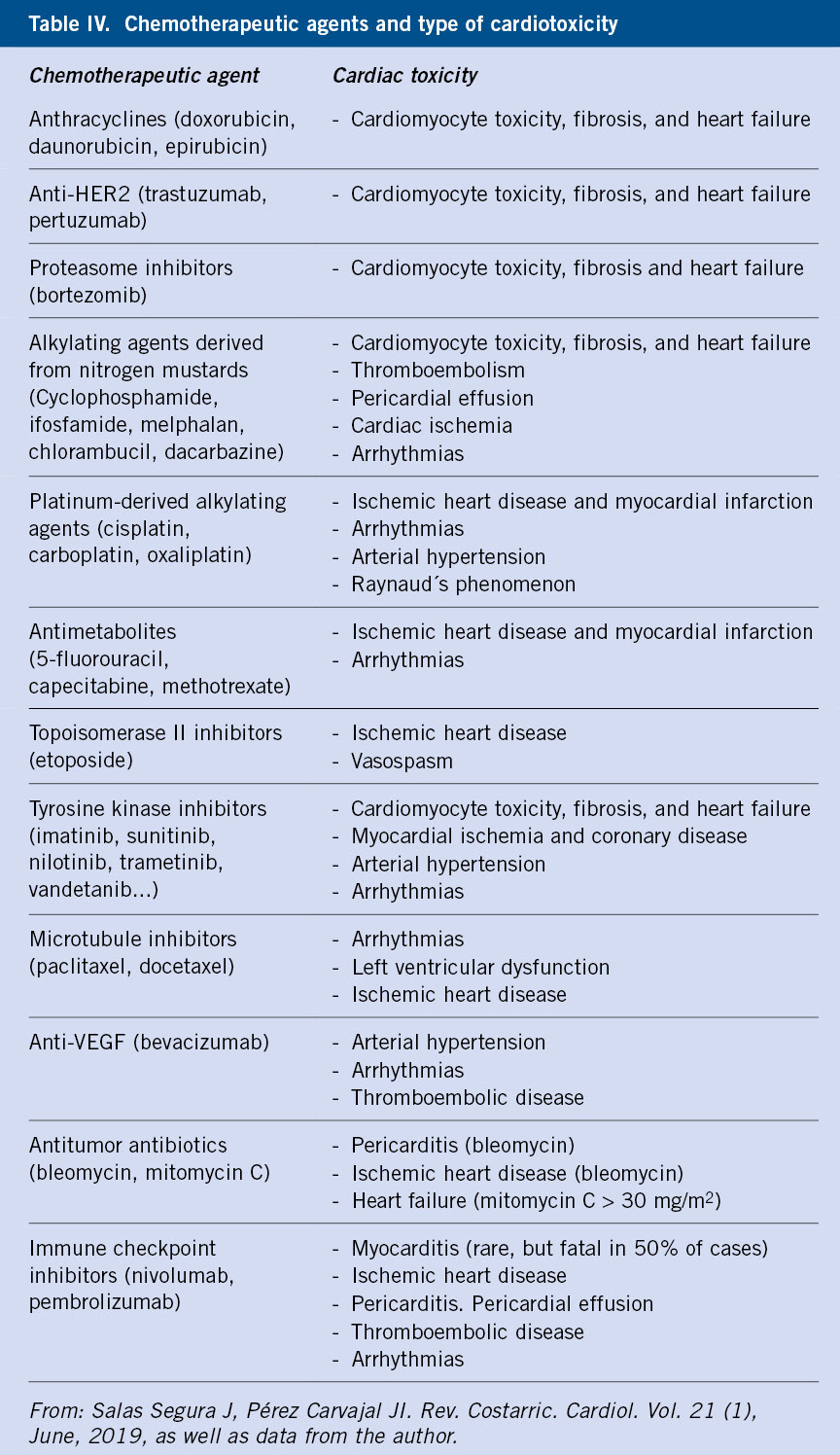
El tratamiento farmacológico de elección para los trastornos de ansiedad son los ISRS. A pesar de que no están aprobados por la Food and Drug Administration (FDA) para el tratamiento de la ansiedad, numerosos estudios demuestran su eficacia y tolerabilidad(19,20). Los ISRS, cuya eficacia ha sido demostrada superior al placebo, son los que se muestran en la tabla IV.
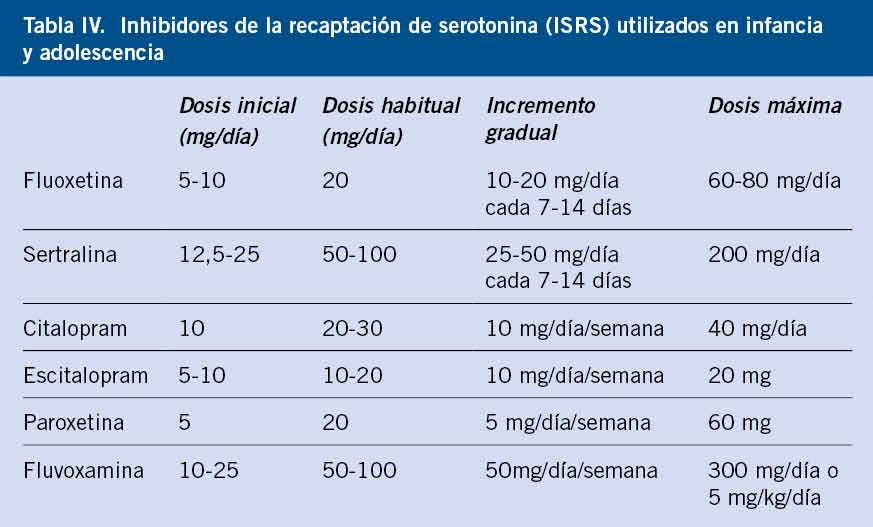
La edad recomendada es por encima de los 7 años para la fluoxetina, de los 6 para la sertralina, de los 8 para la fluvoxamina y de los 12 para el escitalopram. Para el resto, no hay estudios suficientes que recomienden edad de inicio de tratamiento. También son eficaces en el tratamiento de la ansiedad algunos antidepresivos duales, como la venlafaxina (37,5-150 mg/día), la duloxetina (30-60 mg/día) y la mirtazapina (15-30 mg/día), indicados solo en adolescentes y fuera de ficha técnica.
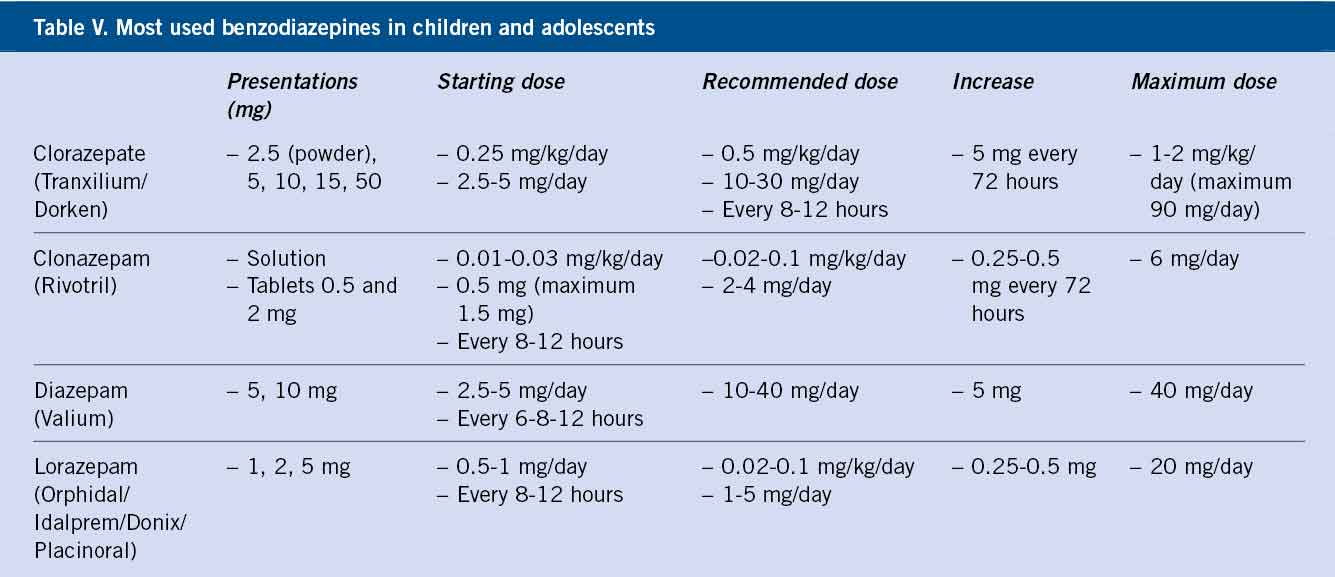
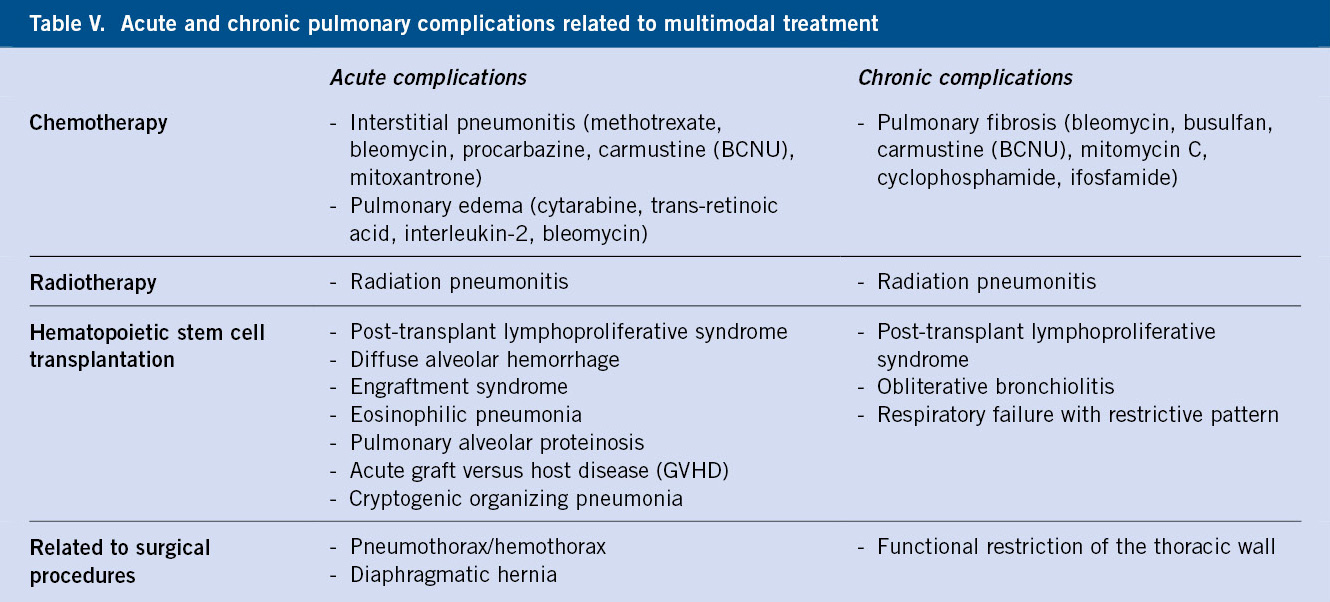
Respecto a los eventos adversos de los ISRS, los más frecuentes son: síntomas gastrointestinales, alteraciones del ciclo sueño-vigilia, inquietud, cefalea, acatisia, cambios en la ingesta alimentaria y disfunción sexual. La venlafaxina puede aumentar ligeramente la tensión arterial, y los niños pueden mostrar empeoramiento de su conducta con mayor impulsividad. Con relación al uso de benzodiacepinas, no han demostrado una eficacia superior a placebo en estudios clínicos controlados en niños y adolescentes con trastornos de ansiedad, por lo que no se consideran psicofármacos de primera elección en estos casos. Sin embargo, se usan con frecuencia en adolescentes con síntomas de ansiedad graves y se debería limitar su indicación en situaciones puntuales o en el inicio del tratamiento con ISRS hasta que estos hagan efecto. Las benzodiacepinas más usadas son las de vida media larga (clorazepato, diazepam o lorazepam) por su mayor tolerancia y su menor riesgo de efecto rebote y de dependencia, como las que se incluyen en la tabla V.

En niños y adolescentes, es conveniente evitar benzodiacepinas como alprazolam o bromazepam. Estos fármacos están contraindicados en adolescentes con abuso de sustancias.
Función del pediatra de Atención Primaria
El pediatra de Atención Primaria es el primer contacto que tiene el niño o adolescente y su familia con el sistema sanitario. Este profesional es el que conoce al niño y su familia desde el nacimiento, pudiendo detectar aquellos temperamentos susceptibles de desarrollar trastornos de ansiedad. Identificar y diagnosticar los trastornos de ansiedad en la edad pediátrica es una tarea compleja. Por una parte, los síntomas ansiosos se van a manifestar como quejas somáticas que van a orientar el diagnóstico hacia patología orgánica. Por otra, la identificación de las preocupaciones en los niños y adolescentes es complicada, debido a la dificultad en la expresión de sentimientos que presentan, propia de su desarrollo madurativo. No olvidemos que, muchos de los trastornos de ansiedad aparecen en asociación con otros trastornos ansiosos o en comorbilidad con otros trastornos mentales. El pediatra de Atención Primaria debe desarrollar la destreza suficiente en la identificación de los síntomas relacionados con la ansiedad y el diagnóstico de los distintos trastornos de ansiedad presentes en la edad pediátrica. Y si se detecta tempranamente, dar las pautas oportunas, tanto al niño/adolescente como a su familia, para iniciar un tratamiento precoz y evitar así enmascaramiento y cronicidad de estos trastornos.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito. Declaración de intereses: ninguno.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1.** Sánchez Mascaraque P, Cohen DS. Ansiedad y depresión en niños y adolescentes. Adolescere 2020; VIII(1): 16-27. Disponible en: https://www.adolescere.es/revista/pdf/volumen-VIII-n1-2020/Adolescere-2020-1-WEB.pdf.
2.*** Guerrero F, Sánchez Mascaraque P. Trastornos por ansiedad y trastorno obsesivo-compulsivo en la infancia y la adolescencia. En: I Curso de psiquiatría del niño y del adolescente para pediatras. Editores: Hidalgo Vicario MI, Rodríguez Hernández PJ; 2019. p.135-62.
3.** Ochando Perales G, Peris Cancio SP. Actualización de la ansiedad en la edad pediátrica. Pediatr Integral. 2017; XXI: 39-46.
4. Riordan DM, Singhal D. Anxiety-related disorders: An overview. J Paediatr Child Health. 2018; 54: 1104-9.
5.** American Psychiatric Association. Manual diagnóstico y estadístico de los trastornos mentales, quinta edición (DSM-5). Médica Panamericana, 2014 (edición original en inglés 2013).
6. Sprang G, Silman M. Posttraumatic Stress Disorder in Parents and Youth After Health Related Disasters. Disaster Med Public Health Prep. 2013; 7: 105-10.
7.** Salud Mental en la Infancia y la Adolescencia en la era del COVID-19. Evidencias y Recomendaciones de las Asociaciones Profesionales de Psiquiatría y Psicología Clínica. Disponible en: https://www.sepypna.com/documentos/2020_InformeCOVID_final.pdf.
8. Zhou S-J, Zhang L-G, Wang L-L, Guo Z-C, Wang J-Q, Chen J-C, et al. Prevalence and socio-demographic correlates of psychological health problems in Chinese adolescents during the outbreak of COVID-19. Eur Child Adolesc Psychiatry. 2020; 29: 749-758. DOI: 10.1007 / s00787-020-01541-4.
9.** Orgilés M, Morales A, Delvecchio E, Mazzeschi C, Espada JP. Immediate psychological effects of the COVID-19 quarantine in youth from Italy and Spain. Front in Psychol. 2020; 11: 579038. DOI:10.3389/fpsyg.2020.579038.
10. Fox NA, Henderson HA, Marshall PJ, Nichols KE, Ghera MM. Behavioral inhibition: linking biology and behavior within a developmental framework. Annu Rev Psychol. 2005; 56: 235.
11. Pérez-Edgar K, Fox NA. Temperament and anxiety disorders. Child Adolesc Psychiatr Clin N Am. 2005; 14: 681.
12.** Rapee RM. Preschool environment and temperament as predictors of social and non-social anxiety disorders in middle adolescence. J Am Acad Child Adolesc Psychiatry. 2014; 53: 320.
13. Meyer A. A biomarker of anxiety in children and adolescents: a review that focuses on error-related negativity (ERN) and anxiety throughout development. Dev Cogn Neurosci. 2017; 27: 58.
14. Hettema JM, Neale MC, Kendler KS. Review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry. 2001; 158: 1568.
15. Ochando G, Peris SP. Actualización de la ansiedad en la edad pediátrica. Pediatría Integral. 2012; XVI: 707-14.
16. Sánchez Mascaraque P. Ansiedad. En: Medicina de la Adolescencia. Atención Integral. Editores: Hidalgo Vicario MI, Rodríguez Molinero L, Muñoz Calvo MT. 3ª Ergon S.A.; 2021. Cap. 120, p.1013-9.
17. San Sebastián J. Trastornos por ansiedad en la infancia y adolescencia. En: Ballesteros MC. ed. Práctica clínica paidopsiquiátrica. Historia clínica. Guías clínicas. Madrid: Adalia. 2006. p. 150-63.
18.** Heiervang ER, Villabo MA, Wergeland GJ. Cognitive behavior therapy for child and adolescents anxiety disorders: an update on recent evidence. Curr Opin Psychiatry. 2018; 31: 484-9.
19. Reinblatt SP, Riddle MA. The pharmacological mangement of childhood anxiety disorders: a review.Psychopharmacology (Berl). 2007; 191: 67-86.
20. Rynn MA, Walkup JT, Compton SN, Sakosky DJ, Sherrill JT, Shen S, et al. Child/adolescent anxiety multimodal study: evaluating safety. J Am Acad Child Adolesc Psychiatry. 2015; 54: 180-90.
Bibliografía recomendada
– Guerrero F, Sánchez Mascaraque P. Trastornos por ansiedad y trastorno obsesivo-compulsivo en la infancia y la adolescencia. En: I Curso de psiquiatría del niño y del adolescente para pediatras. Editores: Hidalgo Vicario MI, Rodríguez Hernández PJ; 2019. p.135-62.
Libro básico de psiquiatría infanto-juvenil para pediatras de Atención Primaria. En el capítulo de ansiedad se expone de una forma práctica el abordaje de los trastornos de ansiedad en la infancia y la adolescencia.
– Sanchez Mascaraque P. Ansiedad. En: Medicina de la Adolescencia. Atención Integral. Editores: Hidalgo Vicario MI, Rodríguez Molinero L, Muñoz Calvo MT. 3ª Ergon S.A.; 2021. Cap. 120, p.1013-9.
Este capítulo de ansiedad se centra en la adolescencia. Es en esta franja de edad donde debutan muchas de las patologías mentales, por eso la detección y el manejo específico de los adolescentes es tan importante para el pediatra que trata a esta población hasta los 14-16 años.
– Rubio Morell B, Moreno Pardillo D, Lázaro García L. Manual de psiquiatría en la Infancia y la Adolescencia. (1ª edición). Elsevier. 2021. ISBN: 978-84-911384-7-1.
Libro que reúne las bases teóricas de la disciplina desde un modelo biopsicosocial e integrador, cuyo contenido se asienta en las últimas y más robustas evidencias científicas. Este enfoque práctico surge como respuesta a la inminente creación de la especialidad de Psiquiatría de la Infancia y la Adolescencia, con el objetivo de que los profesionales dispongan de un acceso a la formación: práctico, rápido y basado en la evidencia.
| Caso clínico |
|
Motivo de consulta: dolor abdominal y pérdida de peso. Enfermedad actual: adolescente de 13 años de edad, la deriva su pediatra porque, durante el confinamiento por la pandemia COVID, se ha venido quejando de dolor abdominal casi diario. Por este motivo ha disminuido la ingesta alimentaria, ha restringido alimentos grasos e hipercalóricos y también come menos cantidades. La razón que da es que así le duele menos. En alguna ocasión ha vomitado tras la comida, no ha sido de forma provocada. Ha perdido unos 5 kg en dos semanas y esto le ha provocado una gran preocupación por su salud. Afirma verse muy delgada y con mal aspecto físico. Mide 1,58 cm y pesa 42 kg. Su índice de masa corporal es 16,8. Menarquia a los 11 años, reglas regulares. Entre sus antecedentes personales, destaca que padece de dolor abdominal desde la infancia y que este se exacerba cuando tiene exámenes y cuando se tiene que enfrentar a situaciones nuevas. Es estreñida de siempre y durante el confinamiento ha empeorado. Siempre ha sido delgada, no consigue engordar. En verano es cuando se encuentra mejor y gana algo de peso. Su pediatra, tras realizar una exploración física exhaustiva y pruebas complementarias de primer nivel (analítica de sangre y orina), no ha encontrado patología física que justifique los síntomas. La exacerbación de su dolor motiva que acuda a urgencias hospitalarias y es ingresada para estudio. No se encuentra ningún hallazgo patológico. Le recomiendan una dieta y medidas higiénico conductuales que sigue escrupulosamente, llegando a la obsesividad en el cumplimiento. Psicobiografía: vive con sus padres y una hermana de 11 años. No tiene antecedentes psiquiátricos. Estudiante brillante, no se conforma con menos de un notable. Muy buena conducta, tanto en casa como en el instituto. En primaria sintió rechazo por sus compañeros, pero en el instituto está contenta. Es una niña tímida, inhibida, insegura, con necesidad de anticipar y controlar lo que hace. Ante situaciones nuevas o cambios en su vida se pone muy nerviosa. En exámenes lo pasa muy mal y, a veces, vomita. Apenas se relaciona con sus iguales con los que no comparte intereses, su meta es estudiar y sacar muy buenas notas. Sus padres la describen como hiperexigente y perfeccionista. Con la pandemia lo está pasando muy mal. Pasa horas estudiando agobiada por los resultados. Las clases on line la han descentrado y ha perdido el control. Tiene mucho miedo a enfermar de COVID por si contagia a sus padres. No quiere salir a la calle, porque la gente no respeta las medidas de protección. Ella relaciona los vómitos con su nivel de ansiedad. Exploración psicopatológica: ánimo triste, importante ansiedad, preocupación excesiva por su rendimiento académico y por su futuro, con una visión pesimista, anticipa fracasos de forma no realista, temores hipocondriacos, miedo a contagiar a sus padres no justificado, insomnio de conciliación, ansiedad ante la comida por miedo a que le genere dolor abdominal y vómitos, no presenta alteración en la percepción de su esquema corporal. Familia: como único antecedente, la madre está diagnosticada de colon irritable.
|