 |
| Temas de FC |
A. Sastre Urgellés, P. Rubio Aparicio
Servicio de Hemato-Oncología Pediátrica. Hospital Universitario La Paz – Hospital Materno-Infantil. Madrid
| Resumen
Los tumores óseos malignos suponen el 6% de todas las neoplasias infantiles. Prácticamente, la totalidad de casos son osteosarcomas o tumores de la familia Ewing. Son neoplasias muy agresivas, que se deben considerar enfermedades sistémicas desde el diagnóstico por la presencia de micrometástasis. Su tratamiento requiere un abordaje multidisciplinar, combinando cirugía, quimioterapia y radioterapia, y debe realizarse en centros con experiencia, consiguiendo así aumentar la supervivencia de estos pacientes, al tiempo que se disminuyen las secuelas y limitaciones físicas debidas a la cirugía, que es una parte esencial del protocolo de tratamiento. Es fundamental diagnosticarlos precozmente, pues existe una gran diferencia en la supervivencia para los pacientes con enfermedad localizada (70-80%) y aquellos que presentan metástasis (20-30%). |
| Abstract
Malignant bone tumors represent 6% of childhood cancers. The two predominant histologies are Osteosarcoma (OS) and Ewing Family of Tumors (EFT). These are aggressive neoplasms, which due to the presence of micrometastasis must be considered as disseminated diseases from diagnosis. For their treatment, a multimodal approach is required, combining surgery, chemo and radiotherapy, and patients should be referred to expert units in order to increase survival, while minimizing sequelae of local therapy, which is an essential part of the management of the disease. It is paramount to perform an early diagnosis, as there is a huge survival gap between localized (70-80%) and metastatic (20-30%) presentations. |
Palabras clave: Osteosarcoma; Sarcoma de Ewing; Rabdomiosarcoma.
Key words: Osteosarcoma; Ewing sarcoma; Rhabdomyosarcoma.
Pediatría Integral 2021; XXV (7): 348 – 356
Tumores óseos. Rabdomiosarcomas
Tumores óseos
Los tumores óseos malignos primarios en la infancia suponen el 8% de las neoplasias en el Registro Español de Tumores Infantiles (RETI)(1), y son principalmente: el osteosarcoma y el sarcoma de Ewing; el condrosarcoma es el sarcoma óseo más frecuente en adultos, pero es muy raro en la edad pediátrica.
Osteosarcoma o sarcoma osteogénico
El osteosarcoma (OS) es un tumor maligno primario del hueso, de origen mesenquimal, compuesto por células neoplásicas productoras de osteoide.
Epidemiología
El osteosarcoma es el tumor óseo maligno más frecuente en la infancia y adolescencia.
Representa el 55% de los tumores óseos en menores de 20 años y en adolescentes es más frecuente que el sarcoma de Ewing (SE), con una incidencia máxima entre los 13 y los 16 años de edad, coincidiendo con el estirón de la adolescencia. Por razones desconocidas, el OS afecta más a los varones (proporción 1,34 a 1) y es más frecuente en la raza negra y en otras razas en comparación con los caucásicos(2). Se localiza preferentemente en las metáfisis de los huesos largos, especialmente: fémur distal, tibia proximal y húmero proximal.
Etiología
No existe una causa conocida, aunque en algunos casos sí se relaciona con varios agentes etiológicos probados.
Muchas veces su aparición se relaciona con un traumatismo reciente, pero no es cierto que este sea la causa, solo pone de manifiesto la enfermedad. La exposición a radiación ionizante sí que es un agente causal verificado, con periodos de latencia de 10-20 años. La administración previa de citostáticos alquilantes también se relaciona con la aparición de OS. Se han descrito asociaciones con enfermedades hereditarias, la más clara con el retinoblastoma hereditario; también con los síndromes de Li-Fraumeni, Rothmund-Thomson, Bloom y la enfermedad de Paget(3).
En el OS se han descrito diversas alteraciones genéticas, siendo las más destacadas las mutaciones en el gen RB, en el cromosoma 13 (gen supresor tumoral del retinoblastoma, codificador de una proteína nuclear que inhibe el crecimiento celular). Otra alteración presente es la mutación homocigota del gen p53, relacionada con el control del crecimiento y del ciclo celular.
Histopatogenia
El OS se caracteriza por la presencia de células mesenquimales malignas del estroma, asociadas a la producción de sustancia osteoide y hueso.
El tipo histológico más frecuente es el OS convencional (intramedular de alto grado, 90% de todos los OS), con tres subcategorías: osteoblástico (50%), fibroblástico (25%) y condroblástico (25%), en función del componente celular predominante. Además de estas 3 subcategorías, se describen dos variantes: OS telangiectásico (quístico, vascularizado) y OS de célula pequeña, muy agresivo y morfológicamente similar al sarcoma de Ewing. Otra categoría son los OS superficiales, en contraposición a los intramedulares, que incluyen: tipo parostal de bajo grado, tipo perióstico de grado intermedio y osteosarcomas superficiales de alto grado. En general, son menos agresivos y su evolución más favorable.
A diferencia de otros sarcomas, los OS no presentan ninguna translocación cromosómica característica.
Clínica
El principal síntoma es el dolor, con más frecuencia localizado en una extremidad y, generalmente, lleva varios meses de evolución antes del diagnóstico.
El paciente lo suele relacionar con el ejercicio físico o un traumatismo. Con el tiempo, aparecerá inflamación local, efecto de masa e impotencia funcional. No suele haber fiebre, pérdida de peso ni otra sintomatología sistémica. El OS tiene predilección por las metáfisis de los huesos largos: el 80% se localiza en las extremidades (fémur 40%, tibia 20%, húmero 10%), creciendo desde la cavidad medular hacia la corteza y los tejidos blandos. Las metástasis aparecen, sobre todo, en el pulmón, sin clínica acompañante. El segundo lugar en frecuencia son otras localizaciones óseas.
Diagnóstico
El diagnóstico del tumor es anatomopatológico y es necesario realizar siempre un estudio de extensión buscando metástasis(4).
• Laboratorio: los hallazgos son inespecíficos, como el incremento de la fosfatasa alcalina, LDH y de la velocidad de sedimentación globular (VSG).
• Radiología (Fig. 1): la primera sospecha surge tras realizar una radiografía simple de la zona afecta.

Figura 1. Estudios radiológicos en los tumores óseos malignos. A. Radiografía simple de un osteosarcoma de tibia: Lesión yuxtacortical en la región diafisometafisaria proximal de la tibia con una matriz grosera calcificada/osificada y que se extiende a las partes blandas. B. Radiografía simple de un sarcoma de Ewing de fémur: lesión intraósea que ocupa el cuello y la región intertrocantérea del fémur, observándose un patrón moteado, alternando áreas líticas con otras más densas. C. Resonancia magnética: osteosarcoma de tibia proximal, que corresponde a la radiografía simple A. D. Tomografía por emisión de positrones (PET) en sarcoma de Ewing de la pared torácica: masa intensamente hipermetabólica que engloba el tercio proximal de la décima costilla izquierda y protruye hacia la cavidad torácica. E. Tomografía computarizada (TC) de tórax: metástasis pulmonares en paciente con sarcoma de Ewing. Se observan numerosos nódulos de diferente tamaño en localización subpleural.
El OS aparece como una masa heterogénea, con zonas osteolíticas y escleróticas, de bordes mal definidos, que crece desde la cavidad medular y progresa hacia la corteza, atravesando y levantando el periostio (que reacciona formando tejido óseo inmaduro, en forma de triángulo: signo de Codman), y pudiendo afectar a los tejidos blandos que rodean al hueso, produciendo imágenes difusas de diferentes densidades. Para valorar la extensión del tumor (ósea y en partes blandas) y la afectación de estructuras contiguas (vasos, nervios…) es necesario realizar una resonancia magnética (RM) o una tomografía computerizada (TC), siendo preferible la RM por su mayor sensibilidad. En ocasiones, existen lesiones satélites (skip) en el propio hueso, sin contigüidad con el tumor principal, por lo que hay que incluir la totalidad del hueso afecto en la técnica de imagen(5).
• Biopsia: el estudio histopatológico del tumor proporciona el diagnóstico de certeza. Debe obtenerse mediante un trócar y dentro de la zona que se marcará, para después ser resecada con el tumor, y es preferible que la realice el mismo equipo de cirujanos que posteriormente efectuará la intervención quirúrgica, o un radiólogo intervencionista con experiencia(4).
• Estudio de extensión: conocido el diagnóstico anatomopatológico, se completará el estadiaje del tumor con un TC pulmonar de alta resolución, para detectar metástasis pulmonares, y una gammagrafía ósea con tecnecio (99Tc) para localizar metástasis óseas. La tomografía por emisión de positrones (PET) se está usando cada vez más para valorar la afectación metastásica, fundamentalmente si se combina con TC. Aunque existen varias clasificaciones para el estadiaje, realmente es suficiente con distinguir entre tumor localizado (L) o con metástasis (M). En el momento del diagnóstico, el OS se presenta como enfermedad metastásica en el 20% de casos.
El proceso diagnóstico aparece desarrollado en la figura 2.

Figura 2. Procedimiento diagnóstico en los tumores óseos.
Diagnóstico diferencial
Principalmente ha de hacerse frente a los tumores de la familia Ewing, metástasis de otros tumores como el neuroblastoma, y con los tumores óseos benignos (Tabla I) (6).

El diagnóstico histopatológico bastará para diferenciar estas entidades.
Factores pronósticos
Son factores de mejor pronóstico: la enfermedad localizada (supervivencia libre de eventos [SLE] a los 5 años de 60-70%, frente a 20-30% para la enfermedad metastásica)(7), volumen tumoral inferior a 200 cc, tumor localizado en extremidades (no axial), necrosis superior al 90% tras la quimioterapia (QT) de inducción y resección con márgenes libres de tumor(7).
Tratamiento
El abordaje debe ser multidisciplinar, con la administración de QT antes y después de la cirugía, siendo esencial conseguir la resección en bloque de todo el compartimento tumoral.
Debido la alta incidencia de micrometástasis, indetectables, pero presentes ya en el momento del diagnóstico, la cirugía por sí sola no consigue curar más allá del 20% de los pacientes. Por ello, resulta esencial administrar QT adyuvante para erradicarlas.
Los protocolos de tratamiento incluyen 3 fases (Fig. 3):
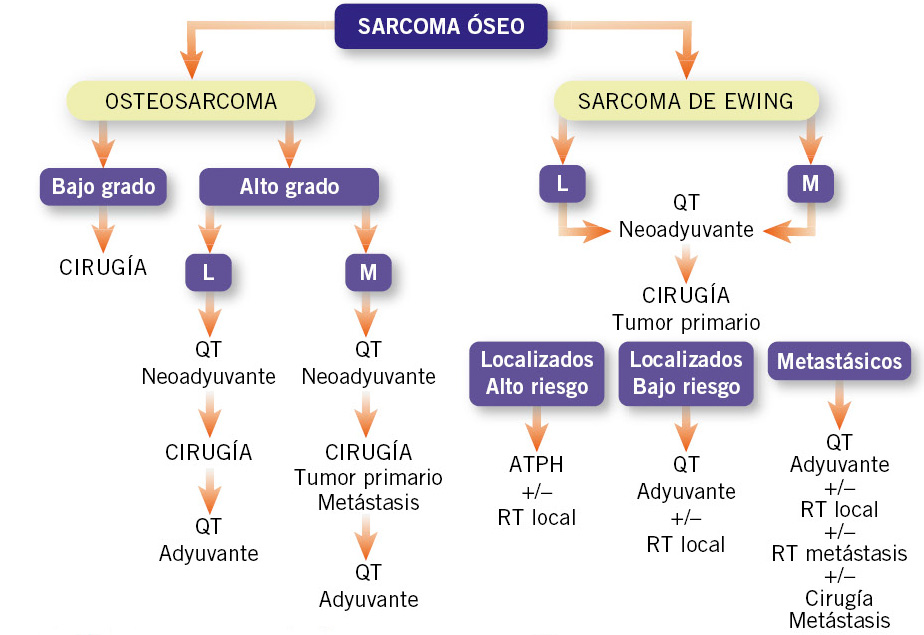
Figura 3. Tratamiento de los tumores óseos malignos. ATPH: megaterapia con autotrasplante de progenitores hematopoyéticos; L: localizado; M: metastásico; QT: quimioterapia; RT: radioterapia.
1. Quimioterapia neoadyuvante en ciclos, combinando metotrexato a altas dosis, cisplatino y doxorrubicina (esquema MAP). Además de eliminar las micrometástasis, consigue una disminución de volumen del tumor facilitando así su resección.
2. Cirugía, que debe conseguir una resección completa del tumor en bloque, incluyendo: el compartimento óseo, las partes blandas circundantes y los trayectos cutáneos y subcutáneos de las biopsias iniciales, y con márgenes de seguridad(4). La reconstrucción del sistema musculoesquelético se consigue mediante injertos óseos (autólogos o alogénicos de cadáver) o prótesis. Son cirugías complejas que requieren ser realizadas por un cirujano ortopeda con experiencia en el tratamiento de tumores óseos pediátricos. Actualmente, se consigue salvar la extremidad afecta en el 80% de los casos(7).
3. Quimioterapia postquirúrgica con los mismos agentes o añadiendo ifosfamida, para eliminar las células tumorales residuales que hayan podido movilizarse con la intervención.
Con esta estrategia se ha conseguido elevar la SLE a los 5 años hasta el 60-70%, pero desde 1990, no se han conseguido mejorías significativas(5,7).
El OS no es un tumor muy radiosensible, por lo que la radioterapia (RT) solo se emplea en localizaciones axiales en las que la cirugía no es posible, en casos de OS multifocal o con fines paliativos para el control del dolor.
Para la enfermedad metastásica se emplean combinaciones de QT con los mismos citostáticos, pero con mayor número de ciclos, además de resecar el tumor primario y las metástasis pulmonares que continúen siendo visibles tras la QT neoadyuvante. A pesar de todo ello, la SLE oscila entre un 16-53%.
Recaídas
Las recaídas ocurren en los 3 primeros años tras el diagnóstico hasta en un 40% de los pacientes con enfermedad localizada, sobre todo, en forma de metástasis pulmonares.
La segunda línea de QT suele consistir en combinaciones de ifosfamida con/sin etopósido, además de resecar las metástasis pulmonares(8). La supervivencia global (SG) tras la recaída oscila entre el 13-57%(9). Se están estudiando diferentes inhibidores de tirosina quinasa de molécula pequeña cuya diana es VEGFR, como: regorafenib, cabozantinib y sorafenib, y otros tratamientos basados en la inmunoterapia, con el empleo de inhibidores de punto de control como el pembrolizumab(10).
Tumores de la familia Ewing
Los tumores de la familia Ewing (TFEw) lo constituyen un grupo de neoplasias anteriormente descritas como entidades diferentes que comparten un origen histológico similar y una translocación cromosómica característica.
Incluye: el SE típico y atípico, el tumor neuroectodérmico primitivo (PNET, anteriormente llamado neuroepitelioma), el tumor de células pequeñas de la región tóraco-pulmonar (tumor de Askin) y el SE extraóseo.
Epidemiología
El SE es el segundo tumor óseo maligno primario por frecuencia en niños y adolescentes, siendo el tumor óseo más frecuente en niños menores de 4 años.
Es raro por encima de los 30 años, y en las razas negra y asiática. Predomina en varones (1,5:1)(4). No es hereditario ni se asocia a síndromes malformativos, y tampoco se relaciona con ningún agente externo causal.
Histopatogenia
Los TFEw comparten la misma anomalía cromosómica, consistente en una translocación entre los cromosomas 11 (gen EWS) y 22 (gen FLI1), originando el gen quimérico EWS-FLI1.
La proteína resultante ocasiona una disregulación en los genes responsables de la proliferación y diferenciación celular, favoreciendo el desarrollo del tumor. Se detecta por FISH o PCR en el 90-95% de los TFEw y, por ello, es un marcador diagnóstico que ayuda a distinguir el SE de otras entidades(11).
El origen histogenético del SE es una cuestión que continúa debatiéndose entre un origen neuroectodérmico o mesenquimal. James Ewing lo describió en 1921 como: un tumor óseo, muy agresivo, sensible a la radioterapia y sin un claro origen histológico. El SE típico está constituido por células redondeadas, pequeñas, azules, con núcleos hipercromáticos y escaso citoplasma. Se ha propuesto que se desarrolle a partir de las células posganglionares parasimpáticas, basándose en que todos los TFEw tienen cierto grado de diferenciación neural (escasa en el SE típico, marcada en el PNET, e intermedia en el SE atípico) y expresan marcadores neurales, como la enolasa neuroespecífica y la S-100. Una teoría alternativa sugiere que los TFEw surgen a partir de células madre mesenquimales. Todos los TFEw expresan niveles altos de una glicoproteína de membrana, CD99, que ayuda a distinguirlos de otros tumores de células redondas y pequeñas de la infancia (neuroblastoma, otros sarcomas, linfomas).
Clínica
Los síntomas son poco alarmantes y muchos pacientes llevan más de 6 meses de evolución antes de que se realice el diagnóstico.
Los TFEw pueden originarse en cualquier hueso y en tejidos blandos. Son algo más frecuentes en el esqueleto axial (54%), sobre todo en los huesos de la pelvis (25%). En extremidades, el lugar más común es el fémur (16%)(12). El dolor local suele ser el síntoma inicial (96%); al comienzo tiene carácter intermitente y paulatinamente gana intensidad. Suele haber afectación de los tejidos blandos adyacentes, apareciendo tumefacción local. A veces, existen síntomas generales, como fiebre (21%). Otra sintomatología dependerá de la localización del tumor: derrame pleural en los tumores torácicos, dolor radicular en tumores vertebrales y problemas de esfínteres en tumores pélvicos.
Diagnóstico
El diagnóstico del SE es anatomopatológico, y es necesario siempre realizar un estudio de extensión buscando metástasis(4).
• Laboratorio: hallazgos inespecíficos, aumento de VSG y de la LDH en relación con la masa tumoral existente.
• Radiología (Fig. 1): en la radiografía simple, el hueso afecto presenta un patrón moteado difuso, con predominio de áreas líticas. Puede existir el triángulo de Codman, y es típica la imagen en “capas de cebolla”, debido a la existencia de múltiples capas de reacción perióstica con neoformación ósea. En huesos planos predominan zonas de esclerosis. La RM es la técnica radiológica de elección para valorar la extensión ósea y extraósea del tumor(4). La TC proporciona información sobre la cortical y los cambios en la estructura ósea.
• Biopsia: el estudio anatomopatológico proporciona el diagnóstico. Se hará con trócar, y con las mismas precauciones y exigencias que lo comentado previamente con el OS(4).
• Estudio de extensión: los TFEw tienen una elevada capacidad de metastatizar a distancia en: pulmón (38%), huesos (31%) y médula ósea (11%). En el momento del diagnóstico, un 20% de los pacientes presentan metástasis visibles, pero la mayoría tienen metástasis subclínicas. Como en el OS, es preciso realizar una TC torácica de alta resolución y una gammagrafía ósea (99Tc). Además, se descartarán las metástasis en médula ósea, obteniendo, al menos, dos biopsias en dos lugares alejados del tumor primario. Como para el OS, para el estadiaje es suficiente con distinguir entre tumor localizado (L) o con metástasis (M).
El proceso diagnóstico aparece desarrollado en la figura 2.
Diagnóstico diferencial
Se hará con otros tumores malignos (OS, metástasis óseas…), osteomielitis, y con tumores benignos óseos (Tabla I)(6).
Factores pronósticos
La supervivencia ha ido mejorando en las últimas décadas gracias a protocolos más agresivos, pero aún la SG se sitúa en torno al 60-75%.
Los factores pronósticos asociados a una mejor supervivencia son: localización no axial, volumen tumoral inicial inferior a 200 cc, grado de necrosis del tejido tumoral tras la QT inicial superior al 90% y, sobre todo, ausencia de metástasis al diagnóstico(13).
Tratamiento
Los protocolos para el tratamiento de los TFEw combinan la QT sistémica con las medidas locales (RT y/o cirugía).
La QT sistémica es imprescindible para curar un TFEw, dada la presencia de micrometástasis desde el comienzo; si únicamente se aplica un tratamiento local, solo sobrevivían el 10% de los pacientes. En los últimos años, la combinación de ciclofosfamida, vincristina y doxorrubicina, alternando con ifosfamida y etopósido, se ha impuesto como la más eficaz y con menos toxicidad que otras combinaciones.
El esquema de tratamiento es similar al del OS(4,14) (Fig. 3):
1. QT inicial neoadyuvante, para reducir el volumen tumoral y eliminar las micrometástasis.
2. El tratamiento local consistirá en la resección quirúrgica, siempre que sea posible, con los mismos criterios que para el OS. Los TFEw son radiosensibles y, si el hueso no es resecable, o la cirugía supone una grave mutilación o deformidad estética, se utiliza la RT como tratamiento local único.
3. En la fase de consolidación tras el tratamiento local, se administra más QT para eliminar tumor residual y RT posquirúrgica, si el grado de necrosis tumoral no es superior al 90%, o existen células tumorales en los márgenes de la resección. En tumores de mal pronóstico tras la cirugía, se puede optar por una QT mieloablativa (megaterapia) con rescate de progenitores hematopoyéticos autólogos. Para el acondicionamiento, se utiliza la combinación de busulfán y melfalán. En caso de persistir metástasis pulmonares, al final del tratamiento se puede administrar RT pulmonar.
Recaídas
La mayoría de los pacientes recaen en los dos primeros años desde el diagnóstico y, sobre todo, tras suspender la quimioterapia.
La localización más frecuente es en pulmón. El pronóstico es malo, con supervivencias del 20-25%. El tratamiento no debe limitarse a actuar sobre el tumor, debe administrarse de nuevo quimioterapia, existiendo actualmente diversos ensayos que intentan establecer cuál es la combinación de citostáticos más eficaz.
Rabdomiosarcoma (Fig.4)
El rabdomiosarcoma (RMS) es un tumor mesenquimal maligno que asemeja músculo estriado(15).

Figura 4. Imágenes en resonancia magnética de rabdomiosarcomas en diferentes localizaciones. A. Rabdomiosarcoma en región orbitaria, que aparece como una masa redondeada localizada en la zona superoexterna y comprime el globo ocular. B. Rabdomiosarcoma de gran tamaño, localizado en la región pélvica, que comprime el recto y tiene su origen en la fosa isquiorrectal. C. Rabdomiosarcoma parameníngeo. Masa centrada en el espacio masticador derecho que invade la fosa craneal media y el seno cavernoso.
Epidemiología
El RMS representa aproximadamente la mitad de los diagnósticos de sarcoma de partes blandas (SPB) en Pediatría.
La incidencia anual es entre 4 y 5 casos por millón. De media, se diagnostican en España cada año 30 RMS(1).
Es un tumor típico del niño pequeño, más frecuente entre 1 y 4 años de edad, habiendo otro pico de incidencia en la adolescencia(16).
Aunque se han descrito factores de riesgo, tanto ambientales (exposición de los padres a drogas de abuso, y de la madre a radiaciones ionizantes) como genéticos (síndromes de hipercrecimiento, polimalformativos y de predisposición a cáncer), el RMS es, como la mayoría de los tumores pediátricos, una neoplasia que surge de novo, sin posibilidad de prevención(15).
Histopatogenia
Desde el punto de vista anatomopatológico, se clasifica en cuatro variantes morfológicas, de las que tres aparecen en la edad pediátrica: embrionario (80%), alveolar (20%) y fusocelular/esclerosante (menos del 1%).
El RMS embrionario se caracteriza por su aspecto inmaduro, mientras que el alveolar se distingue por la observación de septos rodeando formaciones alveolares tumorales(17).
La presentación y agresividad están vinculadas a la biología, en función de la presencia de genes de fusión involucrando FOXO1. Así, los RMS embrionarios, y aproximadamente el 25% de los alveolares, no presentan reordenamiento de FOXO1, que sí está presente en el otro 75% de las formas alveolares(17). Esto es relevante, ya que tanto la presentación como el pronóstico de los RMS alveolares FOXO1 negativo son similares a los embrionarios(18).
Los RMS FOXO1 negativo suelen aparecer en cabeza y cuello, o en abdomen-pelvis, con tropismo por el sistema genitourinario, y raramente presentan diseminación metastásica; en cambio, los casos FOXO1 positivo asientan habitualmente sobre extremidades y tienen mayor potencial metastásico, sobre todo en pulmón.
Clínica
La presentación clínica es variable, dependiendo de la ubicación del tumor. Puede manifestarse por el efecto funcional de la masa (tumefacción, dolor, impotencia funcional) o, en ocasiones, detectarse en una exploración clínica o radiológica rutinaria.
En la tabla II, se especifican las diferentes manifestaciones clínicas que se presentan según la estructura anatómica en la que asiente el tumor.

Diagnóstico
El procedimiento diagnóstico requiere, desde el momento de sospecha, la intervención de una unidad multidisciplinar con alta experiencia en sarcomas pediátricos. Para establecer el diagnóstico, es preciso el estudio anatomopatológico y genético del tumor.
• Laboratorio: los estudios analíticos suelen ser normales, no hay ningún dato que sea indicativo de la presencia o gravedad de la enfermedad.
• Radiología: la evaluación de imagen inicial (radiografía simple, ecografía) se completará con imagen de alta resolución mediante RM del tumor primario, para valorar la extensión del tumor y su relación con las estructuras vecinas.
• Biopsia: es fundamental el estudio anatomopatológico del tumor para establecer el subtipo histológico y, además, hay que determinar la presencia de reordenamientos de FOXO1, pues los casos positivos se clasifican como de alto riesgo y su tratamiento será más agresivo.
• Estudio de extensión: la búsqueda de metástasis incluye una TC torácica de alta resolución y una gammagrafía ósea con 99Tc. En pacientes de riesgo alto (alveolares, FOXO positivo, metastásticos), está recomendado completar el estudio con biopsia bilateral de médula ósea. En los últimos años, la 18F-FdG-PET-TC con contraste está reemplazando al estudio de extensión tradicional, especialmente al diagnóstico, sin haberse podido aún establecer su papel en la evaluación de respuesta(19).
Factores pronósticos
La presencia de metástasis al diagnóstico y el estado FOXO1 positivo condicionan los casos de peor pronóstico.
Otros factores con relevancia para la supervivencia son: edad del paciente al diagnóstico (favorable entre 1 y 10 años), localización del tumor (posibilidades de tratamiento quirúrgico), tamaño tumoral (favorable si es menor de 5 cm de diámetro), la afectación de ganglios regionales y el grado de resección inicial según criterios IRS (Tabla III).

La supervivencia global se sitúa en el momento actual en el 82% a 3 años del diagnóstico(1).
Tratamiento
El RMS es un tumor quimio y radiosensible. Su tratamiento es multidisciplinar, y se basa en el control local del tumor y en eliminar la enfermedad a distancia, visible o no.
Todos los pacientes precisan la administración de quimioterapia sistémica, incluso los resecados completamente, ya que se considera una enfermedad diseminada: el estudio por biopsia líquida detecta ADN tumoral en sangre periférica en 2/3 de los casos localizados(20). La quimioterapia se basa en la combinación de vincristina, actinomicina y ciclofosfamida/ifosfamida, con la introducción en los últimos años de irinotecán. El uso de antraciclinas no parece aumentar la supervivencia en pacientes de riesgo estándar/alto y se reserva solo para casos de riesgo muy alto (metastásicos y FOXO +).
El control local es clave en el RMS. Dado que el RMS puede localizarse en muy diferentes ubicaciones, la cirugía requiere la participación de diferentes especialistas, además de cirujanos pediátricos: oftalmólogos, cirujanos maxilofaciales, otorrinolaringólogos, urólogos… Deben tener experiencia en el tratamiento quirúrgico de estas neoplasias. El control de la enfermedad local idealmente debe obtenerse mediante la cirugía, pero la radioterapia es eficaz y puede considerarse como terapia local única en casos inoperables o con un riesgo quirúrgico inaceptable. El RMS es una de las indicaciones aprobadas de protonterapia. La radioterapia local se administra concomitante con la quimioterapia de consolidación, mientras que la irradiación de las metástasis pulmonares se demora tras esta.
El tratamiento en Europa se basa en los protocolos elaborados por el Grupo Europeo Pediátrico de Sarcomas de Tejidos Blandos (EpSSG: European pediatric Soft tissue Sarcoma Group). Se estratifica a los pacientes en grupos de riesgo en función de los criterios arriba mencionados.
Los pacientes de riesgo bajo, con resección completa inicial (IRS I) y genética favorable, dado que ya se ha conseguido el control local, reciben únicamente quimioterapia de consolidación.
Los pacientes de riesgo intermedio representan el grupo más numeroso. Se trata, en su mayoría, de casos con biopsia inicial o resección incompleta, y genética favorable. Reciben quimioterapia de inducción, seguida de control local (cirugía ± radioterapia) y de nuevo quimioterapia de consolidación.
Los pacientes de riesgo alto/muy alto presentan reordenamiento de FOXO1 y/o enfermedad metastásica. El esquema terapéutico es similar al riesgo intermedio, con las siguientes consideraciones: se recomienda la combinación de cirugía y radioterapia para el control local, y se añade, tras la consolidación, una fase de quimioterapia de mantenimiento (combinando ciclofosfamida oral y vinorelbina intravenosa) durante un mínimo de 6 meses.”.
Recaídas
La tasa de recidiva es baja, y son más frecuentes las recaídas locales que a distancia.
Los pacientes FOXO1 positivo tienen más posibilidades de recidivar. Se recomienda realizar un control estrecho durante, al menos, los dos primeros años, con revisiones trimestrales clínicas, analíticas y pruebas de imagen para detectar precozmente las recidivas, tanto a nivel local como metastásicas.
El tratamiento de la recaída requiere el empleo de quimioterapia, con introducción de nuevos agentes (irinotecán, temozolamida, topotecán…), cirugía más agresiva y radioterapia, si esta no se ha empleado antes.
Funciones del pediatra de Atención Primaria
Los sarcomas son un grupo de neoplasias muy amplio, siendo los más frecuentes en la infancia el osteosarcoma, el sarcoma de Ewing y el rabdomiosarcoma. En el caso de los tumores óseos, el dolor suele ser el síntoma principal, y su persistencia debe ser motivo suficiente para realizar una radiografía simple que, en muchas ocasiones, será el punto de partida que llevará al diagnóstico del tumor. En el caso del rabdomiosarcoma, la sintomatología puede ser muy variable, pero ante cualquier masa dura, indolora, de nueva aparición, se debe realizar una exploración radiológica que permita descartar el diagnóstico de sarcoma.
En cualquier caso, ante la sospecha de un sarcoma en cualquier localización, se debe derivar al paciente con urgencia a un Centro con experiencia en el tratamiento de estas patologías, preferentemente a centros con acreditación CESUR (Centros/Servicios/Unidades de Referencia) en Sarcomas, donde es posible realizar el abordaje multidisciplinar que requiere este grupo de neoplasias en el mínimo tiempo posible.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.** Pardo Romaguera E, Muñoz López A, Valero Poveda S, Porta Cebolla S, Cañete Nieto A, Barreda Reines MS, et al. Cáncer infantil en España. Estadísticas 1980-2019. Registro español de Tumores Infantiles (RETI-SEHOP). 2020 (Valencia: Universitat de València).
2.** Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009; 115: 1531-43.
3.** Fuchs B, Pritchard DJ. Etiology of osteosarcoma. Clin Orthop Relat Res. 2002; 397: 40-52.
4.*** Casali PG, Bielack S, Abecassis N, Aro HT, Bauer S, Biagini R, et al. Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; 29: iv79-iv95.
5.** Zhao X, Wu Q, Gong X, Liu J, Ma Y. Osteosarcoma: a review of current and future therapeutic approaches. Biomed Eng Online. 2021; 20: 24.
6.** Plaza D, Sastre A, P G-M. Tumores Óseos. An Pediatr Contin. 2008; 6: 266-75.
7.** Anderson ME. Update on Survival in Osteosarcoma. Orthop Clin North Am. 2016; 47: 283-92.
8.** Palmerini E, Setola E, Grignani G, D’Ambrosio L, Comandone A, Righi A, et al. High Dose Ifosfamide in Relapsed and Unresectable High-Grade Osteosarcoma Patients: A Retrospective Series. Cells. 2020; 9: 11.
9.*** Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol. 2002; 20: 776-90.
10.** Wedekind MF, Wagner LM, Cripe TP. Immunotherapy for osteosarcoma: Where do we go from here? Pediatr Blood Cancer. 2018; 65: e27227.
11.** Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, et al. The Ewing family of tumors–a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med. 1994; 331: 294-9.
12.** Cotterill SJ, Ahrens S, Paulussen M, Jurgens HF, Voute PA, Gadner H, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000; 18: 3108-14.
13.*** Rodríguez-Galindo C, Liu T, Krasin MJ, Wu J, Billups CA, Daw NC, et al. Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital studies. Cancer. 2007; 110: 375-84.
14.** Anderton J, Moroz V, Marec-Berard P, Gaspar N, Laurence V, Martin-Broto J, et al. International randomised controlled trial for the treatment of newly diagnosed EWING sarcoma family of tumours – EURO EWING 2012 Protocol. Trials. 2020; 21: 96.
15.*** Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019; 5: 1.
16.** Sultan I, Qaddoumi I, Yaser S, Rodríguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009; 27: 3391-7.
17.** Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013; 20: 387-97.
18.** Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010; 28: 2151-8.
19.** Sa R, Liu D, Zhao H, Hou S, Lin Q, Guan F. Utility of [(18)F] Fluoro-Deoxyglucose Positron Emission Tomography/Computed Tomography for Staging and Therapy Response Evaluation in Pediatric Rhabdomyosarcoma: A Case Series and Literature Review. Front Med (Lausanne). 2020; 7: 281.
20.** Gallego S, Llort A, Roma J, Sabado C, Gros L, de Toledo JS. Detection of bone marrow micrometastasis and microcirculating disease in rhabdomyosarcoma by a real-time RT-PCR assay. J Cancer Res Clin Oncol. 2006; 132: 356-62.
21. Muñoz Villa A. Tumores óseos. Rabdomiosarcomas. Pediatr Integral. 2016; XX(7): 458-64.
Bibliografía recomendada
- Casali PG, Bielack S, Abecassis N, Aro HT, Bauer S, Biagini R, et al. Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; 29: iv79-iv95.
Se trata de una guía elaborada conjuntamente por la European Society for Medical Oncology (ESMO) y la Red Europea de Cáncer Pediátrico (Paedcan), que ofrece una visión muy completa y actualizada sobre el diagnóstico y tratamiento de todos los tumores óseos.
- Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019; 5: 1.
Trabajo muy completo y actual sobre el rabdomiosarcoma, que describe su epidemiología, mecanismos de desarrollo y diferentes aspectos sobre el diagnóstico y tratamiento de la enfermedad.
| Caso clínico |
|
Paciente de 4 años de edad que acude a urgencias por hinchazón del ojo izquierdo. No refiere traumatismo ni picadura, ha tenido febrícula los últimos dos días. No se queja de dolor. A la exploración, presenta buen estado general, y se observa proptosis izquierda sin paresia de pares craneales. No se palpan adenopatías. Auscultación y palpación abdominal normales. Valorado por Oftalmología, el fondo de ojo es normal. Se decide realizar una TC craneal urgente, que objetiva una masa retroorbitaria de 3,2 cm de diámetro mayor.
|

 Follow-up of childhood cancer in Primary Care. How to detect late effects
Follow-up of childhood cancer in Primary Care. How to detect late effects