 |
| Temas de FC |
F. Vázquez Gómez, E. Carceller Ortega, Á. Lassaletta Atienza
Servicio de Hemato-Oncología Pediátrica. Hospital Infantil Universitario Niño Jesús. Madrid
| Resumen
Los tumores del sistema nervioso central (SNC) en los niños representan la segunda causa más frecuente de tumores malignos, suponiendo una importante causa de morbi-mortalidad a pesar de los avances conseguidos en su diagnóstico, tratamiento y seguimiento. |
| Abstract
Tumors of the central nervous system (CNS) in children are the second most common cause of malignant tumors, representing an important cause of morbidity and mortality in spite of the advances achieved in their diagnosis, treatment and follow-up. |
Palabras clave: Tumores cerebrales pediátricos; Clasificación; Diagnóstico; Tratamiento.
Key words: Pediatric brain tumors; Classification; Diagnosis; Treatment.
Pediatría Integral 2021; XXV (7): 357 – 366
Tumores cerebrales en niños
Introducción
Los tumores pediátricos del sistema nervioso central (SNC) son la segunda neoplasia infantil más frecuente y el tumor sólido más común en los niños.
El SNC se divide en tres compartimentos principales: la médula espinal, la región infratentorial y la región supratentorial. La región infratentorial incluye el tronco cerebral y el cerebelo, mientras que la región supratentorial incluye: los hemisferios cerebrales, el tálamo, ganglios de la base, diencéfalo, tractos ópticos/región quiasmática y área hipotálamo-hipofisaria.
Los signos y síntomas derivados de estas neoplasias dependen de diversos factores, como la localización del tumor, la tasa de crecimiento del mismo y la edad del niño.
La clasificación de los tumores pediátricos del SNC actual es compleja, sobre todo, debido al conocimiento molecular que se va adquiriendo sobre ellos.
El tratamiento de estos tumores precisa de un manejo multidisciplinar. Aunque hemos asistido a una mejora significativa en su tasa de curación durante los últimos años, como resultado de los avances en: neuroimagen, neurocirugía, radioterapia, biología molecular y quimioterapia, actualmente suponen una importante causa de morbimortalidad.
Epidemiología
Los tumores del sistema nervioso central (SNC) se consideran los tumores sólidos infantiles más frecuentes, siendo la segunda causa de cáncer infantil en niños de 0 a 14 años (aproximadamente un 20% del total) y la tercera causa de cáncer en los adolescentes de 15 a 19 años (aproximadamente un 10% del total)(1,2).
En España, según el Registro Nacional de Tumores (RETI), cada año se registran en torno a 1.300-1.500 casos nuevos de cáncer infantil, de los cuales un 20% corresponden a tumores del SNC(3). La incidencia anual de tumores del SNC en niños es de unos 5 casos por cada 100.000. Son ligeramente más frecuentes en varones con una ratio varón/mujer de 1,5(4,5).
Los tumores cerebrales infantiles se caracterizan por englobar un grupo heterogéneo de histologías y localizaciones, y una conducta biológica, respuesta al tratamiento y pronóstico que los hace diferentes de los diagnosticados en los adultos. Los gliomas de bajo grado son los tumores del SNC más frecuentes en la población pediátrica (30-50% de los casos), seguidos del meduloblastoma (16-25%).
Por la localización, los tumores supratentoriales son más frecuentes en niños hasta los 3 años de edad y tras los 10 años; mientras que entre los 4 y los 10 años, predominan los tumores infratentoriales (Fig. 1).
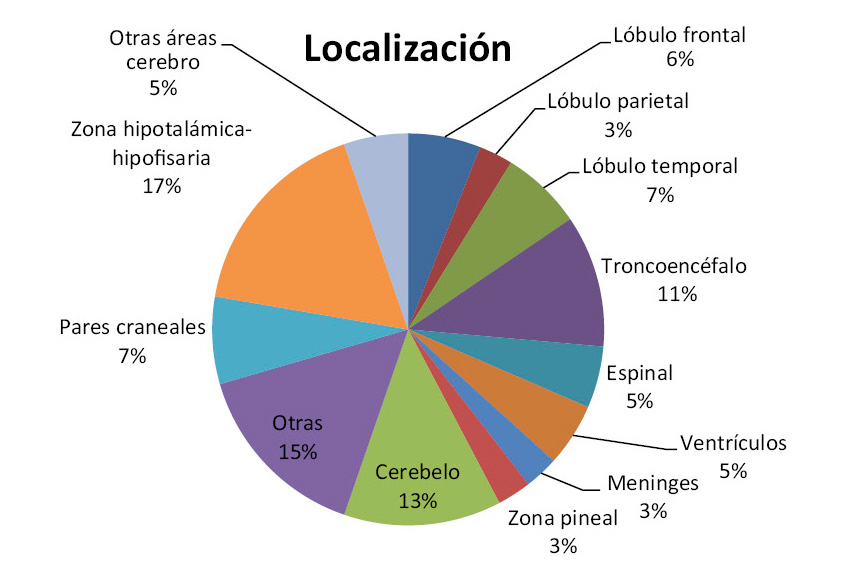
Figura 1. Distribución de los tumores del sistema nervioso central por localización, del Registro Central de Tumores Cerebrales de los EE.UU. (CBTRUS) para pacientes entre 0 a 19 años(5).
A nivel histológico, dentro de los tumores infratentoriales, los más frecuentes son: gliomas cerebelosos y troncoencefálicos, y meduloblastomas, seguidos de los ependimomas. A nivel supratentorial predominan los astrocitomas (Fig. 2).
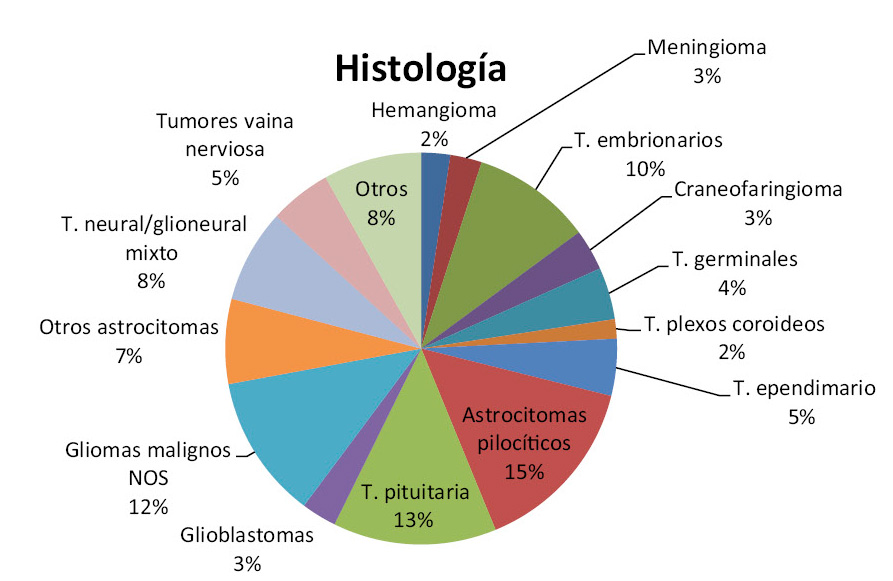
Figura 2. Distribución de los tumores del sistema nervioso central por histología, del Registro Central de Tumores Cerebrales de los EE.UU. (CBTRUS) para pacientes entre 0 a 19 años(5). NOS: no especificados / parcialmente especificados a nivel molecular.
Según los últimos datos publicados, la supervivencia media a cinco años en pacientes de 0 a 14 años diagnosticados de tumores del SNC en España se encuentra en torno al 66 +/- 8% (periodo entre 2010-2014)(6). Ha mejorado considerablemente en los últimos años, debido al diagnóstico más precoz y a los avances en los tratamientos (cirugía, biología molecular, quimioterapia y radioterapia).
Aun así, los tumores del SNC son la primera causa de muerte secundaria al cáncer en niños y adolescentes, dependiendo fundamentalmente de la histología del tumor y de su localización.
Etiología
En la mayoría de los pacientes pediátricos la causa de un tumor del SNC no está clara. Es probable que sea el resultado de una compleja interacción entre la carga genética del niño y la exposición ambiental específica.
A pesar de que la etiología de la mayoría de los tumores del SNC es desconocida, se han identificado como causa factores genéticos, así como la exposición a radiaciones ionizantes.
Factores genéticos
Síndromes de cáncer familiar
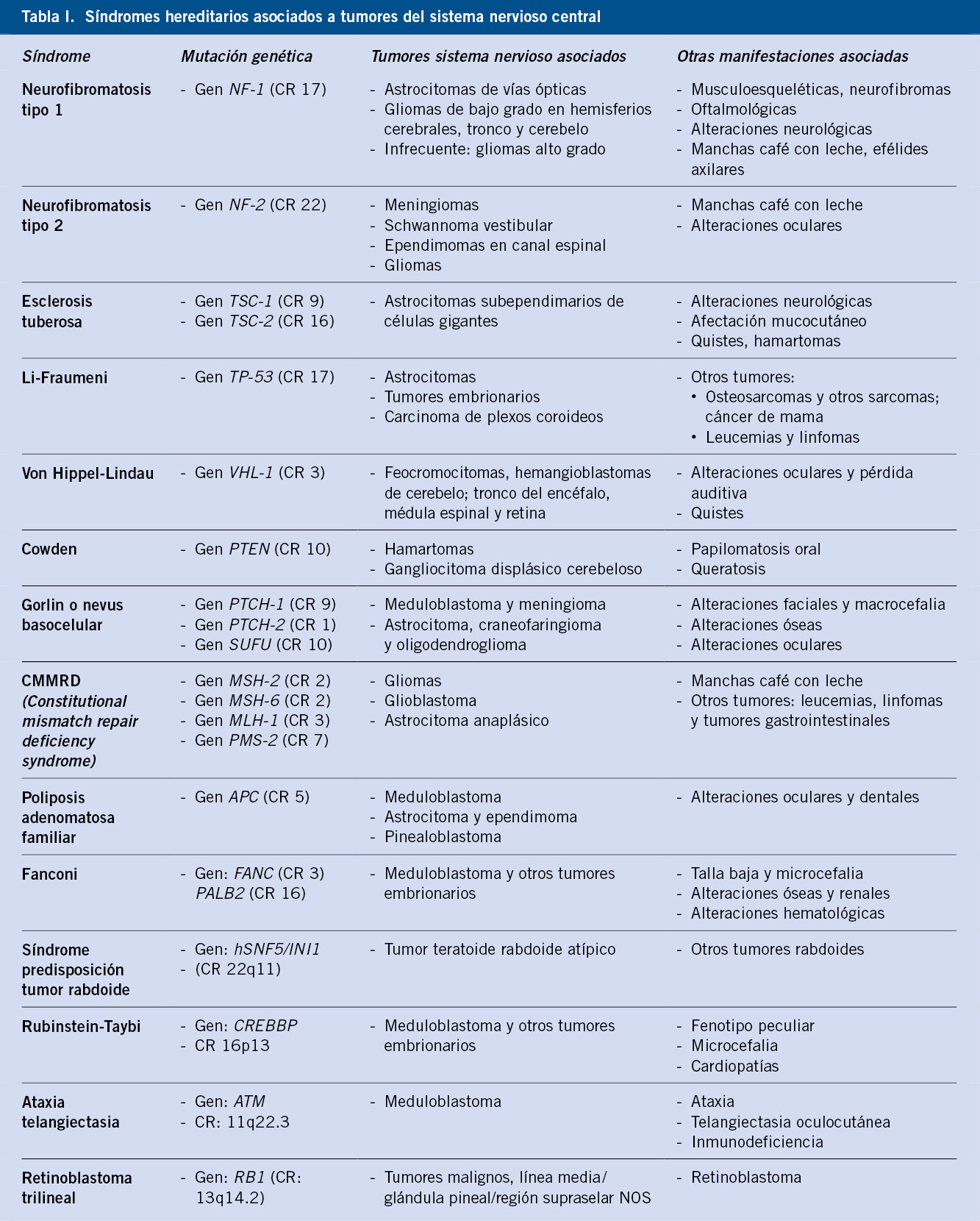
Existen diversos síndromes de cáncer familiar que aumentan la susceptibilidad a padecer tumores cerebrales, aunque menos del 10% de los niños con un tumor cerebral tienen un síndrome hereditario que les coloca en una situación de riesgo aumentado para el desarrollo de este tipo de tumores(7-9).
Estos síndromes se caracterizan por la alteración en oncogenes y genes supresores de tumores, que desencadenan la aparición de neoplasias (Tabla I). Los niños afectados por estas enfermedades hereditarias deben someterse a seguimientos periódicos para detectar precozmente el desarrollo de procesos oncológicos.
Radiaciones ionizantes
La exposición a radiaciones ionizantes intraútero, o directamente al utilizar la radioterapia craneal, es una causa bien documentada de tumores cerebrales en niños. En estos casos, la latencia entre la radioterapia y el desarrollo del tumor cerebral se estima entre 7-9 años, con un mayor riesgo en niños de menor edad(4,9,10).
Clínica
Las manifestaciones clínicas de los tumores cerebrales son variables, pudiendo derivarse de la hipertensión intracraneal o de la infiltración/compresión que pueden provocar.
Los síntomas y signos de los tumores intracraneales en el niño dependen de la edad, de la localización del tumor y de la presencia o ausencia de hipertensión intracraneal, así como de la velocidad de crecimiento del tumor. El diagnóstico de un tumor cerebral puede ser complicado en los niños, sobre todo, en los más pequeños, incapaces de referir sus síntomas. Podemos dividir los síntomas típicos según se deriven de la infiltración tumoral o aumento de presión intracraneal (Tabla II).
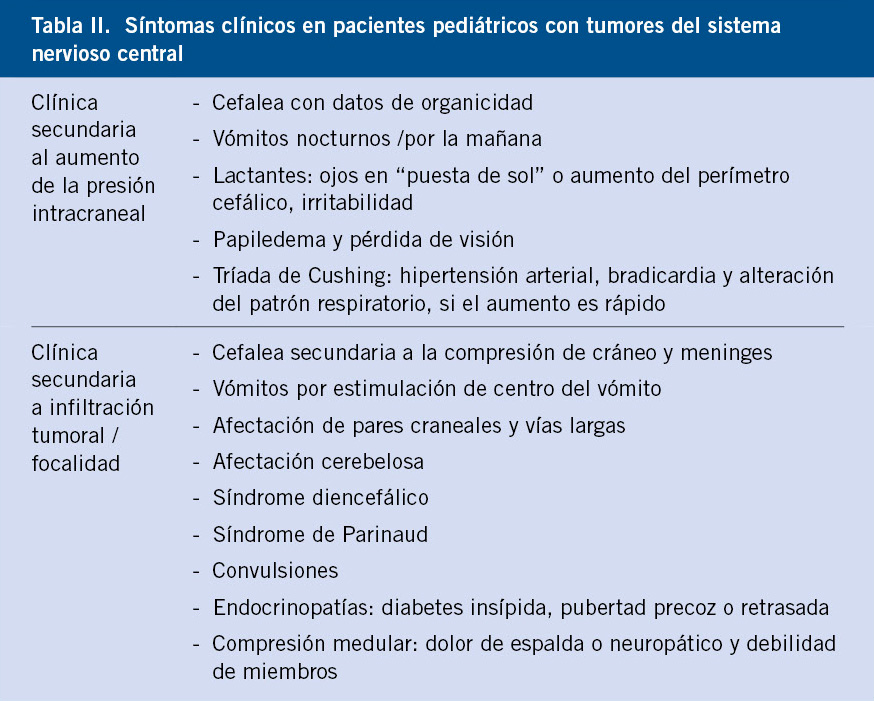
Los síntomas y signos de focalidad son más evidentes cuanto mayor es la edad del niño. La hemiparesia, hipertonía e hiperreflexia son las manifestaciones más frecuentes en los tumores supratentoriales y, en menor proporción, los trastornos de la sensibilidad. En los infratentoriales, los síntomas y signos más habituales son: diplopía, ataxia y nistagmus.
Como características clínicas específicas destacar que la primera manifestación de un tumor de fosa posterior puede ser una tortícolis y en los tumores de tronco puede haber parálisis de pares craneales, ataxia y afectación de vías largas. Las crisis epilépticas también pueden ser manifestación de un tumor cerebral.
También podríamos encontrar síntomas más inespecíficos como manifestaciones de un tumor cerebral: cambios de personalidad, alteraciones emocionales, disminución del rendimiento escolar, así como la detención o el retroceso del desarrollo psicomotor en niños pequeños.
Diagnóstico
La anamnesis y exploración clínica son esenciales para sospechar un tumor del SNC en un paciente y solicitar las pruebas complementarias pertinentes(4,11,12).
Pruebas de imagen
La tomografía computarizada (TC) y la resonancia magnética (RM) son la base del diagnóstico, tratamiento, planificación quirúrgica y seguimiento.
Tomografía computarizada (TC)
La TC proporciona, de forma rápida, datos sobre: presencia o ausencia de tumor, tamaño del mismo, forma y densidad tumoral, localización, comportamiento tras la administración de contraste, presencia de calcificaciones, zonas de necrosis y quistes, edema peritumoral, desplazamientos y herniaciones cerebrales, afectación de estructuras óseas, presencia de hidrocefalia y hemorragia tumoral, entre otras.
Resonancia magnética (RM)
Las principales ventajas de la RM son su capacidad multiplanar y la alta resolución de la imagen, permitiendo un mejor estudio de las características de la lesión, así como diferenciar los distintos tejidos del SNC. Las posibles desventajas de la RM son el largo tiempo de exploración y la degradación de la imagen si existe movimiento, por lo que precisa gran colaboración por parte del paciente o bien su anestesia.
Algunos tumores cerebrales, como: los meduloblastomas y otros tumores embrionarios, los tumores de células germinales y los ependimomas, pueden diseminarse dentro del SNC. Ante la sospecha de estos tumores, se debe ampliar el estudio de neuroimagen al resto del neuroeje.
Se recomienda que la RM de columna se realice antes de la intervención quirúrgica, para disminuir el número de falsos positivos secundarios a restos de sangre.
Si la RM de columna se realiza después de la cirugía, es recomendable esperar, al menos, dos semanas(4).
Punción lumbar
El objetivo es realizar un examen citológico y detectar células tumorales en líquido cefalorraquídeo (LCR) en los tumores con tendencia a diseminar, imposibles de identificar en ninguna prueba de imagen. La presencia de células tumorales en LCR condiciona la asignación a una determinada categoría de tratamiento.
Marcadores tumorales
En determinados tumores localizados en la región pineal/ hipotalámica-hipofisaria o ganglios basales en los cuales podamos sospechar un tumor de células germinales, es obligada la determinación de marcadores tumorales en sangre y LCR, como la alfa-fetoproteína (AFP) y la gonadotropina coriónica (βHCG). Estos son importantes para la orientación sobre el tipo tumoral, la respuesta al tratamiento y la detección de recidivas.
Biopsia/cirugía
Será necesario en la casi totalidad de los tumores, con dos objetivos principales: 1) establecer un diagnóstico de certeza; y 2) reducir el volumen tumoral, esencial para la ulterior eficacia de la radioterapia y/o quimioterapia.
Valoración neuropsicológica
Es esencial para conocer la afectación del paciente por el propio tumor. Posteriormente permite también valorar el daño causado por la resección tumoral y los efectos adversos de los tratamientos coadyuvantes, fundamentalmente de la radioterapia y quimioterapia.
Clasificación y tipos de tumores más relevantes
La clasificación de los tumores cerebrales es compleja, dado que muchos tumores pediátricos del SNC pueden tener múltiples tipos de células.
Las clasificaciones anteriores trataban de identificar el aspecto morfológico por la histología y los estadios de desarrollo dentro del sistema nervioso. Así surge la clasificación de los tumores del SNC de la Organización Mundial de la Salud (OMS) de 2007, que se basó principalmente en el aspecto histológico(13). Sin embargo, debido al mayor conocimiento en la biología de los tumores, en 2016, esta clasificación se modificó para incorporar algunos parámetros moleculares, manteniendo el aspecto histológico(14). Este enfoque molecular ha continuado mejorando, provocando nuevos cambios ya recogidos en la recién publicada clasificación de 2021 de la OMS, permitiendo una mejor identificación de la historia natural de los diversos tipos de tumores a nivel biológico y molecular. Estas clasificaciones actualizadas permitirán a los clínicos conocer mejor el pronóstico y la terapia óptima para los pacientes con tumores del SNC, así como incluir poblaciones más homogéneas de pacientes en ensayos clínicos, lo que facilitará la evaluación de nuevas terapias(15).
Además de la histopatología, los tumores del SNC también se pueden clasificar según su localización en supratentoriales e infratentoriales.
A continuación, se señalan los tumores más relevantes en la edad pediátrica por su frecuencia.
Meduloblastoma (Fig. 3)

Figura 3. Secuencias de RM craneal en paciente diagnosticado de meduloblastoma. A. Secuencia coronal T2. B. Secuencia axial T2.
El meduloblastoma es, por definición, un tumor embrionario de la fosa posterior y se considera el tumor cerebral maligno más frecuente en los niños. Comprende hasta un 20% de todos los tumores cerebrales pediátricos.
Hay una incidencia bifásica, con pico a los 3 o 4 años de edad y de nuevo a los 8 o 9 años. Anteriormente, en la clasificación de la OMS de 2007, el meduloblastoma se dividía en 4 variantes histológicas, incluyendo: la histología clásica, la variante anaplásica/de células grandes, la desmoplásica/nodular y el meduloblastoma con nodularidad extensa(13). En 2016, el sistema de clasificación de la OMS cambió, y el meduloblastoma se subdividió también por subgrupos moleculares: WNT, Sonic Hedgehog (SHH), y grupo no WNT/no SHH, en un sistema de 2 niveles que incorporaba tanto la histología como los hallazgos moleculares(14). En la nueva clasificación de 2021 se mantiene la subdivisión molecular, añadiendo el papel del estado de TP53 en el sub grupo SHH(15,16).
Los tratamientos tradicionales para el meduloblastoma en niños mayores de 3 años incluyen una combinación de cirugía, radioterapia cráneo-espinal y esquemas de quimioterapia basados en platinos, independientemente del subtipo molecular. La resección quirúrgica es el paso inicial en el tratamiento y se conoce que la resección total o casi total del tumor primario se correlaciona con mejores resultados, especialmente en pacientes con enfermedad no diseminada. Tras la resección máxima, los pacientes se estratifican convencionalmente en riesgo estándar o alto, en función de la extensión de la resección quirúrgica y el estado de diseminación en el momento del diagnóstico, dado por la RM cráneo-espinal del diagnóstico y el estudio del LCR realizado como mínimo a los 15 días de la cirugía. La supervivencia libre de enfermedad a los cinco años es superior al 80% en aquellos de riesgo estándar, y del 60% al 65% en los de riesgo alto después de dicho tratamiento.
En los niños más pequeños, principalmente los que tienen menos de 3 o 4 años en el momento del diagnóstico, el tratamiento posquirúrgico suele realizarse con quimioterapia solamente, incluyendo altas dosis con rescate de progenitores hematopoyéticos, debido al impacto que las altas dosis de radiación pueden tener en el cerebro en desarrollo.
Gliomas
Gliomas de bajo grado (LGG-Low grade glioma)
Los LGG son el tumor cerebral más frecuente en los niños y representan aproximadamente el 40% de todos los tumores del SNC en menores de 18 años(5).
Los astrocitomas pilocíticos son los más frecuentes, pero los LGG pediátricos incluyen una amplia gama de subtipos según el patrón histológico/molecular que presenten. Estas subclasificaciones son esenciales para su mejor comprensión clínica, y posterior actitud terapéutica(15).
Los gliomas de bajo grado pueden localizarse en cualquier estructura del SNC. La localización más frecuente es la fosa posterior (20-25%) (Fig. 4).
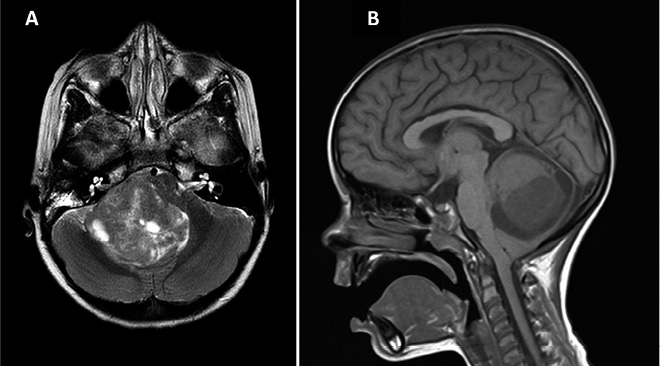
Figura 4. Secuencias RM de paciente diagnosticado de astrocitoma pilocítico de fosa posterior. A. Secuencia axial T2. B. Secuencia sagital T1.
Otras localizaciones típicas son el tronco cerebral o la línea media supratentorial, que incluye: vías ópticas, región supraselar, “tectum” mesencefálico y región talámica. La presentación clínica depende de la localización del tumor. Los gliomas de bajo grado de vía óptica se asocian con frecuencia a neurofibromatosis tipo 1(8). La resección quirúrgica es curativa para los LGG, especialmente los astrocitomas pilocíticos, cuando son resecados totalmente.
El reto terapéutico surge cuando los tumores aparecen cerca de estructuras vitales, lo que dificulta la resección sin causar una morbilidad o un compromiso funcional significativo. En tumores no resecables o con resección incompleta que requieran tratamiento, –por ejemplo si producen síntomas por compresión de estructuras vecinas–, se empleará la quimioterapia como primera opción, con la combinación de carboplatino y vincristina, o vinblastina semanal, intentando no emplear la radioterapia por los daños que pueden provocar a medio-largo plazo(11).
Los avances recientes en secuenciación genética y perfil de expresión génica han derivado en un mejor entendimiento de las vías moleculares que están presentes en la patogenia de los gliomas. Las alteraciones más frecuentes conocidas hasta el momento son las que implican al gen BRAF, destacando el de fusión entre el dominio activador de MAPK y KIAA1549 y la mutación BRAF V600E(17).
Gliomas de alto grado
En los niños, los gliomas de alto grado tienen una incidencia de 0,8 por cada 100.000 niños/año(5). Dentro de ellos destacamos el DIPG (gliomas difusos de la protuberancia). Junto con la clínica e imagen en RM asociada, estos tumores pueden diagnosticarse sin confirmación histológica. Clínicamente, los pacientes suelen presentar múltiples neuropatías craneales, más comúnmente parálisis del sexto y/o séptimo nervio par craneal. El pronóstico es desalentador, ya que más del 90% de los pacientes sucumben a la enfermedad en un plazo de 18 meses, con una supervivencia media de 9 meses. El tratamiento estándar incluye la radioterapia y ha sido el único tratamiento que ha alterado el curso de la enfermedad(11).
Ependimomas
Los ependimomas son el tercer tumor cerebral más frecuente en los niños y representan aproximadamente entre el 8 y el 10% de todos los tumores del SNC infantil. En los niños, la mayoría se producen a nivel intracraneal y dos tercios de ellos se localizan en la fosa posterior (Fig. 5).
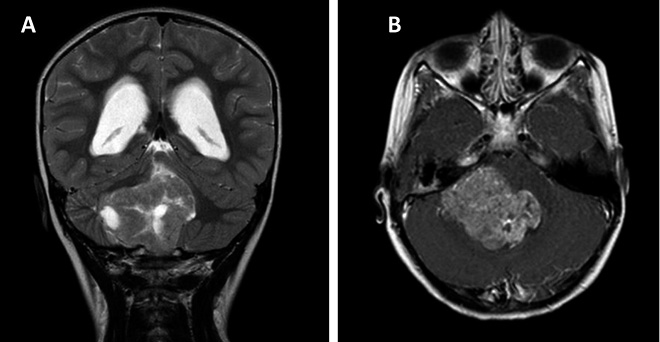
Figura 5. RM de paciente diagnosticado de ependimoma de fosa posterior.
A. Secuencia coronal T2. B. Secuencia axial con gadolinio.
Actualmente, la OMS clasifica los ependimomas en función de su histopatología, localización y alteraciones moleculares (subependimoma, ependimoma mixopapilar, ependimoma de fosa posterior, con subtipos PFA y PFB, ependimoma supratentorial, con subtipos con fusión positiva YAP-1 y ZFTA, así como ependimoma espinal con/sin NMYC amplificado)(15). El estándar de tratamiento sigue siendo la máxima resección quirúrgica segura, seguida de radioterapia postoperatoria adyuvante dirigida al sitio primario (en estos casos se administra a los mayores de un año porque es local). La diseminación en el momento del diagnóstico se observa en menos del 10% de los casos. La utilidad de la quimioterapia es controvertida y se suele utilizar para retrasar el uso de la radioterapia. Su papel se está evaluando actualmente en ensayos clínicos internacionales.
Craneofaringioma
Los craneofaringiomas son tumores epiteliales benignos de crecimiento lento que surgen de restos embrionarios de la bolsa de Rathke en la región supraselar adyacente al quiasma óptico. Suponen entre el 5 y el 10% de los tumores cerebrales pediátricos.
Pueden provocar alteraciones de la visión, panhipopituitarismo por compresión de la glándula pituitaria o del tallo, así como hipertensión intracraneal.
Los tumores son, con frecuencia, grandes en el momento del diagnóstico, clásicamente con calcificaciones en la región supraselar, y con presencia de uno o más quistes. Se dividen en papilares y adamantinomatosos, siendo estos los que tienen mayores tasas de recurrencia.
Aunque se consideran tumores histológicamente benignos, siguen siendo un reto con respecto a las opciones de tratamiento, debido a la proximidad de estructuras vitales y al manejo a largo plazo de los efectos tardíos relacionados con el tratamiento. La transformación maligna es extremadamente rara(18).
Tumores de células germinales del SNC
Los tumores de células germinales (TCG) intracraneales representan aproximadamente el 3% de los tumores cerebrales pediátricos –llegando hasta el 11% en países asiáticos como Japón–. Suelen aparecer en localizaciones de la línea media, como la región pineal o el área supraselar.
El sistema de clasificación de la OMS divide los TCG intracraneales en germinomas –los más frecuentes, entre 50-60% de todos ellos– y TCG no germinomatosos (TCGNG), evidenciando entre estos un grupo heterogéneo, como el carcinoma embrionario, el tumor del seno endodérmico (tumor del saco vitelino), coriocarcinoma, teratoma (maduro e inmaduro) y TCG mixtos(15).
Aunque el examen histológico suele ser necesario para un diagnóstico definitivo, los marcadores tumorales como la AFP o beta-hCG en el LCR o en la sangre, pueden ser diagnósticos y distinguir un germinoma de un TCGNG.
En algunos germinomas secretores de beta-hCG puede observarse una leve elevación de la beta-hCG, pero tienden a ser <50 UI/L. La AFP no está elevada en los germinomas, por lo que una AFP elevada es suficiente para clasificar un tumor como un TCGNG.
Los síntomas que se presentan dependen de la localización del tumor. Los tumores pineales suelen causar hidrocefalia o anomalías neurooftalmológicas. Los localizados en la región supraselar suelen presentar disfunción hipotalámica o hipofisaria, así como déficits visuales por compresión del quiasma o del nervio óptico.
En relación al tratamiento, los germinomas tienen un pronóstico más favorable, son muy sensibles a la radiación, así como quimiosensibles, con una tasa de curación superior al 90% a los 5 años del diagnóstico. Los TCGNG, por otra parte, son menos radiosensibles y presentan peor pronóstico, precisando tratamiento sistémico con diferentes regímenes quimioterápicos junto con radioterapia posterior(19).
Tumores plexos coroideos
Los tumores de plexos coroideos representan menos del 1% de todos los tumores cerebrales y del 3 al 4% de los tumores intracraneales pediátricos.
Son neoplasias papilares intraventriculares derivadas del epitelio del plexo coroideo. Histológicamente, pueden variar desde benignos bien diferenciados (papiloma de plexos coroideos, CPP), hasta carcinomas del plexo coroideo (CPC) altamente agresivos (grado III de la OMS).
El CPC ocurre con mayor frecuencia en pacientes con el síndrome de Li-Fraumeni, aunque la mayoría de los pacientes con CPC no tendrán una mutación en la línea germinal del TP53.
En relación al tratamiento, la resección total suele ser curativa para el CPP, mientras que la naturaleza invasiva del CPC dificulta lograr una resección completa, lo que hace necesario el uso de una terapia adyuvante. El pronóstico sigue siendo malo, con una supervivencia a 5 años inferior al 50% en los pacientes con CPC(20).
Tumores teratoides/rabdoides atípicos (ATRT)
Los ATRT son neoplasias intracraneales malignas infrecuentes –del 1 al 2% de todos los tumores cerebrales pediátricos–, con mayor incidencia en lactantes y niños pequeños. El sello genético de los ATRT son las mutaciones en SMARCB1 en el tejido tumoral. Además, aproximadamente un tercio de los pacientes albergan una mutación en la línea germinal de SMARCB1, o menos comúnmente de SMARCA4.
El tratamiento del ATRT es controvertido. Se están aplicando estrategias con cirugía, quimioterapia, incluyendo altas dosis, más rescate de progenitores hematopoyéticos y radioterapia local, ya que la temprana edad de presentación suele impedir el uso de la radiación craneoespinal. Aunque, en general, los ATRT tienen un pronóstico muy malo, hay algunos con mejores resultados clínicos, lo que sugiere una heterogeneidad molecular intertumoral(21).
Tumores espinales
Los tumores primarios de la médula espinal son tumores raros del SNC en la infancia, con una incidencia anual de menos de 1 por cada 100.000 niños/año y representan menos del 6% de todos los tumores del SNC en la infancia(5). Pueden clasificarse en función de su localización: intramedular, intradural-extramedular y extradural. Los síntomas suelen tener un inicio gradual y pueden ser especialmente difíciles de detectar en niños pequeños.
El subtipo más común a este nivel son los gliomas, seguidos de los ependimomas (Fig. 6).
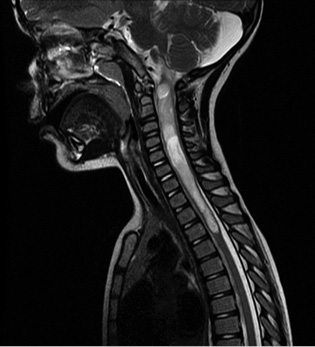
Figura 6. Secuencia RM sagital T2 de paciente con glioma espinal de bajo grado.
La resección quirúrgica sigue siendo el pilar del tratamiento. Cuando no se puede obtener una resección completa, la radioterapia y la quimioterapia suelen ser eficaces en el control de estos tumores, según el diagnóstico histológico, pero normalmente se reservan para los que progresan tras una cirugía extensa.
Tratamiento general
El tratamiento de los tumores del SNC en los niños requiere un abordaje multidisciplinar.
Existen diversas opciones de tratamiento que, fundamentalmente, son: cirugía, quimioterapia y radioterapia, uniéndose recientemente la terapia personalizada en relación a dianas terapéuticas, que pueden encontrarse en el estudio molecular del tumor. El tratamiento local es esencial por la baja penetrancia de la quimioterapia a través de la barrera hematoencefálica.
Hay que destacar que el tratamiento de los tumores del SNC en los niños requiere un abordaje multidisciplinar, coordinando las diversas especialidades que colaboran en el tratamiento mediante comités de Neuro-Oncología pediátrica para decidir la mejor terapia de cada paciente. Es necesaria además la colaboración entre unidades de Oncología pediátrica a nivel nacional e internacional creando protocolos, así como diseñando ensayos clínicos orientados a pacientes con esta patología. Por todo ello, se recomienda que el tratamiento de los tumores cerebrales en niños y adolescentes se realice en unidades de Neuro-Oncología pediátricas de referencia y alta especialización. Deben ponerse a disposición de los pacientes las mejores técnicas neuro-quirúrgicas, de radioterapia y los tratamientos más completos e innovadores (inmunoterapia o tratamientos dirigidos) para alcanzar las mayores tasas de curación, con especial atención a las secuelas de los tratamientos a medio-largo plazo.
En relación a las técnicas de radioterapia actualmente se cuenta con la protonterapia, que permite reducir toxicidad en los pacientes pediátricos, manteniendo las mismas tasas de curación, lo cual posibilita ampliar las indicaciones de la radioterapia convencional con fotones(22).
Efectos a medio-largo plazo tras el diagnóstico y tratamiento
Más de dos tercios de los pacientes supervivientes a largo plazo presentan al menos una complicación médica crónica, siendo las neurocognitivas las más deletéreas(23).
La supervivencia de los pacientes con tumores del SNC ha mejorado de manera significativa en las últimas décadas, asociando un incremento de los efectos tardíos secundarios al tumor, a las técnicas diagnósticas y a los tratamientos(24). Destacan los que se citan a continuación.
Secuelas neurocognitivas
Las alteraciones neurocognitivas son uno de los efectos tardíos más devastadores en los pacientes con tumores cerebrales; varían desde niveles leves a graves de deterioro y pueden persistir y/o empeorar con el tiempo, habiéndose descrito dificultades de atención y concentración, alteraciones de la memoria de trabajo, de la velocidad de procesamiento y del lenguaje.
La radioterapia es el tratamiento considerado más tóxico para el cerebro en desarrollo con respecto al funcionamiento neuropsicológico. Una mayor dosis de radiación y un mayor volumen cerebral irradiado se asocian con peores resultados, especialmente en los niños más pequeños(24).
Alteraciones endocrinas (Tabla III)

El riesgo de determinados efectos endocrino-metabólicos depende de la localización del tumor y de las modalidades de tratamiento que se hayan utilizado, especialmente quimio-radioterapia(23,24).
La evaluación periódica por Endocrinología debe formar parte del seguimiento a largo plazo de los supervivientes de tumores cerebrales infantiles. El diagnóstico precoz de hipotiroidismo leve y/o deficiencia de GH, por ejemplo, permite una intervención temprana para mejorar la velocidad de crecimiento y la calidad de vida.
Secuelas psicológicas/sociales
Los supervivientes de tumores cerebrales pediátricos tienen un mayor riesgo de padecer morbilidad psicológica a largo plazo, como: depresión, ansiedad, menos autoestima, ideas suicidas y problemas de conducta, en comparación con la población general(20). También, tienen peor calidad de vida y experimentan un mayor aislamiento y una peor adaptación social. Tienen menores tasas de obtención de un título universitario, de empleo a tiempo completo y de vida independiente, en comparación con la población general.
Déficits neurosensoriales
Los déficits neurosensoriales pueden producirse como efecto directo del tumor o como efecto tardío de los tratamientos:
• La pérdida de visión se asocia a los tumores de la vía óptica o a la hidrocefalia.
• La pérdida de audición neurosensorial puede producirse tras la quimioterapia y la radioterapia, o surgir y progresar mucho después de la finalización del tratamiento. Los pacientes afectos a este nivel podrían desarrollar un mayor declive en funciones críticas para la competencia lingüística (conciencia fonémica, comprensión lectora y velocidad de procesamiento)(24).
Segundas neoplasias
Las neoplasias secundarias más frecuentes en estos pacientes son tumores del sistema nervioso central, particularmente los astrocitomas malignos. El desarrollo de los segundos tumores es probablemente de origen multifactorial, pero la radioterapia contribuye sin duda a este proceso(23).
Otros efectos tardíos
Los supervivientes de tumores cerebrales, además de las complicaciones mencionadas, presentan riesgo de desarrollar enfermedad cerebrovascular en forma de ictus o accidente isquémico transitorio (AIT), vasculopatía de pequeño vaso o enfermedad de Moya-Moya. Asimismo, es frecuente que estos pacientes presenten secuelas motoras, alteraciones del sueño y alteraciones de la dentición.
Función del pediatra de Atención Primaria
Además de tener un papel esencial en la detección precoz, los profesionales de Atención Primaria deben ser considerados una parte fundamental de los equipos multidisciplinares encargados del seguimiento de los supervivientes de tumores cerebrales.
El pediatra de Atención Primaria ejerce un papel fundamental para conseguir un diagnóstico precoz de la patología neuro-oncológica, sospechándolo en caso de variaciones conductuales, tortícolis, alteraciones en la revisión ocular rutinaria, presencia de vómitos atípicos… En estos casos, la derivación a centros especializados precoz es crucial. De igual modo, tras el final del tratamiento de los pacientes, los pediatras de Atención Primaria deberían recibir un informe detallado sobre el diagnóstico y el tratamiento recibido, así como del plan de seguimiento.
Debe concentrarse en la monitorización de los efectos tardíos, y vigilar la posibilidad de recurrencias o de aparición de segundas neoplasias. Además, promover hábitos de bienestar y conductas de salud positivas es muy importante, fomentando hábitos, como evitar el consumo de tabaco, limitar la ingesta de alcohol, realizar ejercicio físico de forma regular y minimizar la exposición solar, especialmente de zonas que han recibido radioterapia(25).
La colaboración continua entre los especialistas en Oncología y los equipos de Atención Primaria es fundamental para realizar un correcto diagnóstico, tratamiento y seguimiento, así como apoyar la transición de estos pacientes a la edad adulta.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014; 64: 83-103.
2.*** Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol. 2015; 33: 2986-98.
3. Registro Nacional de Tumores Infantiles RNTI-SEHOP. Cáncer infantil en España: preguntas y datos. Disponible en: https://www.uv.es/rnti/.
4. Reynolds R, Grant G. General approaches and considerations for pediatric brain tumor. En: Winn HR, ed. Youmans neurological surgery. Elsevier; 2011. p 2040-46.
5. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol. 2020; 22: 1-96. Disponible en: doi:10.1093/neuonc/noaa200.
6.*** Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikši? M, et al. CONCORD Working Group. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018; 391: 1023-75.
7. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015; 373: 2336-46.
8.*** Villani A, Malkin D, Tabori U. Syndromes Predisposing to Pediatric Central Nervous System Tumors: Lessons Learned and New Promises. Curr Neurol Neurosci Rep. 2012; 12: 153-64.
9. Johnson KJ, Cullen J, Barnholtz-Sloan JS, Ostrom QT, Langer CHE, Turner MC, et al. Childhood brain tumor epidemiology: a brain tumor epidemiology consortium review. Cancer Epidemiol Biomarkers Prev. 2014; 23: 2716-36.
10. Ron E, Modan B, Boice JD Jr, Alfandary E, Stovall M, Angela Chetrit A, et al. Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med. 1988; 319: 1033-9.
11.*** Udaka YT, Packer RJ. Pediatric Brain Tumors. Neurol Clin. 2018; 36: 533-56. doi: 10.1016/j. ncl.2018.04.009.
12. Villarejo Ortega F, Aransay García A, Márquez Pérez, T. Tumores cerebrales en niños. Pediatr Integral. 2016; XX(6): 401-11.
13. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114: 97-109.
14. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. WHO classification of tumours of the central nervous system, revised. 4th edition. World Health Organization; 2016.
15.*** Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021; 23: 1231-51. doi: 10.1093/neuonc/noab106.
16.*** Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017; 547: 331-7.
17. Kilday JP, Bartels UK, Bouffet E. Targeted therapy in pediatric low-grade glioma. Curr Neurol Neurosci Rep. 2014; 14: 441.
18. Müller HL, Merchant TE, Warmuth-Metz M, Martínez-Barbera JP, Puget S. Craniopharyngioma. Nat Rev Dis Primers. 2019; 5: 75. doi: 10.1038/s41572-019-0125-9.
19. Denyer S, Bhimani AD, Patil SN, Mudreac A, Behbahani M, Mehta AI. Treatment and survival of primary intracranial germ cell tumors: a population-based study using SEER database. J Cancer Res Clin Oncol. 2020; 146: 671-85. doi: 10.1007/s00432-019-03088-7.
20. Dudley RW, Torok MR, Gallegos D, Liu AK, Handler MH, Hankinson TC. Pediatric choroid plexus tumors: epidemiology, treatments, and outcome analysis on 202 children from the SEER database. J Neurooncol. 2015; 121: 201-7. doi: 10.1007/s11060-014-1628-6.
21. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016; 29: 379-93.
22. Thomas H, Timmermann B. Paediatric proton therapy. Br J Radiol. 2020; 93: 20190601. doi: 10.1259/bjr.20190601.
23. Bouffet E, Scheinemann K. Pediatric Neuro-oncology. Springer. 2015.
24. Rey-Casserly C, Diver T. Late effects of pediatric brain tumors. Current opinion in pediatrics. 2019; 31: 789-96.
25. Mendoza Sánchez MC. Seguimiento en Atención Primaria del niño oncológico. Cómo detectar las secuelas tardías. Pediatr Integral. 2016; XX: 475-84.
Bibliografía recomendada
- Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol. 2015; 33: 2986-98.
Artículo de revisión en el que se muestran los importantes avances en el conocimiento molecular de los tumores cerebrales pediátricos y cómo estos conocimientos están transformando el diagnóstico e incluso en el tratamiento de los mismos.
– Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikši? M, et al. CONCORD Working Group. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018; 391: 1023-75.
Interesante registro de los tumores en Europa, clasificado por países en los que observamos como los resultados para los tumores cerebrales pediátricos difieren mucho según los países del estudio.
– Villani A, Malkin D, Tabori U. Syndromes Predisposing to Pediatric Central Nervous System Tumors: Lessons Learned and New Promises. Curr Neurol Neurosci Rep. 2012; 12: 153-64.
Excelente revisión de los síndromes de predisposición al cáncer en relación con los tumores cerebrales pediátricos.
– Udaka YT, Packer RJ. Pediatric Brain Tumors. Neurol Clin. 2018; 36: 533-56. doi: 10.1016/j. ncl.2018.04.009.
Revisión exhaustiva de los tumores cerebrales pediátricos, firmada por el prestigioso neuro-oncólogo pediátrico norteamericano Roger Packer. Muy recomendable para una primera lectura general, para entender mejor estos tumores.
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017; 547: 331-7.
El meduloblastoma es el mayor ejemplo de la transformación tan importante que han tenido los tumores cerebrales pediátricos en cuanto al conocimiento de su biología y su nueva clasificación a nivel molecular.
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021; 23: 1231-51. doi: 10.1093/neuonc/noab106.
Artículo esencial para conocer la actual clasificación de la OMS en tumores del SNC, aunando la histopatología con la biología molecular y genética.
| Caso clínico |
|
Paciente de 12 años, varón, que acude a urgencias por clínica de vómitos (2-3 vómitos diarios de predominio matutino y con cambios posturales), astenia y cefalea ocasional de 2 semanas de evolución, junto con tortícolis derecha en los últimos 3 días. Antecedentes personales Embarazo controlado normal. Parto vaginal instrumentado (fórceps) a la 39+1 semanas. Apgar: 9/10. No alergias conocidas. Calendario vacunal correcto, incluida vacuna contra neumococo y rotavirus. Antecedentes familiares Sin interés para el proceso actual. Exploración física inicial Saturación de oxígeno: 99%; peso: 46 kg; pulso: 110 latidos por minuto; temperatura: 36,8ºC; tensión sistólica: 119 mmHg; tensión diastólica: 72 mmHg. Buen estado general. Bien perfundido e hidratado, buen relleno capilar. Sin exantemas ni petequias. Auscultación cardio-pulmonar normal sin dificultad respiratoria. Abdomen blando, depresible, sin masas ni megalias, no doloroso a la palpación. Exploración neurológica GCS (Glasgow Coma Scale): 15/15. Consciente y orientado. Lenguaje coherente y fluido. Pupilas normorreactivas e isocóricas. Movimientos oculares conservados. Pares craneales sin alteraciones. Tono muscular normal. Fuerza 5/5 en ambos MMSS y MMII. ROT presentes y simétricos. RCP (respuesta cutánea plantar) flexor bilateral. Clonus negativo. Sensibilidad conservada por dermatomas. Ataxia y dismetría en miembro superior derecho. Evolución Ante la clínica referida y los hallazgos en la exploración física, se realiza analítica completa de sangre (hemograma, bioquímica y coagulación normal), así como tomografía computerizada (TC) craneal, objetivando lesión ocupante de espacio, compatible con tumor de fosa posterior e hidrocefalia. Se realiza ventriculostomía endoscópica de suelo del tercer ventrículo, con gran mejoría de síntomas; se completa estudio de imagen con resonancia magnética (RM) cráneo espinal, mostrando tumoración de fosa posterior sin signos de diseminación craneoespinal (Fig. 6), y se inicia terapia corticoidea sistémica. Tras ello, se realiza cirugía que transcurre sin incidencias, comprobándose resección macroscópicamente completa en RM craneal a las 48 horas de la cirugía. La anatomía patológica es compatible con meduloblastoma desmoplásico nodular subgrupo molecular Sonic Hedgehog (SHH).
|

 Follow-up of childhood cancer in Primary Care. How to detect late effects
Follow-up of childhood cancer in Primary Care. How to detect late effects