 |
| Temas de FC |
A.I. Benito Bernal, R. Vila de Frutos
*Servicio de Hemato-Oncología Pediátrica y Trasplante Hematopoyético. Hospital Infantil Universitario Niño Jesús. Madrid
| Resumen
El neuroblastoma es una neoplasia derivada de la cresta neural. Representa el tumor sólido extracraneal más frecuente de la edad pediátrica, siendo responsable de hasta el 15% de la mortalidad por cáncer en los menores de 15 años. |
| Abstract Neuroblastoma is a malignant neoplasia derived from the neural crest. It is the most frequent extracranial solid tumor in children, accounting for 15% of cancer mortality below the age of 15 years of age.
|
Palabras clave: Neuroblastoma; Ganglioneuroblastoma; Cresta Neural; NMYC; Dinutuximab.
Key words: Neuroblastoma; Ganglioneuroblastoma; Neural Crest; MYCN Oncogen; Dinutuximab.
Pediatría Integral 2021; XXV(7): 340.e1-340.e16
Neuroblastoma y tumores relacionados
Introducción
El neuroblastoma, segundo tumor sólido más frecuente de la infancia después de los tumores del sistema nervioso central, se caracteriza por su heterogeneidad clínica y biológica.
El neuroblastoma (NBL) es el tumor sólido extracraneal más frecuente de la edad pediátrica, diagnosticándose el 90% de los casos en niños < 5 años de edad(1,2). Es una enfermedad compleja y heterogénea donde diversos factores como: la edad al diagnóstico, el estadio, la localización del tumor primario y sus características histológicas y moleculares, determinan el pronóstico y condicionan el tratamiento(1-10).
Los tumores neuroblásticos (neuroblastoma, ganglioneuroblastoma y ganglioneuroma) derivan de las células de la cresta neural (CN) comprometidas hacia el desarrollo del sistema nervioso simpático (SNS) y de las células gangliónicas de la médula adrenal(11-14). Su localización anatómica es diversa; se pueden originar a cualquier nivel en los ganglios simpáticos paravertebrales, desde el cuello hasta la pelvis, o en las glándulas suprarrenales.
Su presentación clínica es también variable, condicionada por la localización del tumor primario y la extensión de la enfermedad, presentando metástasis hasta el 50% de los pacientes al diagnóstico(7-9). Así, las familias pueden consultar con el pediatra de Atención Primaria o en el servicio de urgencias de Pediatría, por síntomas tan diversos como: una masa abdominal, estreñimiento, dolor abdominal, fiebre, dificultad respiratoria o síntomas neurológicos compatibles con compresión medular o, con menor frecuencia, un síndrome opsoclunus-mioclonus(7-10,15,16).
Su historia natural es también heterogénea. En neonatos y lactantes < 1 año de vida es, por lo general, un tumor de buen pronóstico con características biológicas favorables y que, en muchos casos, regresa espontáneamente; mientras que, en otros, madura a un ganglioneuroma benigno. En cambio, en niños > 18 meses de edad, suele tener un pronóstico desfavorable a pesar del tratamiento multimodal intensivo(7-10,13,15,16).
Los avances en el estadiaje de la enfermedad, conseguidos gracias a las nuevas técnicas de imagen y de genética molecular, han facilitado la estratificación de los pacientes en grupos de riesgo con criterios definidos de tratamiento(5,6,17). Esto ha mejorado la supervivencia global a largo plazo, que se sitúa entre el 85-90%, para los pacientes de riesgo bajo o intermedio.
A pesar del tratamiento intensivo con quimioterapia, cirugía y radioterapia, seguidos de trasplante autólogo, ácido 13-cis retinoico e inmunoterapia, la supervivencia de los pacientes de alto riesgo no ha mejorado sustancialmente, siendo < 10% tras una recaída o en pacientes con enfermedad refractaria(7-9,13,16,18). En el futuro, el desarrollo de estrategias terapéuticas basadas en la inmunoterapia y/o dirigidas contra alteraciones moleculares específicas del tumor, contribuirán a mejorar la curación de la enfermedad, reduciendo al mismo tiempo los efectos secundarios a corto y largo plazo de los tratamientos actuales(8,13,18).
Epidemiología
Es una enfermedad propia de la infancia, siendo la mediana de edad al diagnóstico de 17 meses. Representa el 8-9% de todos los tumores pediátricos y es responsable de hasta el 15% de la mortalidad por cáncer infantil.
El NBL es la neoplasia más frecuente en el primer año de vida, doblando en incidencia a la leucemia. Es el primer tumor sólido extracraneal diagnosticado entre 0-14 años de edad, ocupando el cuarto lugar en frecuencia de todas las neoplasias infantiles después de las leucemias, los tumores del sistema nervioso central y los linfomas(1,2,8).
Representa, aproximadamente, el 8-9% de todos los canceres pediátricos, con una incidencia anual en España de 13 casos por 106 niños(2). Entre 1.100 casos nuevos de cáncer diagnosticados anualmente en nuestro país en niños < 15 años, aproximadamente 95-100 corresponden a tumores neuroblásticos(2).
La mediana de edad al diagnóstico es de 17-18 meses con una discreta predominancia en varones (ratio 1,2:1). Aproximadamente, el 30-40% de los casos se diagnostican durante el primer año de vida, el 90% antes de los 5 años de edad y el 98% antes de los 10 años(1,8,10). La enfermedad es muy rara en adolescentes y adultos jóvenes, en los que tiene un comportamiento más indolente, pero más letal(19).
Además, es responsable del 12-15% de la mortalidad por cáncer en la edad pediátrica, con una supervivencia global a los 5 años del 70-80% y solo del 50% en las formas diseminadas de alto riesgo(1,2,9,10,18).
Etiología
La etiología del NBL es desconocida. El 1-2% de los casos son familiares y se asocian a mutaciones germinales en el gen ALK y con menor frecuencia en PHOX2B.
La etiología del NBL es desconocida no habiéndose relacionado claramente con la exposición a ningún factor ambiental pre o postnatal.
Aunque la mayoría son esporádicos, el 1-2% de los casos tienen historia familiar. Con un patrón de herencia autosómica dominante y de penetrancia variable, se presentan tumores tanto benignos como malignos dentro de la misma familia, generalmente multifocales y en edades tempranas. El 75% de los NBL familiares se relacionan con mutaciones en línea germinal del gen ALK (Anaplastic Lymphoma Kinase), que determinan la activación anormal del gen(9-13). En cambio, solo 6-10% de los casos esporádicos presentan mutaciones activadoras de ALK.
También se han identificado mutaciones con pérdida de función en PHOX2B (Paired Like Homeobox 2B) en el 10% de NBL familiares y en cerca del 4% de NBL esporádicos de alto riesgo(8,11-13,19). Estos últimos parecen asociarse con otras enfermedades de la CN (neurocrestopatías) como la enfermedad de Hirschsprung y el síndrome de hipoventilación congénita central de Ondine.
También se ha encontrado cierta asociación entre NBL y enfermedades relacionadas con mutaciones de NRAS (síndrome de Costello, síndrome de Noonan, Neurofibromatosis tipo 1) o de TP53 (síndrome de Li Fraumeni), el síndrome de Beckwith-Wiedeman y el síndrome de paraganglioma/feocromocitoma familiar, entre otros(8).
Etiopatogenia
El NBL es un tumor embrionario que deriva de los precursores simpático-adrenales de la cresta neural, a consecuencia de la desregulación de su proceso normal de diferenciación, maduración y apoptosis.
El NBL es un tumor embrionario que se origina a partir de los progenitores simpático-adrenales (PSA) de la cresta neural (CN), un tejido primitivo transitorio derivado del neuroectodermo(11-14). A partir de la 5ª-6ª semana de gestación, las células de la cresta neural (CCNs) migran hacia ambos lados de la aorta dorsal, formando las cadenas simpáticas paravertebrales. Algunas de estas células migran por delante de la aorta y dan lugar a los plexos celíaco y mesentérico. En la 7ª semana del desarrollo, células del ganglio simpático suprarrenal (originado en la CN) atraviesan la corteza y forman la médula suprarrenal(11,12,14). Las células de la médula también se diferencian hacia células cromafines, que contienen catecolaminas y otras sustancias (neuropéptido Y, sustancia P, serotonina, etc.) (Fig. 1).
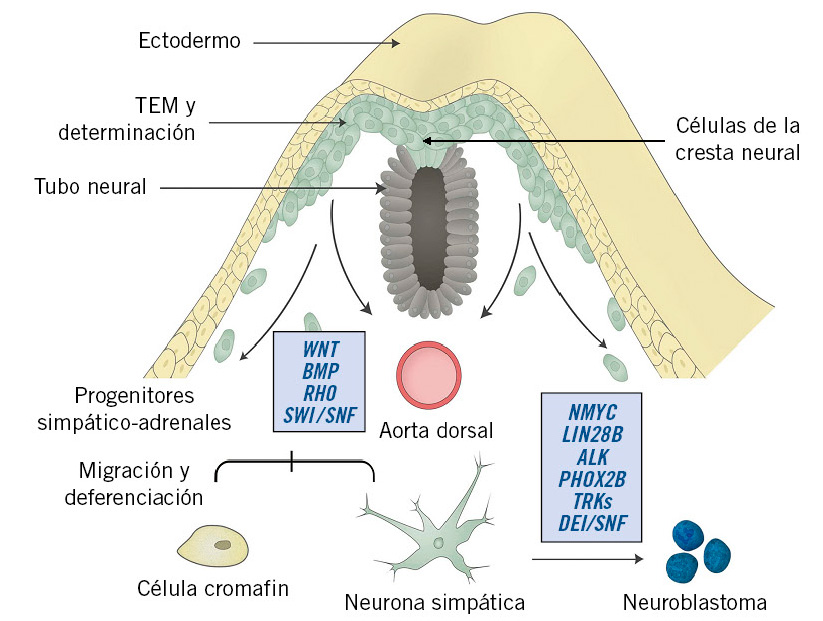
Figura 1. Desarrollo del neuroblastoma a partir de la cresta neural y factores reguladores más importantes. Durante la embriogénesis las CCNs entran en un proceso de TEM, adquiriendo la capacidad de separarse de la CN, migrar y diferenciarse, dando lugar, entre otras, a las células del SNS. Un complejo sistema de señales de activación y de transcripción y de modificaciones epigenéticas regulan este proceso. La alteración del mismo a cualquier nivel puede inducir cambios que a posteriori son relevantes en la oncogénesis, pero solo las mutaciones activadoras de ALK y la sobreexpresión de NMYC tienen un papel directo en el desarrollo del neuroblastoma (NBL). La familia de proteínas BMP, la vía de señalización Wnt y el FGF habilitan a las CCNs para desplazarse, siendo cruciales, al igual que Rho, en la migración y diferenciación de los PSA. La sobrexpresión de NMYC es el mecanismo oncogénico más importante, ya que induce la proliferación e inhibe la apoptosis de los PSA. Lin28B es un inductor en la génesis del NBL: controla la expresión de NMYC y mantiene la indiferenciación de los PSA. ALK participa en la regulación de la proliferación neuronal en etapas tempranas del desarrollo, acelerando la transformación maligna y el desarrollo del NBL en presencia de otros insultos genéticos. PHOX2B, controlado por BMP, es el regulador más importante de la determinación de los PSA; mutaciones en este gen bloquean su diferenciación, proliferando los precursores neurales inmaduros que secundariamente adquirieren lesiones preneoplásicas. SWI/SNF, codificados por ATRX, actúan como supresores tumorales coordinando la remodelación de la cromatina y la reparación del ADN. CCNs: células de la cresta neural; TEM: transición epitelial-mesenquimal; CN: cresta neural; SNS: sistema nervioso simpático; ALK: Anaplastic Lymphoma Kinase; BMP: Bone Morphogenic Proteins; FGF: Fibroblast Growth Factor; Wnt: Wingless; PSA: progenitores simpático-adrenérgicos; PHOX2B: Paired-like homeobox 2B; SWI/SNF: SWItch/Sucrose Non Fermentable; ATRX: Sporadic α-thalassaemia/Mental Retardation Syndrome X-Linked. Adaptado de: Johnsen JI et al; Front Mol Neurosci 2019.
Un complejo sistema de señales de transducción y de factores epigéneticos controla el desarrollo normal de la CN, de modo que la detención o desregulación del proceso de maduración en cualquiera de sus fases podría iniciar la transformación maligna de los neuroblastos(12-14). La figura 1 ilustra este proceso y en ella se muestran los factores más relevantes implicados en el desarrollo del NBL, siendo clave la acción de los oncogenes NMYC y ALK(14). Es razonable pensar que, cuando estas alteraciones ocurren en un estadio precoz del desarrollo, los tumores son más indiferenciados y de peor pronóstico, y viceversa.
Se cree que todos los NBL se originarían del mismo precursor en la CN, pero debido a diferentes tipos de inestabilidad genómica, se producirían alteraciones diversas y tumores con diferente comportamiento clínico(8,11,16) (Fig. 2).
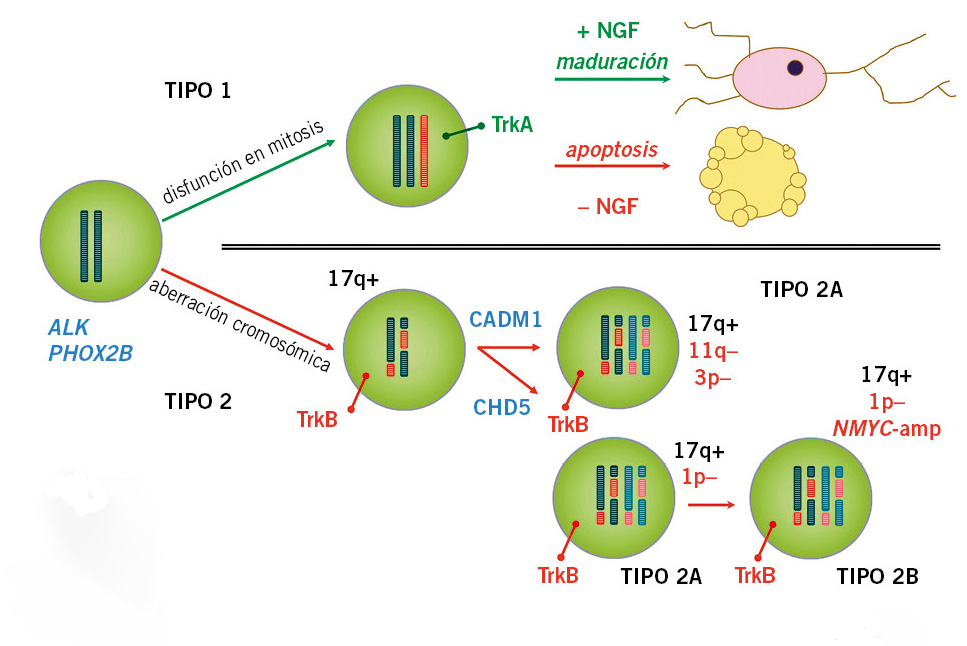
Figura 2. Modelo genómico de oncogénesis en el neuroblastoma (NBL). Según el tipo de alteración genómica se distinguen dos tipos de NBL, cada uno con un comportamiento clínico diferente.
Tipo 1 o perfil NCA: triploides o casi-triploides con ganancia o pérdida de cromosomas enteros, sin alteraciones estructurales, expresan TrkA. En presencia de agentes diferenciadores (+NGF), maduran a tejido ganglionar y, en su ausencia (-NGF), entran en apoptosis. Son tumores localizados de buen pronóstico en < 1 año de edad, sensibles a 13-cis retinoico y con tendencia a madurar o a regresar espontáneamente. Tipo 2 o perfil SCA: diploides o cuatriploides con cambios estructurales (ganancia de 17q, deleciones de 1p o 11q y/o NMYC-amp). Expresan TrkB y su ligando, BDNF, que favorecen la supervivencia de las células tumorales. Se asocian a enfermedad avanzada y de mal pronóstico en > 1 año de edad. El subtipo 2A, sin NMYC-amp y con deleciones de 3p y 11q o con deleción de 1p, tiene un curso insidioso con sucesivas recaídas y supervivencia más prolongada. El subtipo 2B, con deleción 1p y NMYC-amp, presenta frecuentes recaídas precoces y pobre supervivencia a corto plazo. NCA: Numerical Chromosomal Aberrations; SCA: Segmental Chromosomal Aberrations; TrkA/B: Tropomyosina Receptor Kinasa; NGF: Neural Growth Factor; BDNF: Brain-Derived Neurotrophic Factor; CADM1: Cell Adhesion Molecule 1; CHD5: Chromodomain Helicase DNA Binding Protein 5. Adaptado de: Brodeur GM. Nat Rev Ca. 2003; Pediatr Integral. 2016; XX (7): 434-446.
Los receptores de tropomiosín kinasa: TrkA, TrkB y TrkC (codificados por los genes NTRK1, NTRK2 y NTRK3, respectivamente), tienen elevada afinidad para NGF (Neural Growth Factor) y otras neurotrofinas, siendo fundamentales para el desarrollo normal y el mantenimiento del SNC y periférico(11-14). La diferente expresión de TrkA y TrkB en el NBL explicaría en parte la heterogeneidad de la enfermedad.
Presentación clínica
La presentación clínica del NBL es heterogénea y depende de la localización del tumor primario y sus metástasis. Los pacientes pueden estar asintomáticos o presentar cuadros clínicos graves con afectación del estado general.
Los tumores neuroblásticos pueden originarse en cualquier lugar del SNS, por lo que los signos y síntomas de presentación dependen de la localización del tumor primario, de su extensión y de la presencia o no de enfermedad diseminada. Mientras que los pacientes con NBL localizados suelen estar asintomáticos, los niños con enfermedad metastásica presentan síntomas sistémicos (Tablas I y II).

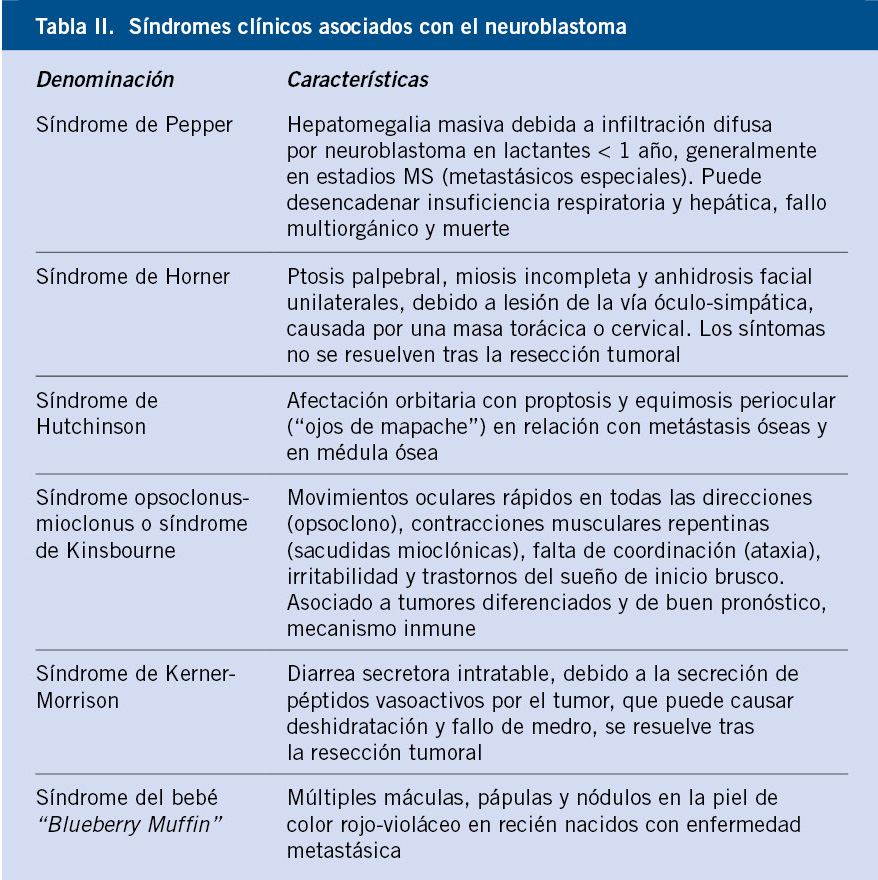
• Enfermedad localizada: el abdomen es el lugar de origen más frecuente (65% de los casos) con > 40% localizados en la glándula suprarrenal. En recién nacidos y lactantes es común el hallazgo casual de una masa suprarrenal en una ecografía. Los NBL retroperitoneales suelen presentarse como una masa abdominal asintomática detectada por los padres o por el pediatra de Atención Primaria durante una revisión de rutina. Los tumores abdominales de gran tamaño pueden causar: importante distensión abdominal, estreñimiento, dolor subagudo o agudo secundario a hemorragia intratumoral y/o hipertensión arterial (HTA) por compresión de los vasos renales(7-10,13,16,20). La producción de catecolaminas por el tumor también puede causar rubor, taquicardia e HTA.
Otras localizaciones frecuentes son: cuello (5%), tórax (15%) y pelvis (5%). Los NBL cervicales pueden debutar con un síndrome de Horner por la lesión del ganglio estrellado o cervicotorácico (Tabla II). Los NBL torácicos generalmente son un hallazgo casual en una radiografía de tórax realizada en un paciente con tos persistente o dificultad respiratoria. Pueden comprimir y desplazar la vía aérea o el parénquima pulmonar y, ocasionalmente, producir un síndrome de vena cava superior. A nivel pélvico, pueden causar síntomas compresivos, como retención urinaria y estreñimiento(7-10,13,16,20). Cuando se originan en los ganglios paraespinales, pueden presentar síntomas neurológicos compatibles con un síndrome de compresión medular (dolor de espalda, debilidad muscular, alteraciones sensitivas), al extenderse a lo largo de las raíces nerviosas y a través del foramen neural.
• Enfermedad metastásica: alrededor del 55% de los pacientes presentan metástasis al diagnóstico (80% de los niños mayores y 40% de los lactantes). La enfermedad se disemina a los ganglios linfáticos locorregionales o por vía hematógena a huesos y médula ósea(7-10,20). Las metástasis óseas causan: dolor, irritabilidad, inflamación y cojera. La infiltración medular puede causar anemia y trombocitopenia. Las metástasis en hígado y piel son más frecuentes en lactantes < 18 meses de edad (Tablas I y II).
• Enfermedad 4S o MS (S = “Special”): es una forma de presentación especial en lactantes con metástasis limitadas a hígado, médula ósea y piel, y un tumor primario de pequeño tamaño y con tendencia a la regresión espontánea. La infiltración hepática masiva puede progresar rápidamente y causar insuficiencia hepática y coagulopatía junto con una gran distensión abdominal (Tabla II). Esta puede producir dificultad respiratoria, insuficiencia renal y edema escrotal y de miembros inferiores, por compresión de la vena cava inferior, especialmente en < 3 meses de edad(7-10,20).
• Síndromes paraneoplásicos (Tabla II): generalmente, asociados a NBL localizados y diferenciados. La secreción de péptido intestinal vasoactivo (VIP) por el tumor puede causar una diarrea secretora intratable. El síndrome opsoclonus-mioclonus (SOM) se presenta en el 2-3% de pacientes y, aunque el pronóstico del tumor suele ser favorable, el 70-80% de los niños presentarán déficits neurológicos a largo plazo(7-10,15,16).
Patobiología
Las características histopatológicas y las alteraciones genéticas conforman los pilares del diagnóstico patológico del NBL, con importantes implicaciones pronósticas y terapéuticas.
Clasificación histológica
El NBL es un tumor de células pequeñas, redondas y azules. La clasificación histopatológica del INPC (International Neuroblastoma Pathology Classification) tiene valor pronóstico.
El NBL es un tumor de células pequeñas, redondas y azules, características que definen un grupo de neoplasias indiferenciadas propias de la edad pediátrica, entre las que se incluyen: sarcoma de Ewing, linfoma no Hodgkin, tumor neuroectodérmico primitivo (PNET), rabdomiosarcoma o tumor de Wilms(8).
Los tumores neuroblásticos están formados por dos tipos celulares, células gangliónicas o neuroblastos y células reactivas, denominadas estroma de Schwann. La disposición de los neuroblastos alrededor de un centro de tinción eosinofílico se conoce como roseta de Homer-Wright, característica del tumor. Las células de Schwann, en respuesta a sustancias sintetizadas por el tumor, inhiben la proliferación e inducen la maduración de los neuroblastos. Entre los marcadores histoquímicos que ayudan al diagnóstico, se encuentran: enolasa neuronal específica (ENE), sinaptofisina, gangliósido GD2, receptores de TrK y la cromogranina A(8).
Existen cuatro subtipos morfológicos clasificados según su composición y diferenciación celular(3,4,8): ganglioneuroma (GN) con predominio de estroma schwanniano, ganglioneuroblastoma (GNBL) entremezclado (rico en estroma), ganglioneuroblastoma nodular (con zonas ricas en estroma y otras pobres en estroma) y neuroblastoma (pobre en estroma schwanniano). Dentro de este último, se incluyen tres tipos: indiferenciado, pobremente diferenciado y en diferenciación. Estos tipos morfológicos representan distintos estadios de maduración de la misma enfermedad, siendo el GN un NBL completamente maduro y diferenciado.
La clasificación histopatológica del NBL utilizada en la actualidad es la del INPC (International Neuroblastoma Pathology Classification), que es una modificación de la clasificación original de Shimada(3,4) y tiene impacto pronóstico ajustado a la edad (Tabla III).

Alteraciones genéticas
Mutaciones somáticas recurrentes en genes como ALK son raras en el NBL, mientras que alteraciones cromosómicas, como la amplificación de NMYC, la ganancia de 17q y la deleción de 1p y 11q se observan con frecuencia.
Las alteraciones citogenéticas identificadas en el NBL incluyen la amplificación de oncogenes, pérdidas y ganancias parciales o completas de cromosomas, alteraciones en el índice de ADN y mutaciones(8-14) (Tabla IV).
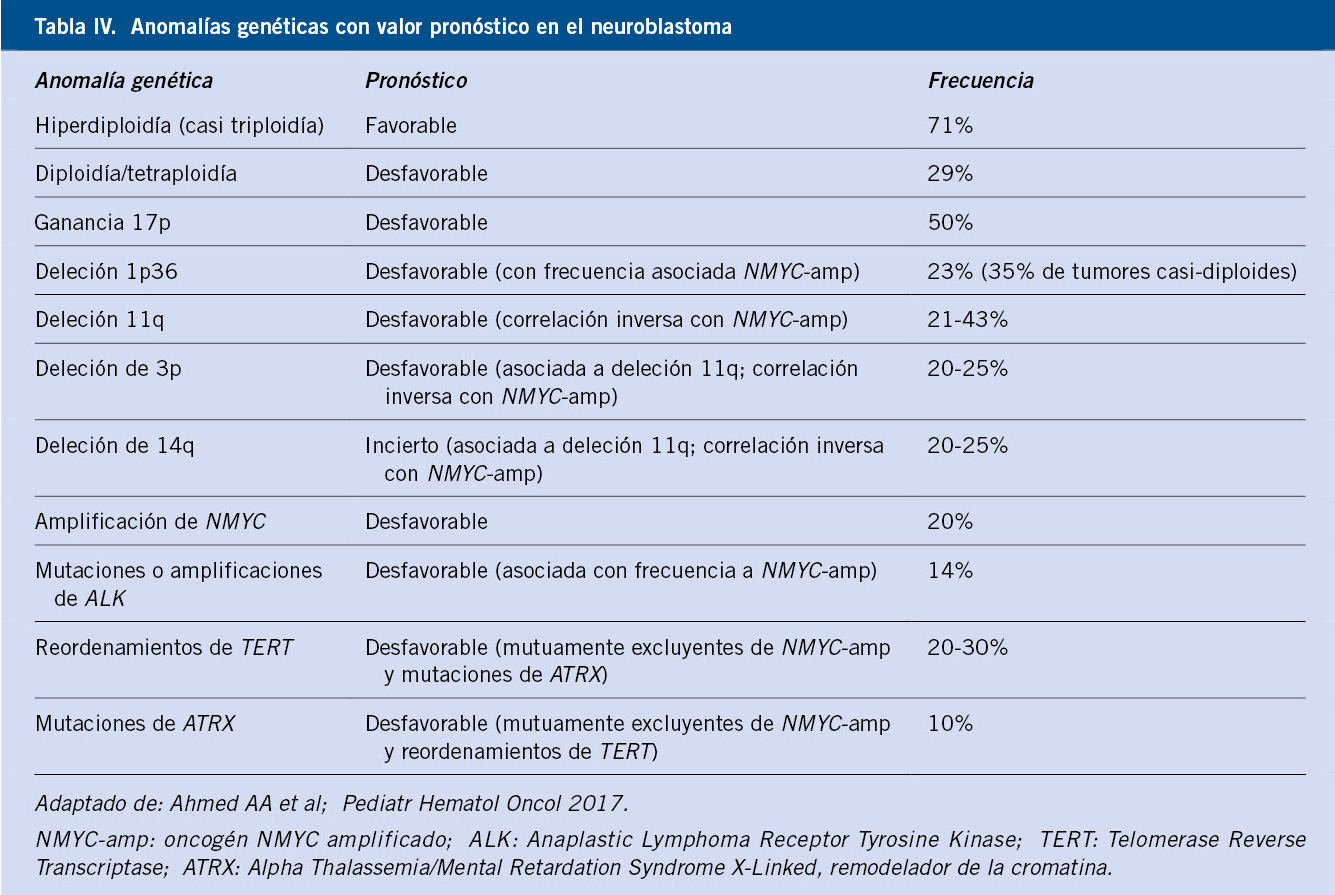
• Amplificaciones: el oncogén NMYC (cromosoma 2p24), está amplificado (> 10 copias) en 20-25% de los casos. Se asocia a mal pronóstico, usándose de rutina para la asignación del riesgo en el sistema INRG (International Neuroblastoma Risk Group)(5,6). En el grupo de bajo riesgo (localizados y MS), el 8-10% tienen NMYC-amp, lo que empeora claramente su pronóstico. Su influencia es menos relevante en estadios 4 o M (30% con NMYC-amp), ya que acumulan otros factores de mal pronóstico(8-14). ALK (cromosoma 2p23.2), es el siguiente gen en frecuencia amplificado en el NBL (2-4%). Es también un factor de mal pronóstico y se asocia a NMYC-amp.
• Alteraciones cromosómicas segmentarias (SCA – Segmental Chromosomal Alterations): la pérdida de heterocigosidad (LOH, Loss of Heterozygosity) en el cromosoma 1, implica la deleción de la región 1p36. Se asocia a un pronóstico desfavorable y a otras características de alto riesgo, como mayor edad, NMYC-amp y enfermedad diseminada(8-14). Se ha relacionado con la pérdida de CHD5 (Chromodomain Helicase DNA Binding Protein 5), un gen supresor tumoral localizado en 1p36.31. La deleción de 11q también se asocia a mal pronóstico, generalmente en tumores sin NMYC-amp, y se ha correlacionado con la pérdida de CADM1 (Cell Adhesion Molecule 1), un gen de supresión tumoral localizado en 11q23.3(8-14). Son también frecuentes las deleciones de 3p, 4p, 9p y 14q. La alteración genómica más frecuente en el NBL es la ganancia de la porción distal de 17q, detectada en el 50% de los casos, asociada a mal pronóstico y a otros factores de riesgo, como mayor edad, NMYC-amp y deleción de 1p. También son frecuentes las ganancias de 1 p y 2p(8-14).
• Ploidía (NCA – Number of Chromosome Copies): la ganancia o pérdida de cromosomas enteros modifica el contenido de ADN o ploidía tumoral. Esta tiene impacto pronóstico, especialmente en pacientes con otros datos favorables, como lactantes < 12-18 meses de edad con estadio MS y sin NMYC-amp o con enfermedad localizada y NMYC-amp(8-14). En estos pacientes, la hiperdiploidía (índice de ADN > 1 y < 2) se asocia con enfermedad de bajo riesgo, buena respuesta a quimioterapia y evolución favorable, especialmente si no tienen NMYC-amp. Los tumores con contenido diploide/tetraploide (índice de ADN = 1 o ≥ 2) se asocian con enfermedad avanzada, NMYC-amp y un comportamiento desfavorable. Los tumores hiperdiploides en niños mayores también tienen SCA y mal pronóstico(8-14).
• Mutaciones somáticas: se han descrito mutaciones activadoras en el dominio tirosín kinasa de ALK (8-10% de NBL), así como mutaciones con pérdida de función en PHOX2B (4%) y en ATRX (10%). Este último juega un papel importante en la regulación epigenética (alargamiento de los telómeros) y se detecta en el 50% de NBL del adolescente y del adulto joven, pero no en lactantes o en tumores con NMYC-amp(8-14).
Marcadores tumorales
Actualmente, sin valor pronóstico, apoyan el diagnóstico y pueden correlacionarse con enfermedad avanzada. La cuantificación de catecolaminas en orina es útil en el diagnóstico y seguimiento de la enfermedad.
La ENE (enolasa neuronal específica) es un marcador inespecífico de NBL, aunque niveles elevados se relacionan con enfermedad avanzada, del mismo modo que la LDH (Lactato Deshidrogenasa). También es frecuente encontrar niveles elevados de ferritina en pacientes con enfermedad avanzada y de mala evolución(8-14).
El NBL es un tumor productor de catecolaminas, siendo los ácidos homovanílico (HVA) y vanilmandélico (VMA) los metabolitos con mayor sensibilidad y especificidad para su detección. La cuantificación de estas catecolaminas en orina es parte de los estudios iniciales diagnósticos del NBL, así como del seguimiento de la enfermedad(9,10,13,17).
Diagnóstico
El diagnóstico del NBL se basa en la combinación de varias pruebas: estudios de imagen, marcadores tumorales, catecolaminas en orina y el análisis histológico y genético del tumor.
La biopsia del tumor primario o de una lesión metastásica de partes blandas es necesaria para el diagnóstico(9,10,13,17). En pacientes inestables en los que el riesgo quirúrgico no es asumible, se puede realizar el diagnóstico demostrando la infiltración tumoral en médula ósea y la elevación de catecolaminas en orina (Tabla V).

Las catecolaminas (HVA y VMA y dopamina) se encuentran elevadas en orina en el 90% de los niños con NBL(8,9,13) (Tabla VI).
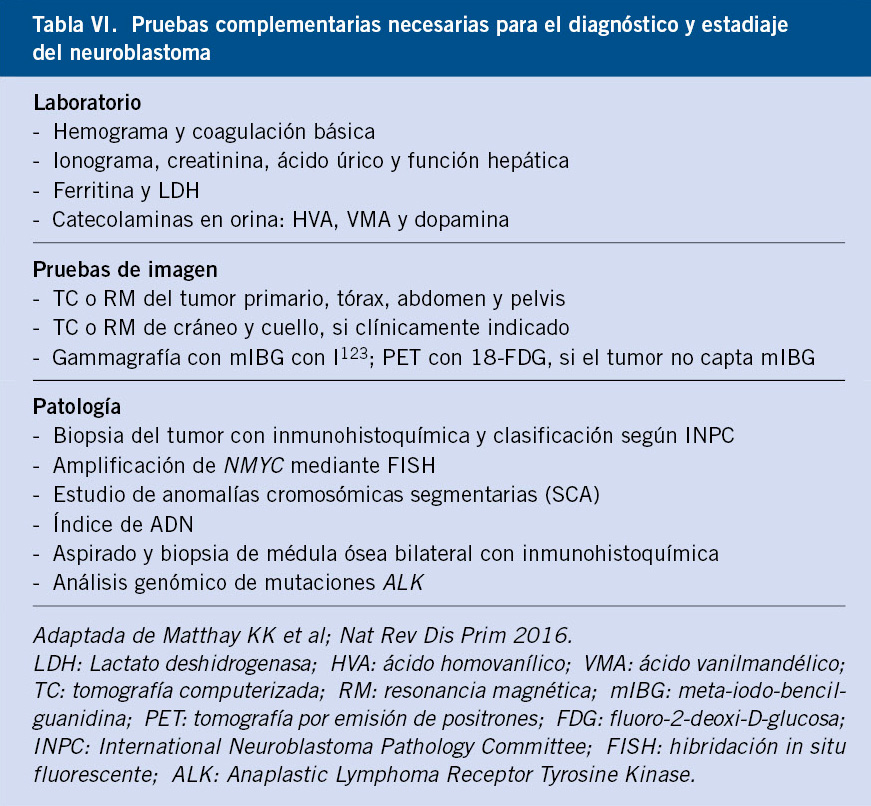
Además de las radiografías convencionales y ecografías iniciales, se debe evaluar el tumor primario mediante RM o TC(8,9,13), determinando su localización, extensión, afectación ganglionar, relación con estructuras vecinas y diseminación metastásica (Fig. 3). Se incluirá el tumor primario, así como tórax, abdomen y pelvis (Tabla VI).
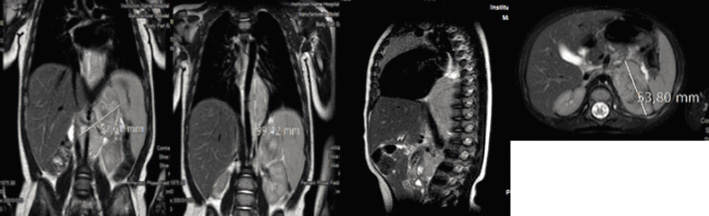
Figura 3. RM tóraco-abdominal en paciente de 10 meses de edad con neuroblastoma en estadio INRG L2. Tumoración sólida en región suprarrenal izquierda que se extiende por el hiato abdominal con prolongación en “reloj de arena” desde retroperitoneo a mediastino posterior. Cranealmente, contacta con la pared posterior del ventrículo izquierdo y, caudalmente, se extiende hasta la bifurcación aórtica. La masa adrenal izquierda engloba a la aorta abdominal y la arteria renal izquierda invadiendo el hilio renal izquierdo. Contacta con la cava abdominal, el origen del tronco celiaco y mesentérica superior, permaneciendo estos vasos permeables. Según el estadiaje INRG, hay múltiples factores de riesgo definidos por imagen, incluyendo la afectación de dos compartimentos anatómicos.
La MIBG (Meta-Iodo-Benzil-Guanidina) es una molécula marcada radiactivamente, generalmente con I123, de estructura similar a la de la norepi-nefrina, captada por el transportador de norepinefrina en el 90% de los NBL. La gammagrafía con MIBG es altamente específica para NBL (Fig. 4) y se debe realizar siempre al diagnóstico para determinar la presencia de metástasis, así como para evaluar la respuesta al tratamiento(8,9,21). En casos dudosos, la correlación entre MIBG y SPECT/TC (Single Photon Emission Computed Tomography) mejora la precisión diagnóstica. Los NBL que no captan MIBG (10%) se estudiarán mediante PET-TC con 18-FDG (18F-fluoro-2-deoxi-D-glucosa).
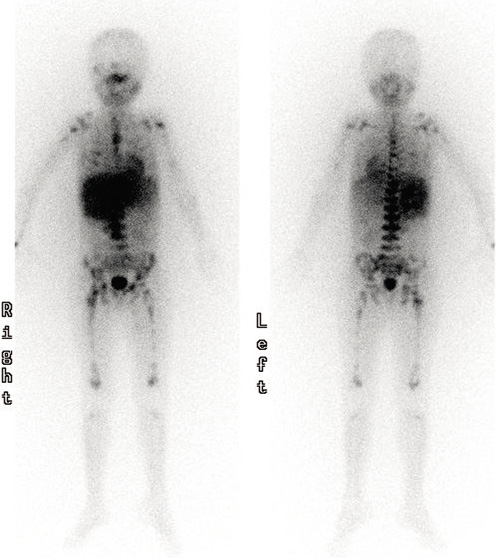
Figura 4. Gammagrafía con MIBG en un paciente de 6 años de edad, que muestra una captación ávida en el neuroblastoma primario suprarrenal y diseminación metastásica ósea generalizada.
También se realizarán aspirados y biopsias de médula ósea bilaterales al diagnóstico y a intervalos regulares durante el tratamiento y en el seguimiento de los pacientes de alto riesgo (Tabla VI).
Varios estudios han demostrado la utilidad de la monitorización de la enfermedad mínima residual (EMR), tanto en sangre periférica (SP) como en médula ósea (MO) para el seguimiento de la enfermedad(22). El método más sensible y especifico es la RT-PCR (Real-Time PCR) de marcadores específicos de NBL, como la tirosina hidroxilasa (TH), principal enzima de la síntesis de catecolaminas. En el NB de alto riesgo, la EMR persistente en MO al final de la quimioterapia de inducción, predice la recaída y/o muerte en los siguientes 24 meses de media. La cuantificación de la GD2 sintetasa mediante anticuerpos monoclonales antiGD2 permite la detección de células tumorales ocultas en MO y SP, y es un marcador muy útil y precoz para evaluar la eficacia de la inmunoterapia con dinutuximab.
Diagnóstico diferencial
Desde el punto de vista histológico, se deben descartar otros tumores pediátricos indiferenciados de células pequeñas, redondas y azules y con tendencia a infiltrar la médula ósea, como el rabdomiosarcoma, el sarcoma de Ewing/PNET, el linfoma de Hodgkin y determinadas leucemias (megacarioblástica)(8).
El diagnóstico diferencial de una masa abdominal se hará con otros tumores retroperitoneales, principalmente con el nefroblastoma. A diferencia de este, el NBL es una masa dura y fija, que cruza la línea media y que, en ocasiones, se asocia con síntomas constitucionales. En cuanto a las masas suprarrenales, en neonatos y lactantes, se deben considerar la hemorragia suprarrenal, la hiperplasia adrenal congénita, el secuestro pulmonar (subdiafragmático extralobar) y el quiste broncogénico(8,23).
La diarrea secretora se puede confundir con infecciones gastrointestinales o enfermedad inflamatoria intestinal y el SOM puede simular un trastorno neurológico. La hepatomegalia en un lactante puede hacer pensar inicialmente en una enfermedad metabólica o de depósito(8,24). Además, el NBL diseminado será considerado dentro del diagnóstico diferencial de enfermedades infecciosas o inflamatorias, como la osteomielitis o la artritis reumatoide(8).
Estadiaje y clasificación
El estadio de la enfermedad, la edad al diagnóstico, la histología y la biología del tumor son criterios necesarios para clasificar a cada paciente en su grupo de riesgo y asignarle el tratamiento más adecuado.
• Estadio tumoral: entre los diversos sistemas de estadiaje del NBL existentes antes del 2010, el más utilizado era del INSS (International Neuroblastoma Staging System)(17). La extensión de la resección quirúrgica al diagnóstico y la presencia de metástasis define los cuatro estadios de este sistema (1 a 4), además del estadio 4S (Tabla VII). Los pacientes con enfermedad en estadio 1 y 2A son de bajo riesgo, mientras que los demás pertenecen al riesgo alto o intermedio. La resecabilidad del tumor y, por tanto, el estadiaje, dependía de la experiencia de equipo quirúrgico, haciendo difícil comparar resultados entre los diferentes centros.

En 2009, el grupo de trabajo del INRG (International Neuroblastoma Risk Group Staging System) diseñó un nuevo sistema de estadiaje y de clasificación del riesgo, tras analizar los datos de más de 8.800 pacientes diagnosticados entre 1990 y 2002 en EE.UU, Europa, Japón y Australia(5,6) (Tabla VII). Este sistema define dos estadios localizados (L1 y L2) y dos metastásicos (M y MS). La presencia de metástasis y de factores de riesgo del tumor, definidos en los estudios de imagen (IDRFs-Image Defined Risk Factors) antes de la cirugía o de cualquier tratamiento, determinan la extensión de la enfermedad (Tabla VIII).
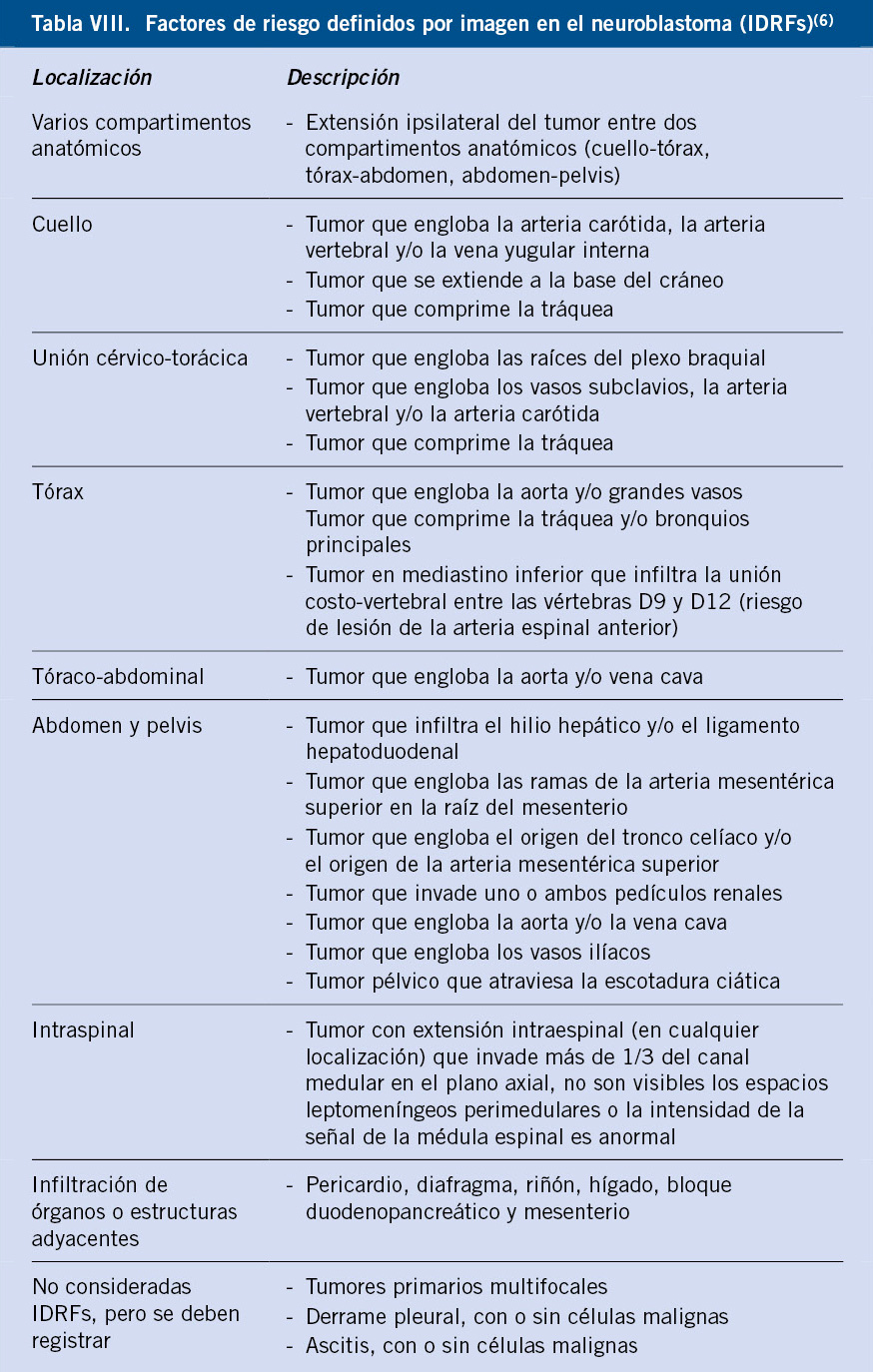
Los IDRFs son características del tumor primario que suponen una amenaza para la vida del paciente o que incrementan el riesgo quirúrgico, condicionando una peor evolución.
• Clasificación en grupos de riesgo: actualmente la clasificación más utilizada es la del INRG que estratifica a los pacientes en 16 grupos de riesgo pretratamiento (Tabla IX)(6).

Para ello, tiene en cuenta siete factores pronósticos: el estadio, la edad, la histología (tipo y grado de diferenciación) y la biología del tumor (amplificación de NMYC, ploidía, deleción 11q)(5,6). Según la supervivencia libre de enfermedad (SLE), a los 5 años los pacientes se agrupan en cuatro categorías de riesgo: muy bajo (SLE > 85%), bajo (75-85%), intermedio (50-75%) y alto (< 50%)(5,6).
En el futuro, los nuevos conocimientos sobre las alteraciones genómicas y moleculares del NBL permitirán redefinir de forma más precisa estos grupos de riesgo y aplicar estrategias basadas en la medicina personalizada(10).
Bases del tratamiento del neuroblastoma
Las modalidades tradicionales de tratamiento del NBL son la cirugía, la quimioterapia y la radioterapia, a las que, en los últimos años, se han incorporado la inmunoterapia y la terapia diferenciadora.
La cirugía tiene un papel fundamental en el NBL, tanto en el diagnóstico como en el tratamiento. La biopsia inicial es crucial para establecer el diagnóstico inequívoco del tumor y para el estudio de sus características biológicas. En la enfermedad en recaída, la biopsia puede aportar información adicional sobre la biología tumoral y, mediante las nuevas técnicas de secuenciación masiva, posibilitar la detección de nuevos marcadores moleculares para la aplicación de terapias personalizadas.
El abordaje quirúrgico, ya sea mediante cirugía abierta o técnicas menos invasivas (laparoscopia, toracoscopia, biopsia percutánea), dependerá de la localización, extensión y resecabilidad de la lesión. Los tumores localizados sin IDRFs se resecarán en la cirugía inicial(8,9,13). En los demás casos, la biopsia garantizará la obtención del material suficiente para realizar todos los estudios y se valorará extirpar el resto tumoral de forma diferida. El riesgo de complicaciones quirúrgicas es mayor en los lactantes, sobre todo en < 2 meses de edad, en los que la enfermedad tiene mejor pronóstico; por lo que, en estos casos, se debe valorar cuidadosamente la indicación de la cirugía.
El NBL es un tumor radiosensible, pero la elevada prevalencia de metástasis óseas hace que la radioterapia no sea curativa(8). La radioterapia está indicada como tratamiento de la enfermedad residual en algunos tumores localizados, en el control de la enfermedad local y de metástasis refractarias en pacientes de alto riesgo, así como en pacientes en fase terminal, para mitigar el dolor asociado a las metástasis óseas(8). Actualmente, el uso de radioterapia urgente en la compresión medular o en la infiltración hepática masiva es excepcional, siendo preferible la quimioterapia, tanto por su eficacia como por el menor riesgo de efectos secundarios a largo plazo.
La quimioterapia está indicada en pacientes de riesgo intermedio o alto, así como en algunos pacientes de bajo riesgo con compromiso vital, variando la intensidad de la misma según el riesgo.
Tratamiento ajustado al riesgo
El tratamiento del NBL depende del grupo de riesgo al que pertenezca el paciente, basado en las diferencias pronósticas relacionadas con la edad y la biología tumoral, por lo que las estrategias terapéuticas son muy variadas.
El tratamiento dentro de cada grupo de riesgo, especialmente en el intermedio, difiere en cierta medida entre los distintos grupos colaborativos(9,13,24). A continuación, se describen las recomendaciones terapéuticas del grupo de NBL de la Sociedad Internacional de Oncología Pediátrica Europea o SIOPEN (SIOP-Europa-Neuroblastoma) (Tabla X y Fig. 5).

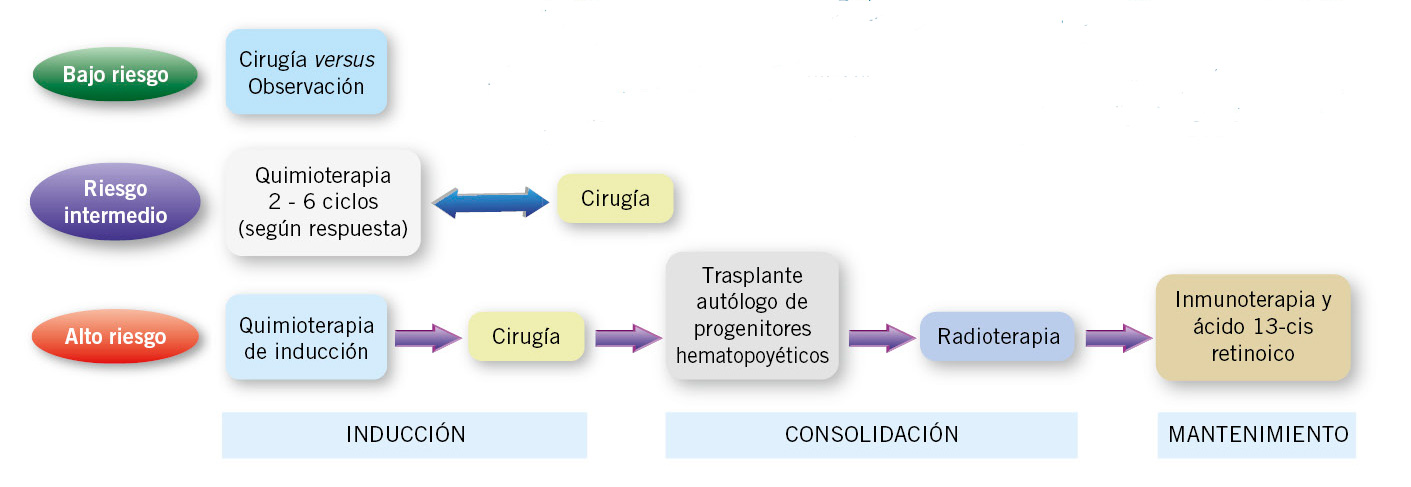
Figura 5. Tratamiento del neuroblastoma adaptado al riesgo. Los pacientes de bajo riesgo generalmente se curan solo con cirugía; los pacientes asintomáticos con tumores con alta probabilidad de regresión, se manejan solo con observación. En el riesgo intermedio, los pacientes reciben quimioterapia, con un nº de ciclos variable en función de la respuesta y resección quirúrgica. Los pacientes de alto riesgo necesitan tratamiento intensivo que incluye: quimioterapia, cirugía, altas dosis de quimioterapia, radioterapia, inmunoterapia y terapia diferenciadora.Adaptado de: Tolbert VP et al, Cell Tissue Res. 2018.
Tratamiento del NBL de muy bajo y de bajo riesgo
En los pacientes de bajo riesgo y asintomáticos, la resección quirúrgica o la observación en tumores con tendencia a la regresión espontánea, son suficientes para el control de la enfermedad.
Dentro de SIOPEN, se consideran de bajo riesgo los estadios INRG L1, L2 y MS con edad y características biológicas favorables (Tabla X). Su pronóstico es excelente, con SLE > 90% y supervivencia global (SG) cercana al 100%. La mayoría de estos pacientes se curan solo con cirugía y en algunos casos sin ningún tratamiento, dada la tendencia de estos tumores a la regresión espontánea(8,9,13,20).
Pacientes ≤ 12 meses, estadio MS y sin NMYC-amp alcanzan SG > 85% solo con observación clínica y seguimiento ecográfico. La quimioterapia se reserva para los pacientes con síntomas amenazantes para la vida (SAVs), como compresión medular o distrés respiratorio secundario a infiltración hepática masiva (Tabla XI).

Los fármacos utilizados son: etopósido, carboplatino, ciclofosfamida y doxorrubicina, y se administrarán solo los ciclos necesarios para el control de síntomas(2-4).
La mayoría de los NBL neonatales son masas adrenales en estadios INRG L1/L2 de histología favorable, que suelen regresar espontáneamente. En los niños < 3 meses de edad y asintomáticos, con tumores < 5 cm de diámetro, se realiza solo observación y, en caso de progresión, se realiza cirugía y el estudio histológico y molecular del tumor(8,9,13,20).
La cirugía es de elección en tumores localizados (INRG L1 y L2), resecables y biológicamente favorables. Algunos pacientes progresan después, pero se pueden rescatar con cirugía y/o quimioterapia sin un impacto significativo en la SG. La observación está indicada en los pacientes con INRG L2 (INSS 2A y 2B) con características biológicas favorables y con IDRFs, recibiendo quimioterapia en caso de progresión o presencia de SAVs, seguida de exéresis tumoral si fuera posible. Los niños con NBL en estadio L2 sin NMYC-amp, pero con perfil biológico SCA (antes incluidos en el grupo de alto riesgo), también reciben quimioterapia (4 ciclos) seguida de cirugía(8,9,13,20).
Tratamiento del NBL de riesgo intermedio
Los pacientes de riesgo intermedio se tratan con quimioterapia cuya duración depende de la respuesta, así como cirugía del tumor primario.
Es un grupo muy heterogéneo con tumores de características biológicas variables, pero con tasas de SG > 80%, cuando se combinan quimioterapia y cirugía. Incluye niños > 18 meses de edad con enfermedad en estadio INRG L2 (INSS 3) y ≤ 12 meses en estadio M, ambos sin NMYC-amp (Tabla X). La resección del tumor primario se realiza después de 4-6 ciclos de quimioterapia, siempre que sea posible. Si la histología es desfavorable, los pacientes reciben después radioterapia local y terapia diferenciadora. Con esta estrategia, la SG a los 5 años es > 90% en los lactantes en estadio M y del 70% en niños > 18 meses de edad con NBL en estadio L2(8,9,13,20).
Los pacientes con NBL estadio L1 y NMYC-amp son un tipo especial dentro del grupo de riesgo intermedio y reciben quimioterapia (6 ciclos), inmediatamente después de la cirugía del tumor primario, ya que sin tratamiento suelen presentar recaídas metastásicas en los meses siguientes.
Tratamiento del NBL de alto riesgo
Los pacientes de alto riesgo reciben tratamiento multimodal intensivo con: quimioterapia, cirugía, radioterapia, inmunoterapia y terapia diferenciadora con ácido 13-cis retinoico.
En este grupo se incluyen los pacientes con estadios INRG > L1 (L2, M y MS) con NMYC-amp y los ≥ 12 meses en estadio M y MS sin NMYC-amp. Presentan recaídas frecuentes y alta mortalidad, con una probabilidad de SG a los 5 años del 50%.
El tratamiento se divide en 4 fases (Fig. 5): inducción (quimioterapia y cirugía), consolidación (trasplante autólogo y radioterapia) y mantenimiento (inmunoterapia y ácido 13-cis-retinoico). El objetivo de la quimioterapia de inducción es conseguir la máxima reducción del tumor primario y de las metástasis. La intensidad de dosis se relaciona con la respuesta y la supervivencia. El régimen recomendado es el “Rapid COJEC” (vincristina, carboplatino, etopósido, cisplatino y ciclofosfamida), que administrado en un periodo de tiempo muy ajustado (3-4 meses) trata de evitar el desarrollo de resistencias farmacológicas(8,9,13,20). Al final de la inducción, la persistencia de infiltración medular y de metástasis óseas en la mIBG (score SIOPEN) son factores independientes de mal pronóstico.
En los pacientes con respuesta adecuada, se procede a la extirpación quirúrgica del tumor primario, finalizando con ello el tratamiento en los pacientes > 12 meses, estadio M y sin NMYC-amp. Los demás pacientes continúan con la fase de consolidación, que consiste en quimioterapia mieloablativa (busulfan y melfalan) seguida de rescate con trasplante autólogo y radioterapia local. El tratamiento de mantenimiento incluye ácido 13-cis-retinoico, que induce la diferenciación de las células tumorales residuales e inmunoterapia con el anticuerpo monoclonal anti-GD2, dinutuximab(8,9,13,20,25).
Tratamiento de enfermedad refractaria y las recaídas
El NLB refractario o en recaída es difícilmente curable, no habiéndose definido aún el tratamiento óptimo de los pacientes de alto riesgo en esta situación.
Alrededor del 20% de los pacientes con NBL de alto riesgo son refractarios al tratamiento de primera línea y el 60% de los que completan el tratamiento recaen (mediana de 1,5 años desde el diagnóstico). La SG después de una recaída es < 10%, siendo mejor en pacientes en estadio L1 (70%) comparado con aquellos en estadio L2 (40%) o M (2%). No se ha establecido aún el tratamiento estándar de estos pacientes. La quimioterapia de rescate incluye combinaciones de temozolamida con irinotecan y/o bevacizumab (BEACON) y topotecan con temozolomida o ciclofosfamida o con doxorrubicina y vincristina en perfusión continua (TVD), con resultados variables(9,13,20). La terapia con 131I-mIBG es otra alternativa, con un 30-40% de respuestas globales.
Actualmente, se encuentran en investigación varios fármacos dirigidos contra dianas moleculares como: los inhibidores de ALK (crizotinib), inhibidores de Aurora A kinasa (desestabilizan N-MYC), de histona deacetilasa (vorinostat) o de Trkb (entrectinib). Otros ensayos clínicos incluyen: inhibidores de check-point, inmunoterapia (anti-GD2 con células NK, nalixamab con GM-CSF) y terapia celular con CAR-T (Chimeric Antigen Receptors -T cells) contra GD2(9,13,20).
Efectos secundarios a largo plazo
Los pacientes que sobreviven a un neuroblastoma de riesgo intermedio o alto que han recibido tratamiento con quimioterapia, radioterapia y cirugía, tienen un riesgo elevado de desarrollar efectos secundarios a largo plazo.
Los pacientes que reciben quimioterapia y radioterapia tienen riesgo de presentar efectos secundarios a largo plazo como: ototoxicidad, cardiotoxicidad, osteoporosis, complicaciones endocrinas (talla baja, hipotiroidismo), diarrea crónica, hiperplasia nodular focal hepática, segundos tumores (cáncer de tiroides, renal y de partes blandas y leucemia aguda) e infertilidad(8,9). Además, al diagnóstico, los pacientes pueden presentar complicaciones neurológicas, como compresión medular o SOM, que a largo plazo pueden determinar secuelas neuromusculares como: paraplejia, incontinencia, escoliosis, déficits neurocognitivos y alteraciones del desarrollo(15).
Feocromocitoma y paraganglioma
Son tumores secretores de catecolaminas derivados de las células cromafines de la médula suprarrenal (feocromocitoma) o de los ganglios del SNS (paraganglioma).
Son tumores neuroendocrinos originados a partir de las células cromafines derivadas de la CN (Fig. 1). El 80% se localizan en la médula suprarrenal y se denominan feocromocitomas y el resto, los paragangliomas, asientan en los ganglios simpáticos prevertebrales y paravertebrales de cabeza y cuello, mediastino, abdomen y pelvis.
Predominan en población adulta, y solo el 10-20% se presentan en niños. Pueden ser esporádicos, familiares (mutaciones en las subunidades de la succinato-dehidrogenasa, SDHD) o asociarse a síndromes genéticos, como la enfermedad de Von Hippel Lindau (la más frecuente), la neoplasia endocrina múltiple tipo 2 y la neurofibromatosis tipo 1(26). Los tumores multifocales, extraadrenales o bilaterales en pacientes pediátricos sugieren una causa genética.
Ambos tumores sintetizan y secretan catecolaminas (dopamina, norepinefrina, epinefrina) y sus metabolitos (HVA, normetanefrina y metanefrina). Por ello, se manifiestan con: hipertensión, taquicardia, sudoración, cefalea, temblor y ansiedad. La invasión local o la existencia de metástasis a distancia (hueso, hígado, pulmón y ganglios linfáticos) determinan su malignidad, siendo, por lo general, tumores > 5 cm, paragangliomas y familiares(26).
El diagnóstico se realiza mediante TC o RM y la cuantificación de catecolaminas y sus metabolitos en sangre y orina. El estudio de extensión se completa con la 123I-MIBG, aunque otros radiotrazadores como análogos de somatostatina marcados con galio, 18-fluorodopa o 18-fluoro-dihidroxifenilalanina, también son de utilidad(26).
El tratamiento de elección es la resección quirúrgica. La complicación más frecuente es la hipotensión arterial grave intraoperatoria y postoperatoria, por lo que es prioritario el control de la TA previo a la cirugía, mediante bloqueo adrenérgico (primero alfa y después beta) y prevenir la hipovolemia. Los pacientes metastásicos se pueden tratar con: quimioterapia (ciclofosfamida/vincristina/dacarbazina o temozolamida), radioterapia, embolización, ablación con radiofrecuencia o radionucleótidos (o 131I-MIBG o 177Lu-DOTATATE)(26). También se han ensayado los inhibidores de tirosín kinasa (sunitinib) con resultados variables.
Función del pediatra de Atención Primaria
Es fundamental el papel del pediatra de Atención Primaria en el diagnóstico de sospecha del neuroblastoma. Evitar demoras en la atención de estos pacientes en unidades especializadas de Oncología Pediátrica, tiene gran impacto en el pronóstico y calidad de vida de los supervivientes.
Por sus síntomas de presentación, se considera al neuroblastoma como un gran simulador, por lo que se debe incluir en el diagnóstico diferencial de enfermedades infecciosas y reumatológicas, cuadros digestivos (dolor abdominal, estreñimiento, anorexia, fallo de medro) y renales (hipertensión arterial idiopática).
Así mismo, es crucial identificar datos de alarma, como debilidad muscular, rechazo de la marcha o alteración del control de esfínteres, que pueden indicar una compresión medular o síntomas compatibles con un SOM, como alteraciones del comportamiento, ataxia y mioclonias.
En los casos en los que se sospeche un tumor de la cresta neural, la aproximación inicial en un paciente estable mediante analítica, ecografía y radiografía de tórax, es de gran utilidad. No se debe olvidar la posible aparición de citopenias en el hemograma o de alteraciones bioquímicas, como la elevación de la ferritina y de la LDH, que pueden simular una leucemia.
Los casos con alto índice de sospecha deben remitirse de inmediato a centros especializados con Unidades de Oncología Pediátrica.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1. Yan P, Qi F, Bian L, Xu Y, Zhou J, Hu J, et al. Comparison of incidence and outcomes of neuroblastoma in children, adolescents, and adults in the united states: A surveillance, epidemiology, and end results (SEER) program population study. Med Sci Monit; 2020. p. 26.
2. Cañete Nieto A, Pardo Romaguera E, Muñoz López A, Valero Poveda S, Porta Cebolla S, Barreda Reines M, et al. Cáncer infantil en España. Estadísticas 1980-2020.Registro Español de Tumores Infantiles (RETI-SEHOP). Valencia. 2021. Disponible en: https://www.uv.es/rnti/informes.html.
3. Shimada H, Ambros IM, Dehner LP, Hata JI, Joshi VV, Roald B, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999; 86: 364-72.
4. Peuchmaur M, D’Amore ES, Joshi VV, Hata J, Roald B, Dehner LP, et al. Revision of the International Neuroblastoma Pathology Classification. Cancer. 2003; 98: 2274-81.
5. Cohn SL, Pearson ADJ, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG task force report. J Clin Oncol. 2009; 27: 289-97.
6. Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The international neuroblastoma risk group (INRG) staging system: An INRG task force report. J Clin Oncol. 2009; 27: 298-303.
7.** Maris JM. Recent Advances in Neuroblastoma. N Engl J Med. 2010; 362: 2202-11.
8. Park J, Hogarty M, Bagatell R, Schleiermacher G, Mosse Y, Maris JM. Neuroblastoma. In: Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. 8th ed. Philadelphia, PA, EE.UU.: Wolters Kluwer; 2020. p. 647-72.
9.*** Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Prim; 2016. p. 2.
10.** Ahmed AA, Zhang L, Reddivalla N, Hetherington M. Neuroblastoma in children: Update on clinicopathologic and genetic prognostic factors. Pediatr Hematol Oncol. 2017; 34: 165-85.
11.** Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003. 2003; 3: 203-16.
12. Cheung NK V., Dyer MA. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013; 13: 397-411.
13.*** Nakagawara A, Li Y, Izumi H, Muramori K, Inada H, Nishi M. Neuroblastoma. Jpn J Clin Oncol. 2018; 48: 214-41.
14.*** Johnsen JI, Dyberg C, Wickström M. Neuroblastoma—A neural crest derived embryonal malignancy. Front Mol Neurosci. 2019; 12: 1-11.
15. Hero B, Schleiermacher G. Update on pediatric opsoclonus myoclonus syndrome. Neuropediatrics. 2013; 44: 324-9.
16. Rubio Aparicio PM, Rosich Del Cacho B. Tumores de la cresta neural. Pediatr Integr. 2016; 20(7): 434-46.
17. Brodeur GM, Pritchard J, Berthold F, Carlsen NLT, Castel V, Castleberry RP, et al. Revisions of the International Criteria for Neuroblastoma Diagnosis, Staging, and Response to Treatment Purpose and Methods: Based on preliminary experience, there was a need for modifications and clarifications in the International Neuroblastoma Stagin. J Clin Oncol. 1993; 11: 1466-77.
18. Ladenstein R, Pötschger U, Valteau-couanet D, Luksch R, Castel V, Ash S, et al. Investigation of the role of dinutuximab beta-based immunotherapy in the siopen high-risk neuroblastoma 1 trial (HR-NBL1). Cancers (Basel). 2020; 12: 1-19.
19. Mosse YP, Deyell RJ, Berthold F, Nagakawara A, Ambros PF, Monclair T, et al. Neuroblastoma in Older Children, Adolescents and Young Adults: A Report From the International Neuroblastoma Risk Group Project. Pediatr Blood Cancer. 2014; 61: 627-35.
20.*** Tolbert VP, Matthay KK. Neuroblastoma: clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018; 372: 195-209.
21. Ladenstein R, Lambert B, Pötschger U, Castellani M-R, Lewington V, Bar-Sever Z, et al. Validation of the mIBG skeletal SIOPEN scoring method in two independent high-risk neuroblastoma populations: the SIOPEN/HR-NBL1 and COG-A3973 trials. Eur J Nucl Med Mol Imaging. 2018; 45: 292-305.
22. Uemura S, Ishida T, Kyae K, Thwin M, Yamamoto N. Dynamics of Minimal Residual Disease in Neuroblastoma Patients. Front Oncol. 2019; 9: 455.
23. Fisher JPH, Tweddle DA. Neonatal neuroblastoma. Semin Fetal Neonatal Med. 2012; 17: 207-15.
24. Chung C, Boterberg T, Lucas J, Panoff J, Valteau-Couanet D, Hero B, et al. Neuroblastoma. Pediatr Blood Cancer. 2021; 68: 1-8.
25. Yu AL, Gilman AL, Ozkaynak MF, Kreissman SG, London WB, Chen HX, et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N Engl J Med. 2010; 363: 1324-34.
26.** Antonio K, Valdez MN, Mercado-Asis L, Taieb D, Pacak K. Pheochromocytoma/paraganglioma: recent updates in genetics, biochemistry, immunohistochemistry, metabolomics, imaging and therapeutic options. Gland Surg. 2020; 9: 105-23.
Bibliografía recomendada
– Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Prim; 2016. p. 2.
Revisión detallada sobre el neuroblastoma y su fisiopatología, con una exposición enfocada a la práctica clínica sobre el diagnóstico, estadiaje y tratamiento de la enfermedad, sin olvidar aspectos relativos a la calidad de vida y efectos secundarios a largo plazo en los supervivientes.
– Nakagawara A, Li Y, Izumi H, Muramori K, Inada H, Nishi M. Neuroblastoma. Jpn J Clin Oncol. 2018; 48: 214-41.
Revisión exhaustiva sobre los conocimientos actuales del neuroblastoma, con especial detalle a sus características genéticas y moleculares y su papel en la oncogénesis. Actualiza aspectos relativos al diagnóstico, estadiaje y tratamiento.
– Hero B, Schleiermacher G. Update on pediatric opsoclonus myoclonus syndrome. Neuropediatrics. 2013; 44: 324-9.
Actualización sobre el síndrome de opsoclonus-mioclonus en Pediatría y su relación con el neuroblastoma, con recomendaciones para su diagnóstico y tratamiento.
– Fisher JPH, Tweddle DA. Neonatal neuroblastoma. Semin Fetal Neonatal Med. 2012; 17: 207-15.
Revisión pormenorizada sobre el neuroblastoma en neonatos y lactantes, describiendo las características específicas del tumor a esta edad, en cuanto a presentación clínica, diagnóstico diferencial, manejo y evolución.
– Shohet J, Foster J. Neuroblastoma. BMJ. 2017; 357: j1863.
Breve revisión de lectura rápida, enfocada a la práctica clínica y basada en la evidencia sobre el manejo del neuroblastoma.
– Pearda L, Costb NG, Saltzman AF. Pediatric pheochromocytoma: current status of diagnostic imaging and treatment procedures. Curr Opin Urol. 2019, 29: 493-9.
Revisión actualizada y concisa sobre los aspectos básicos del feocromocitoma y paraganglioma, con especial detalle a su presentación clínica y síndromes genéticos relacionados en pacientes pediátricos y al diagnóstico por imagen y su tratamiento.
| Caso clínico |
|
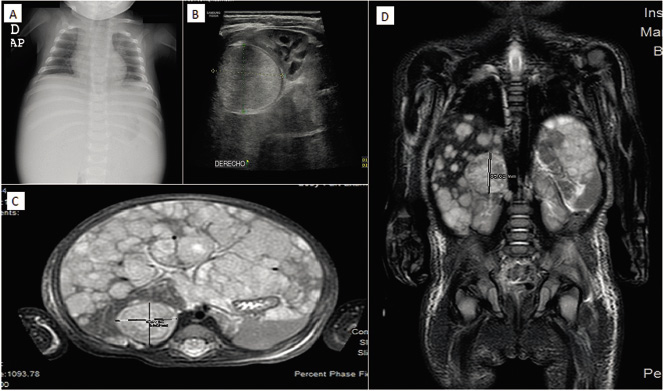
Anamnesis Lactante de 2 meses de vida que consulta por distensión abdominal progresiva de 2 semanas de evolución, con empeoramiento de su reflujo gastroesofágico y cólicos. No refiere irritabilidad ni rechazo de las tomas, y se encuentra afebril. Hábito intestinal normal y diuresis conservada. Embarazo controlado y parto a término, eutócico. Apgar 9/10. Alimentado con lactancia materna exclusiva. Desarrollo psicomotor normal para la edad. Pauta de vacunación que corresponde a los 2 meses. Exploración física Afebril, con tensión arterial y frecuencia cardíaca normales para su edad. Buen estado general, bien hidratado y perfundido. Sin lesiones cutáneas y con auscultación cardiopulmonar normal. Destaca a la palpación abdominal, una masa pétrea que se extiende desde región subcostal hasta genitales, con molestias a la exploración, aunque sin signos de irritación peritoneal. Perímetro abdominal de 41,5 cm y genitales masculinos normales. Pruebas complementarias En la ecografía abdominal se objetiva una masa redondeada en región suprarrenal derecha de 3,5 x 2,9 x 3 cm y hepatomegalia masiva, encontrándose el hígado ocupado en su totalidad por lesiones nodulares (Fig. 6).
Figura 6. Imágenes del caso clínico. A. Radiografía simple (PA), en la que se aprecia importante distensión abdominal. B. Ecografía abdominal que muestra la masa suprarrenal. C y D. Imágenes de RM, en la que además de la masa suprarrenal, se aprecia la infiltración hepática masiva con múltiples imágenes nodulares (síndrome de Pepper). Se completa el estudio de imagen con una radiografía de tórax que es normal y una resonancia magnética abdominal, donde se confirma la presencia de una masa redondeada hipointensa en T1 e hiperintensa en T2, localizada en región suprarrenal derecha. El parénquima hepático está sustituido en su práctica totalidad por innumerables lesiones nodulares, de una señal similar a la masa suprarrenal, sin lesiones a otros niveles (Fig. 6). El hemograma y la bioquímica son normales, salvo elevación de LDH de 1172 U/L (180-430), sin otros datos de lisis tumoral, y de enolasa neuronal específica (ENE) hasta 134 ug/L (0-18). Evolución Tras el ingreso, la situación clínica del paciente empeora, desarrollando hipertensión arterial que requiere tratamiento antihipertensivo y presenta datos de coagulación intravascular diseminada, por lo que ingresa en la unidad de Cuidados Intensivos Pediátricos.
|

 Follow-up of childhood cancer in Primary Care. How to detect late effects
Follow-up of childhood cancer in Primary Care. How to detect late effects