 |
| Temas de FC |
M.D. Corral Sánchez*, B.M. Tarabini-Castellani Ciordia**
*Servicio de Hemato-Oncología Pediátrica. Hospital Universitario La Paz. Madrid. **Sección de Hemato-Oncología Pediátrica. Servicio de Pediatría. Hospital Universitario Donostia. San Sebastián.
Ambas autoras igual contribución
| Resumen
Los tumores renales en la edad pediátrica suponen aproximadamente el 6% de las neoplasias. Dentro de ellos, el más frecuente es el tumor de Wilms (aproximadamente un 90%). Se presenta típicamente entre los 3 y los 5 años de edad. El tumor de Wilms puede aparecer de forma aislada o en el contexto de síndromes genéticos con predisposición al cáncer, asociando o no malformaciones. En el 5-8%, se puede presentar como enfermedad bilateral. Cada vez se conoce más de la genética de estos tumores, y su asociación con el comportamiento tumoral. |
| Abstract
Renal tumors account for approximately 6% of the tumors in the pediatric age. The most frequent one is Wilms tumor (approximately 90%). Most of the cases are diagnosed between 3 and 5 years of age. Wilms tumor appears as a sporadic disease or it may occur as part of genetic syndromes with cancer predisposition, with or without malformations. 5 to 8% of the cases are bilateral. There is an increasing knowledge of the genetics of these tumors and their correlation with tumoral behavior. |
Palabras clave: Wilms; Renal; Masa abdominal; Pediatría.
Key words: Wilms; Kidney; Abdominal mass; Pediatrics.
Pediatría Integral 2021; XXV (7): 341 – 347
Tumor de Wilms y otros tumores renales
Introducción
Los tumores renales suponen aproximadamente el 6% de los tumores malignos en la edad pediátrica(1). El tumor renal más frecuente (90%) es el tumor de Wilms, con una prevalencia de 1 caso entre 10.000 niños menores de 15 años(2). Otros tumores renales menos frecuentes son: nefroma mesoblástico, sarcoma de células claras, tumor rabdoide-teratoide atípico y carcinoma renal (Tabla I)(1).

Tumor de Wilms
El tumor de Wilms es el tumor renal más frecuente en la infancia. Se puede desarrollar a partir de lesiones precursoras llamadas restos nefrogénicos.
El tumor de Wilms aparece con una edad típica de presentación entre los 3 y 5 años de edad, siendo el carcinoma renal más frecuente en el grupo de 15 a 19 años y el nefroma mesoblástico en los primeros meses de vida(3).
Se desarrolla a partir de restos nefrogénicos o tejido metanéfrico persistente. Estos restos están presentes en el 1% de los niños al nacimiento, pero suelen regresar durante la infancia. Se reconocen en todos los casos de tumor de Wilms bilateral y en el 35% de los tumores unilaterales(4) y se consideran lesiones precursoras.
El pronóstico del tumor de Wilms ha mejorado en los últimos años, variando según el estadio y la histología, con unos porcentajes de supervivencia a 5 años del 85-90% en los casos favorables. Los pacientes con estadios avanzados y con histología desfavorable tienen peor pronóstico, con una supervivencia a 5 años del 38-84%)(5).
Bases genéticas del tumor de Wilms
Aproximadamente, un tercio de pacientes con tumor de Wilms presentan mutaciones en: WT1, CTNNB1 o WTX. El subtipo anaplásico se caracteriza por mutaciones en TP53. El gen WT1 se localiza en el brazo corto del cromosoma 11 (11p13). Es imprescindible para el desarrollo normal genitourinario(3).
Síndromes asociados al tumor de Wilms
El tumor de Wilms puede aparecer en el contexto de un síndrome genético de predisposición a cáncer, que puede asociar o no anomalías congénitas.
Aproximadamente, el 10% de los casos de tumor de Wilms aparecen en niños con anomalías o síndromes congénitos, entre los que se incluyen los siguientes:
• Síndrome WAGR (tumor de Wilms, aniridia, anomalías genitourinarias y retraso mental). Estos pacientes presentan un riesgo del 50% de desarrollar tumor de Wilms a una edad más temprana y con mayor incidencia de tumores bilaterales. Se produce por una deleción del gen WT1 (11p13)(2-4).
• Síndrome Denys-Drash: se caracteriza por enfermedad renal progresiva, pseudohermafroditismo y tumor de Wilms. La enfermedad renal comienza en edad temprana con proteinuria, que progresa a síndrome nefrótico y, posteriormente, a fallo renal. El 90% de los individuos afectos desarrollan tumor de Wilms. Se produce por una mutación puntual en el gen WT1(4).
• Síndrome de Beckwith-Wiedemann: es un síndrome producido por mutaciones en la región 11p15.5. Estos pacientes presentan: macrosomía, hemihipertrofia, macroglosia, onfalocele y predisposición a desarrollar tumores, tales como: tumor de Wilms, hepatoblastoma y rabdomiosarcoma (Tabla II). Aproximadamente el 10% de estos pacientes desarrollarán un tumor de Wilms(3).
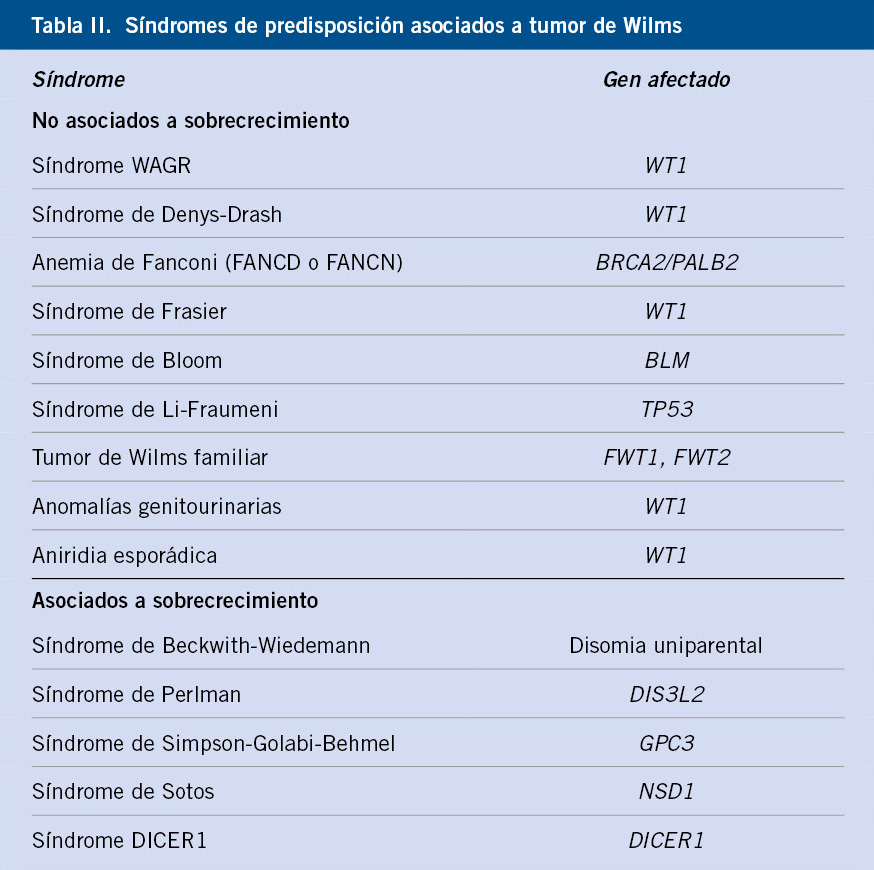
• Síndromes de sobrecrecimiento, tales como: síndrome de Sotos, Perlman, Simpson-Golabi-Behmel o la hemihipertrofia aislada.
• Malformaciones genitourinarias aisladas, como hipospadias o criptorquidia en varones, o malformaciones uterinas en mujeres, se asocian a tumor de Wilms.
• Tumor de Wilms familiar: es poco frecuente (1-2% de los casos) y se asocia a mutaciones en los genes FTW1 y FTW2. Además, también se ha descrito mayor predisposición a tumor de Wilms en el síndrome de Li-Fraumeni o en pacientes con mutaciones en BRCA2 (Tabla II).
• Anemia de Fanconi con mutaciones en BRCA2 (FANCD1) o PALB2 (FANCN). Ambos fenotipos de anemia de Fanconi presentan un riesgo muy aumentado de tumores en la edad pediátrica, entre ellos el tumor de Wilms. Estos genes participan en la reparación del DNA(3).
En los pacientes con síndromes de sobrecrecimiento, así como en los síndromes WAGR y Denys-Drash, se recomiendan controles ecográficos trimestrales para despistaje de tumor de Wilms, al menos, hasta los 8 años de edad, así como en los pacientes con restos nefrogénicos(3).
Nefroblastoma bilateral
En el 5-8% de los casos, el tumor de Wilms puede presentarse como enfermedad bilateral. Supone un reto terapéutico preservar la función renal en estos pacientes.
El nefroblastoma bilateral ocurre en el 5-10% de los pacientes y es más frecuente cuando existe un síndrome de predisposición o mutación germinal en WT1. Se asocia con la presencia de restos nefrogénicos. Estos restos son detectables por histología en el tumor de Wilms unilateral, mientras que en los casos bilaterales, a menudo, se detectan por imagen(2) (Fig. 1).

Figura 1. Nefroblastomatosis bilateral.
La enfermedad bilateral puede ser sincrónica o metacrónica en los primeros 4-5 años del diagnóstico del primer tumor y se presenta a una edad más temprana que la unilateral, habitualmente en los primeros 2 años de vida(2). El pronóstico de estos pacientes ha mejorado con los protocolos actuales de tratamiento, alcanzando supervivencia del 80%(6).
Clínica
La presentación más frecuente del tumor de Wilms es el hallazgo casual de una masa abdominal.
La presentación clínica más frecuente del tumor de Wilms es el hallazgo casual de una masa abdominal a la exploración sin otros síntomas acompañantes. Puede ser detectada en la exploración rutinaria por el pediatra o por los propios padres al vestir o bañar al niño(3,4).
El dolor se da en un 30-40% de los casos y la hematuria, que es menos frecuente, se presenta de forma intermitente como microhematuria (24%) o, en ocasiones, macrohematuria (18%). También puede presentarse de forma más infrecuente con afectación del estado general: fiebre, vómitos y otros síntomas constitucionales(3,4).
La tríada clásica de dolor, hipertensión y hematuria es poco frecuente en este tipo de tumores, apareciendo en aproximadamente una cuarta parte de los pacientes. La hipertensión es debida a la secreción de renina por parte del tumor, aunque también puede darse por compresión de vasos renales.
A la palpación, se trata de una masa generalmente de bordes bien definidos que se localiza en uno de los flancos. Rara vez, sobrepasa la línea media abdominal.
Una vez detectada, hay que realizar la exploración física de manera cuidadosa, puesto que se corre el riesgo de presentar una rotura tumoral.
Una forma de presentación más infrecuente es en forma de abdomen agudo, tras la rotura del tumor. Esto puede suceder de forma espontánea o tras un traumatismo abdominal, y cursa con hemoperitoneo, siendo una urgencia quirúrgica.
En el diagnóstico de tumor de Wilms, hay que buscar los signos clínicos típicos de síndromes de predisposición genética, por si no hubieran sido previamente detectados.
La diseminación tumoral se puede producir por contigüidad a través de la cápsula renal, o de una forma hematógena. Aunque presentar metástasis al diagnóstico es poco frecuente (12% de los casos), en caso de aparecer, la localización más frecuente es el pulmón (80%). Otras localizaciones metastásicas menos frecuentes son: el hígado (15%), y aún más raras, el hueso, la médula ósea y el sistema nervioso central(6).
Diagnóstico
Aunque el diagnóstico definitivo es anatomopatológico, en la mayoría de los casos, mediante radiología, se puede realizar un diagnóstico que permite iniciar el tratamiento sin precisar cirugía inicial.
Radiología
Inicialmente, y dada la rápida disponibilidad de la misma, la primera prueba diagnóstica a realizar tras la exploración es una ecografía(3,4,7).
Mediante la ecografía, se puede determinar si la masa se origina en el riñón. También permite determinar la presencia de trombos intravasculares o de infiltración vascular por parte del tumor mediante el empleo de Doppler. La limitación principal de esta prueba es que es operador dependiente, y que puede verse artefactada por la presencia de gas en el abdomen o de obesidad(7,8).
Una vez detectada una masa renal, para determinar la anatomía de la misma y su extensión, se debe realizar otra prueba de imagen, para lo cual la mayor parte de niños precisa sedación. Preferiblemente, se realizará una RMN abdominal. En caso de que esta prueba no esté disponible en un tiempo razonable o si se trata de un abdomen agudo, se puede realizar también una TAC abdominal (siendo preferible la RMN por la radiación que implica la realización de la TAC)(4,7). Con estas pruebas, además de la extensión y naturaleza de la masa detectada, se puede detectar la presencia de otras masas abdominales y determinar si existe afectación del riñón contralateral(7,8) (Fig. 2).

Figura 2. Masa renal izquierda en imagen de TAC.
Además, se debe realizar una radiografía basal de tórax, así como una TAC torácica para detectar metástasis pulmonares(3,7,8).
En los casos en los que la clínica, la edad del paciente y la imagen radiológica son claramente sugestivas de tumor de Wilms, en Europa se inicia tratamiento con quimioterapia preoperatoria sin confirmación histológica(4,7).
Sin embargo, en los casos en los que existen dudas tras realizar las pruebas diagnósticas pertinentes, puede ser necesaria la realización de una extirpación quirúrgica total de la masa para determinar la naturaleza de la misma, en caso de ser posible. En el caso de tratarse de una masa irresecable, puede ser necesaria la realización de una biopsia(7).
Los pacientes con otro tipo de tumores renales pueden precisar otro tipo de pruebas como son la RMN cerebral y la gammagrafía ósea en el tumor rabdoide y en el sarcoma de células claras(7).
Laboratorio
Además de la radiología, también es necesaria la realización de pruebas de laboratorio. Al diagnóstico, se realiza: hemograma completo, ionograma (incluyendo calcio), función renal y hepática, análisis de orina y estudio de coagulación(3,4,7).
Hasta un 2% de los pacientes con tumor de Wilms presentan enfermedad de von Willebrand adquirida, que se detecta mediante el análisis de la coagulación.
Estadiaje y grupos de riesgo
El estadiaje se basa en el estadio local y la histología, y se obtiene tras la resección quirúrgica una vez administrada la quimioterapia preoperatoria.
Los tumores de Wilms se dividen en tres grupos de riesgo en función de la histología(7):
• Riesgo bajo: nefroma mesoblástico y nefroma quístico parcialmente diferenciado.
• Riesgo intermedio: nefroblastoma no anaplásico y sus variantes (epitelial, estromal, mixto y regresivo); nefroblastoma con anaplasia focal.
• Riesgo alto: nefroblastoma con anaplasia difusa; blastematoso; sarcoma renal de células claras; y tumor rabdoide renal.
Estadios
Tras la cirugía, se estratifica el riesgo según el subtipo histológico y el estadio local. Estos datos, en combinación con el volumen tumoral inicial, permiten clasificar a los pacientes en grupos de riesgo(1,7).
• Estadio I: el tumor se encuentra limitado al riñón, no ha sobrepasado la cápsula tumoral y se ha realizado extirpación quirúrgica completa en la cirugía.
• Estadio II: el tumor se ha extendido del riñón a las estructuras vecinas, pero también se ha realizado extirpación completa en la cirugía.
• Estadio III: el tumor se ha extendido a las estructuras vecinas del riñón, sin extirpación completa; o presenta afectación linfática abdominal o ha habido rotura tumoral; o hay afectación vascular. En los casos en los que se ha precisado biopsia abierta, también se considera estadio III.
• Estadio IV: implica diseminación a distancia; ya sea por presencia de metástasis hematógenas o afectación ganglionar fuera del abdomen.
• Estadio V: tumor bilateral. Precisa estadiaje de cada tumor de forma independiente.
Diagnóstico diferencial
El diagnóstico diferencial del tumor de Wilms incluye otros tumores renales y el neuroblastoma.
Para diferenciar entre tumor de Wilms y neuroblastoma, la radiología suele ser suficiente para distinguir el origen del tejido tumoral. Sin embargo, en ocasiones, puede llegar a ser necesaria la realización de una MIBG (gammagrafía con metayodobencilguanidina que resulta positiva en el 90% de los neuroblastomas), catecolaminas en orina o de una biopsia para confirmación histológica en casos de serias dudas(3,8).
Otros tumores renales
Hasta en un 10% de los casos existen otros tumores renales, con histología y comportamiento muy diferente(4,7,9).
• Sarcoma renal de células claras: es el segundo tumor renal más frecuente en la edad pediátrica (3- 5%). Se presenta a la misma edad que el tumor de Wilms. Es más frecuente en varones. Casi siempre es unilateral. En el 15% de los casos presenta recaídas, que pueden ser tardías. Puede presentar metástasis óseas(7,9).
• Nefroma mesoblástico congénito: Es el tumor más frecuente en el periodo neonatal y durante los primeros 3 meses de vida. El 90% de estos tumores se diagnostica en el primer año de vida. Es más frecuente en varones. En un 15-20% de los casos, se diagnostica de forma prenatal mediante ecografía. Tiene muy buen pronóstico, con una supervivencia a 5 años del 95%(7,9).
• Tumor rabdoide renal: es un tumor poco frecuente, de mal pronóstico. El 80% se diagnostica en los 2 primeros años de vida. Presenta metástasis al diagnóstico en un 22-38% de los casos. En un 10-15% de los casos, puede asociar la presencia de un tumor teratoide rabdoide cerebral atípico (ATRT). En un 90%, presenta inactivación bialélica de SMARCB1. En estos casos, hay que descartar la mutación en línea germinal, puesto que puede tratarse de un síndrome de predisposición rabdoide. Este tumor presenta una mortalidad de hasta el 80% en el primer año del diagnóstico(7,9).
• Carcinoma de células renales: es un tumor raro en la edad pediátrica, siendo algo más frecuente en la adolescencia. Se presenta de forma más diseminada que en la edad adulta. Puede tratarse de un segundo tumor tras haber recibido quimioterapia o radiación abdominal(9).
• Carcinoma renal medular: se da de forma casi exclusiva en los pacientes con anemia de células falciformes. Es muy invasivo, se presenta con metástasis precoces y tiene una alta mortalidad(9).
Tratamiento
Los pilares del tratamiento del tumor de Wilms son: la quimioterapia y la cirugía, precisando, además, radioterapia un subgrupo de pacientes.
Se relacionan con peor respuesta los siguientes factores(3,4): histología anaplásica(3), estadio III-IV, edad mayor de 2 años y hallazgos moleculares en el tumor, como pérdida de heterocigosidad de 1p, 11p15 y 16q o la ganancia de 1q, alteraciones que se presentan en el 28% de los tumores(10,11).
Actualmente, en Europa, se siguen las directrices de tratamiento del protocolo SIOP-UMBRELLA-RTSG2016 (Renal Tumour Study Group of the International Society of Pediatric Oncology), mientras que en los países americanos, se tratan según protocolo COG (Children’s Oncology Group). La principal diferencia entre ambos es la cirugía inicial que recomienda COG, mientras que en SIOP, se administra quimioterapia precirugía con el objetivo de reducir las posibles complicaciones derivadas de la misma, principalmente rotura tumoral, con la consiguiente menor necesidad de radioterapia. Ambos protocolos son comparables en términos de supervivencia(1,7).
De acuerdo con el protocolo SIOP(6,7), se administra quimioterapia preoperatoria a todos los pacientes entre los 6 meses y 16 años de edad. En niños menores de 6 meses es frecuente el nefroma mesoblástico congénito, cuyo tratamiento es únicamente quirúrgico. Por ello, en esta edad, se debe valorar individualmente el riesgo de rotura tumoral frente a la toxicidad de la quimioterapia. En pacientes mayores de 16 años, se debe descartar carcinoma de células renales mediante cirugía inicial, siempre que sea posible.
Para tumores localizados, la quimioterapia preoperatoria consiste en la combinación de vincristina y actinomicina durante 4 semanas. La cirugía se debe realizar entre las semanas 5 y 6, y está recomendada la nefrectomía radical con toma de muestras de adenopatías. En los metastásicos se añade doxorrubicina y el tratamiento inicial se prolonga 6 semanas(6,7). Con este régimen, el 61-67% de pacientes presentan remisión de las metástasis previamente a la cirugía(6).
El tratamiento posquirúrgico, según estadio y subtipo histológico, es el siguiente(7):
• Estadio I:
- Histología de bajo riesgo: no requiere tratamiento posterior.
- Histología de riesgo intermedio: vincristina y actinomicina 4 semanas.
- Histología de alto riesgo: vincristina, actinomicina y doxorrubicina 27 semanas.
• Estadios II y III:
- Histología de riesgo bajo/intermedio: vincristina y actinomicina 27 semanas.
- Histología de riesgo alto: régimen HR1 34 semanas (combina ciclofosfamida, doxorrubicina, etopósido y carboplatino).
En caso de enfermedad metastásica al diagnóstico(6), el tratamiento posquirúrgico combina vincristina, actinomicina y doxorrubicina, además de radioterapia pulmonar en caso de nódulos mayores de 3 mm. Siempre que sea posible, se recomienda la extirpación de nódulos para la confirmación histológica(7). Los nódulos menores de 3 mm no se consideran metástasicos(6).
En el caso de estadio IV en pacientes con histología de alto riesgo, se recomienda un régimen basado en: vincristina, irinotecán, ciclofosfamida, carboplatino, etopósido, y doxorrubicina, seguido de megadosis de quimioterapia con rescate autólogo de progenitores hematopoyéticos(12).
En la enfermedad bilateral, uno de los objetivos primordiales es preservar la mayor funcionalidad renal, evitando en lo posible la nefrectomía total(6). El tratamiento inicial es vincristina y actinomicina durante 6 semanas con reevaluaciones periódicas hasta poder realizar cirugía. Si no se objetiva respuesta tras 12 semanas de tratamiento, se cambia el régimen quimioterápico a carboplatino/etopósido. La cirugía debe intentar preservar el mayor tejido funcional posible (“cirugía conservadora de nefronas”). El tratamiento posquirúrgico debe ser el correspondiente a la histología de más riesgo y el estadio más avanzado(2,7).
Tratamiento de las recaídas
El riesgo de recaída es del 15% en pacientes con histología de riesgo intermedio y del 50% en pacientes con anaplasia o tipo blastematoso. El 50-60% de las recidivas ocurren a nivel pulmonar, el 30% a nivel abdominal y el 10-15% en otros sitios como hueso o sistema nervioso central(7).
Según el protocolo SIOP-UMBRELLA, los pacientes en recaída se clasifican en tres grupos, según la histología inicial y el tratamiento recibido en primera línea(6,7).
• Riesgo estándar (Grupo AA) (30%): corresponden a los pacientes con estadio inicial I-II, histología de riesgo bajo o intermedio, tratados con vincristina/actinomicina, sin radioterapia: se tratan con ciclos de doxorrubicina/ciclofosfamida y carboplatino/etopósido. La supervivencia en este grupo es del 70-80%.
• Alto riesgo (grupo BB) (45-50%): corresponde a los pacientes que han recibido, en primera línea, tratamiento con 3 o más fármacos, con o sin radioterapia. La supervivencia en este grupo es del 40-50%. Estos pacientes reciben altas dosis de melfalán, con rescate autólogo de progenitores hematopoyéticos, precedido de quimioterapia, que incluye: carboplatino, etopósido, ciclofosfamida e ifosfamida(13).
• Muy alto riesgo (grupo CC) (10-15%): corresponde a los pacientes con anaplasia o con histología de tipo blastematoso, que han recibido inicialmente tratamiento con 4 fármacos, además de los pacientes que recaen tras el tratamiento de rescate. La supervivencia en este grupo es solo del 10%. En estos pacientes, se recomienda tratamiento con irinotecán/vincristina e inclusión en ensayos clínicos, siempre que sea posible. Se realizará megadosis de quimioterapia con rescate autólogo en los pacientes que presenten respuesta.
Tratamiento con radioterapia
Está indicado el tratamiento con radioterapia en los siguientes casos(6,7):
• Los pacientes con estadio III e histología de riesgo intermedio o alto, y los pacientes con estadio II e histología de riesgo alto recibirán radioterapia local.
• Los pacientes con estadio III por rotura tumoral pre o intraoperatoria o depósitos peritoneales macroscópicos recibirán radioterapia abdominal.
• En ambos casos, la radioterapia se inicia 2-4 semanas tras la cirugía.
• Durante la radioterapia, se debe evitar o reducir la dosis de actinomicina D y de doxorrubicina.
• Recibirán radioterapia pulmonar todos los pacientes con histología de alto riesgo y metástasis pulmonares. En caso de histología de riesgo intermedio, la recibirán si persisten tras quimioterapia.
Toxicidad de los tratamientos
Hasta el 25% de los supervivientes de un tumor de Wilms presentan efectos adversos a largo plazo que pueden comprometer su calidad de vida y su supervivencia.
Los supervivientes de un tumor renal tienen un riesgo incrementado respecto a la población general de enfermedades crónicas, incluyendo: segundos tumores, afectación renal, cardiaca o pulmonar o problemas de fertilidad, entre otros(7,14,15).
• Segundos tumores: presentan segundos tumores el 0,5-1% a los 10 años y el 2-3% a los 30 años. Los más frecuentes son: cáncer de mama, sarcomas y linfomas.
• Problemas cardiacos: asociados a la administración de doxorrubicina, con mayor riesgo cuando se supera la dosis acumulada de 250 mg/m2 y cuando se asocia a radioterapia.
• Insuficiencia renal: ocurre en el 1% de tumores unilaterales y en el 10% de casos bilaterales. Se relaciona con la cirugía y con el tratamiento citostático.
• Fibrosis pulmonar: directamente relacionada con la radioterapia pulmonar.
• Ototoxicidad: se relaciona directamente con la administración de carboplatino.
• Toxicidad gonadal: los pacientes no suelen presentar problemas de fertilidad, salvo en los casos que han recibido radioterapia abdominal y agentes alquilantes.
Seguimiento a largo plazo
El seguimiento de estos pacientes debe incluir la detección de recidivas y de los efectos adversos a largo plazo.
El riesgo de recidiva en los pacientes con tumor de Wilms ocurre principalmente en los primeros 2-3 años tras finalizar el tratamiento(1).
Por ello, se recomienda exploración física (incluyendo tensión arterial), ecografía abdominal, radiografía de tórax y analítica de sangre y orina cada 3 meses, durante los primeros dos años de seguimiento. Durante el tercer y cuarto año de seguimiento, estas revisiones se pueden espaciar a cada 4-6 meses; y tras el quinto año, se recomienda repetirlas anualmente(7).
El seguimiento de efectos secundarios a largo plazo incluye: valoración cardiológica, audiometría y valoración endocrinológica de crecimiento y desarrollo puberal, y de la función tiroidea en pacientes que han recibido radioterapia torácica.
Función del pediatra de atención primaria
Es imprescindible la exploración abdominal en las revisiones del niño sano o cuando se valora a un paciente por otro motivo, para detectar posibles masas abdominales asintomáticas. En el caso de descubrir una masa se debe derivar al paciente lo más rápidamente posible a un centro hospitalario especializado. Es importante tomar la tensión arterial previa a la derivación. No se debe olvidar que, una vez detectada la masa, existe riesgo de rotura tumoral, por lo que se deberá realizar la exploración cuidadosamente.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1.** Vujani? GM, Gessler M, Ooms AHAG, Collini P, Coulomb-l’Hermine A, D’Hooghe E, et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol. Noviembre de 2018; 15: 693-701.
2.** Charlton J, Irtan S, Bergeron C, Pritchard-Jones K. Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med. 2017; 19: e8.
3.*** PDQ Pediatric Treatment Editorial Board. Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®): Health Professional Version. En: PDQ Cancer Information Summaries (Internet). Bethesda (MD): National Cancer Institute (US); 2002 (citado el 19 de mayo de 2021). Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK65842/.
4.*** Leslie SW, Sajjad H, Murphy PB. Wilms Tumor. En: StatPearls (Internet). Treasure Island (FL): StatPearls Publishing; 2021 (citado el 19 de mayo de 2021). Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK442004/.
5.** Aldrink JH, Heaton TE, Dasgupta R, Lautz TB, Malek MM, Abdessalam SF, et al. Update on Wilms tumor. J Pediatr Surg. 2019; 54: 390-7.
6.*** On behalf of the International Society of Paediatric Oncology – Renal Tumour Study Group (SIOP-RTSG), van den Heuvel-Eibrink MM, Hol JA, Pritchard-Jones K, van Tinteren H, Furtwängler R, et al. Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat Rev Urol. 2017; 14: 743-52.
7.*** Protocolo SIOP-UMBRELLA RTSG 2016. EudraCT number: 2016-004180-39. Versión 2.0. Octubre 2018.
8.** Watson T, Oostveen M, Rogers H, Pritchard-Jones K, Olsen Ø. The role of imaging in the initial investigation of paediatric renal tumours. Lancet Child Adolesc Health. 2020; 4: 232-41.
9.** Dome JS, Gooskens SL, van den Heuvel-Eibrink MM. Non-Wilms Pediatric Renal Tumors. En: Pritchard-Jones K, Dome JS, editores. Renal Tumors of Childhood: Biology and Therapy (Internet). Berlin, Heidelberg: Springer; 2014 (citado el 15 de junio de 2021). p. 249-69. (Pediatric Oncology). Disponible en: https://doi.org/10.1007/978-3-662-44003-2_14.
10.* Perlman EJ, Grundy PE, Anderson JR, Jennings LJ, Green DM, Dome JS, et al. WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children’s oncology group study. J Clin Oncol Off J Am Soc Clin Oncol. 2011; 29: 698-703.
11.* Grundy PE, Breslow NE, Li S, Perlman E, Beckwith JB, Ritchey ML, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol Off J Am Soc Clin Oncol. 2005; 23: 7312-21.
12.** Kremens B, Gruhn B, Klingebiel T, Hasan C, Laws H-J, Koscielniak E, et al. High-dose chemotherapy with autologous stem cell rescue in children with nephroblastoma. Bone Marrow Transplant. 2002; 30: 893-8.
13.* Ha TC, Spreafico F, Graf N, Dallorso S, Dome JS, Malogolowkin M, et al. An international strategy to determine the role of high dose therapy in recurrent Wilms’ tumour. Eur J Cancer Oxf Engl 1990. 2013; 49: 194-210.
14.** van Dijk IWEM, Oldenburger F, Cardous-Ubbink MC, Geenen MM, Heinen RC, de Kraker J, et al. Evaluation of late adverse events in long-term wilms’ tumor survivors. Int J Radiat Oncol Biol Phys. 2010; 78: 370-8.
15.** Termuhlen AM, Tersak JM, Liu Q, Yasui Y, Stovall M, Weathers R, et al. Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. 2011; 57: 1210-6.
16. Llort Sales A, Gros Subias L. Tumores renales en la infancia y adolescencia. Pediatr Integral. 2016; XX(7): 447-57.
Bibliografía recomendada
– Leslie SW, Sajjad H, Murphy PB. Wilms Tumor. En: StatPearls (Internet). Treasure Island (FL): StatPearls Publishing; 2021 (citado el 19 de mayo de 2021). Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK442004/.
Revisión sobre aspectos generales de tumor de Wilms.
– Protocolo SIOP-UMBRELLA RTSG 2016. EudraCT number: 2016-004180-39. Versión 2.0. Octubre 2018.
Protocolo de diagnóstico, tratamiento y seguimiento vigente en Europa para tumores renales.
– PDQ Pediatric Treatment Editorial Board. Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®): Health Professional Version. En: PDQ Cancer Information Summaries (Internet). Bethesda (MD): National Cancer Institute (US); 2002 (citado el 19 de mayo de 2021). Disponible en: Http://www.ncbi.nlm.nih.gov/books/NBK65842/.
Revisión sobre aspectos generales de tumor de Wilms.
– Fernández CV, Geller JI, Ehrlich PF, Hill DA, Kalapurakal JA, Grundy PE, et al. Cap. 29: Renal Tumors. En: Principles and Practice of Pediatric Oncology, 7th ed, Pizzo P, Poplack D (Eds), Lippincott Williams & Wilkins; 2016.
– Acha García T. Cap. 50: Tumores renales. En: Hematología y Oncología pediátricas. 3ª edición. Madero L, Lassaletta A. Sevilla J. Ergon; 2015. p. 609-20.
Ambos capítulos de libros de Hemato-Oncología Infantil con aspectos generales de los tumores renales.
| Caso clínico |
|
Paciente de 3 años de edad derivado a nuestro centro por masa abdominal, detectada por ecografía tras palpación por parte de sus padres. Refieren astenia y palidez de 6 meses de evolución, y dolor abdominal intermitente. A la exploración física, destaca masa en hipocondrio izquierdo. Resto sin hallazgos. A su llegada, se realiza analítica (normal) y ecografía abdominal, en la que se visualiza una masa sólida que ocupa la porción central del riñón izquierdo y mide aproximadamente 10 x 8,5 x 7,5 cm (Fig. 3).
Figura 3. Se realiza TC tóraco-abdominal (Fig. 4), confirmando la existencia de tumoración renal dependiente de tercio medio del riñón izquierdo, con bordes bien definidos, y comportamiento predominantemente sólido, con captación heterogénea de contraste y área en su porción más inferior de aspecto necrótico. La lesión mide 8,5 x 8,2 x 8,7 cm (de ejes AP x T x CC) (vol. 317 cc). En tórax presenta dos micronódulos en pulmón derecho (2 mm).
Figura 4. Ante la sospecha de tumor de Wilms, se incluye en protocolo SIOP-RTSG UMBRELLA 2016 y, tras canalización de vía central, inicia tratamiento de inducción con vincristina y actinomicina. Tras completar inducción, se realiza nefrectomía izquierda. La anatomía patológica confirma el diagnóstico de tumor de Wilms de riesgo intermedio: nefroblastoma tipo regresivo, con márgenes quirúrgicos libres de infiltración tumoral (Estadio I). Recibe tratamiento poscirugía con vincristina y actinomicina 4 semanas. En la revisión de fin de tratamiento, se objetiva remisión abdominal, pero recidiva pulmonar, con nódulos en pulmón derecho, por lo que se inicia tratamiento según grupo de riesgo AA con doxorrubicina/ciclofosfamida alterno con etopósido/carboplatino y radioterapia pulmonar.
|



 Follow-up of childhood cancer in Primary Care. How to detect late effects
Follow-up of childhood cancer in Primary Care. How to detect late effects