 |
| Temas de FC |
V. Galán Gómez*, A. Pérez Martínez**
*Médico Adjunto Servicio de Hemato-Oncología Pediátrica. **Jefe del Servicio de Hemato-Oncología Pediátrica. Hospital Infantil Universitario La Paz. Madrid
| Resumen
El síndrome hemofagocítico es una entidad clínica poco frecuente. Existen formas primarias (a menudo, cuadros graves que se presentan a edades tempranas) y formas adquiridas, de gravedad variable, secundarias a inmunodeficiencias, metabolopatías, infecciones, neoplasias… El origen de este cuadro se sustenta en una respuesta inmune exagerada, mediada por linfocitos T citotóxicos (CTL) y células natural killer (NK), hiperestimulados de manera continua por un ambiente inflamatorio. El espectro clínico de presentación es variado, pudiendo mimetizar, en ocasiones, otras patologías graves, como pueden ser el fallo hepático agudo o una meningoencefalitis. El diagnóstico se realiza en función de criterios genéticos, clínicos y analíticos. A menudo, no todos los criterios se presentan a la vez, apareciendo de manera progresiva a lo largo del tiempo, por lo que es necesario mantener un nivel de sospecha continuo. Sin tratamiento, el pronóstico de la enfermedad es desfavorable. El objetivo terapéutico es el control de la inflamación a través de potentes inmunosupresores. El trasplante de progenitores hematopoyéticos (TPH) es el tratamiento de elección en las formas primarias. No existen medidas preventivas que se hayan demostrado efectivas para disminuir la frecuencia de este fenómeno. En las formas primarias en las que exista una alteración conocida, el consejo genético está indicado. |
| Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an unusual clinical entity. There are primary forms, oftentimes aggressive presenting in the first weeks of life, and those secondary to immunodeficiencies, metabolic diseases, infections, neoplasms… with variable severity. An exuberant immune response mediated by cytotoxic T lymphocytes and natural killer (NK) cells, continuously hyperstimulated by an inflammatory environment, is the origin of the disease. The clinical spectrum of presentation is varied, and can sometimes mimic other serious diseases such as acute liver failure or meningoencephalitis. The diagnosis is based on genetics, clinical and analytical criteria. Frequently, not all criteria are present at the same time, appearing progressively, thus a continuous level of suspicion is recommended. Without treatment, prognosis of the disease is unfavorable. The therapeutic objective is to control inflammation by immunosuppression. Hematopoietic stem cell transplantation (HSCT) is the treatment of choice for primary forms. There are no preventive measures that have been proved effective in reducing the frequency of this phenomenon. In primary forms where there is a known alteration, genetic counselling is indicated. |
Palabras clave: Síndrome hemofagocítico; Linfohistiocitosis hemofagocítica; Hiperinflamación; Inmunomodulación; Trasplante de progenitores hematopoyéticos.
Key words: Hemophagocytic syndrome; Hemophagocytic lymphohistiocytosis; Hyperinflammation; Immunomodulation; Hematopoietic stem cell transplantation.
Pediatr Integral 2021; XXV (6): 326.e1 – 326.e9
Síndrome hemofagocítico
Introducción
El síndrome hemofagocítico es una patología grave resultante de una desregulación inmunológica que genera un estado hiperinflamatorio.
Se denomina linfohistiocitosis hemogafocítica (HLH) o síndrome hemofagocítico a un cuadro hiperinflamatorio sistémico derivado de la desregulación del sistema inmune, en el que la activación permanente de CTL, células NK y macrófagos tisulares desarrollan el papel principal.
Estas alteraciones inmunológicas pueden aparecer de manera constitucional en el paciente (formas primarias), o por el contrario, ser desencadenadas por factores externos (formas secundarias)(1-4).
Es importante conocer su existencia de manera precoz, puesto que es una entidad amenazante para la vida por su rápida capacidad de generar afectación multiorgánica, y la instauración de un tratamiento eficaz en una fase temprana, condicionará el pronóstico del paciente.
El diagnóstico de esta entidad puede ser inicialmente complejo, debido a que para realizarlo deben cumplirse una serie de criterios clínico-analíticos que detallaremos más adelante, no siempre presentes al debut del cuadro clínico.
La terminología clásica de síndrome hemofagocítico se fundamenta en la presencia de macrófagos fagocitantes de las tres series hematológicas, así como de progenitores, presente en gran parte de los pacientes.
Epidemiología
En su mayoría son diagnosticadas formas secundarias, aunque cada vez conocemos más sobre las formas primarias.
A pesar de los avances en el conocimiento de esta enfermedad, el síndrome hemofagocítico se trata de una entidad a menudo infradiagnosticada, por lo que disponer de cifras que representen la realidad es un reto ambicioso(5).
Por otro lado, la heterogeneidad de este cuadro, debida a la gran diversidad genética, al factor étnico y a la variedad de elementos trigger o desencadenantes, condicionan de manera importante la incidencia de la enfermedad.
No existen datos pertenecientes a grandes series de pacientes que analicen específicamente incidencia, etiología y tasas de supervivencia. Además, un número importante de los datos publicados corresponden a estudios realizados sobre series asiáticas(6,7).
Un estudio sueco, realizado entre los años 2007 y 2011, reveló que la incidencia de las formas primarias se encontraba alrededor de los 1,5 casos por millón de recién nacidos vivos (superior a los datos reportados en esa misma población con anterioridad, debido a un mayor conocimiento de la enfermedad y a la modificación de los criterios diagnósticos, lo que permitió un mayor reporte de casos nuevos diagnosticados)(8).
Los datos publicados por el Texas Children’s Hospital situaban la incidencia del síndrome hemofagocítico en 1/3.000 ingresos(5,9).
En un estudio retrospectivo unicéntrico realizado en nuestro país, se recogieron un total de 22 pacientes con síndrome hemofagocítico. El 27,3% correspondía con formas primarias, mientras que el 72,7% lo hacía con formas secundarias. Dentro de este último grupo: el 68,7% fue asociado a infección (VEB el más frecuente, seguido del VHS y la leishmania); el 18,7% asociado a neoplasias hematológicas; y el 12,65% asociado a enfermedades autoinmunes(10).
Fisiopatología
La hiperestimulación del sistema inmune y las alteraciones en la citotoxicidad en las células efectoras, así como la ausencia de apoptosis de las mismas, son los pilares de la patogenia.
Independientemente del origen, el mecanismo final común que genera este cuadro es: la ausencia de actividad citotóxica frente a las células diana, la hipersecreción de citocinas y, por ende, la persistencia de los estímulos inflamatorios(1).
Es conocido el papel fundamental en la patogenia de este cuadro, que supone la actividad citotóxica de los CTL (CD8 positivos) y las células NK activadas. Estos dos linajes celulares, en condiciones fisiológicas, se encargan del control mediante su eliminación de las células infectadas o tumorales.
Las células presentadoras de antígeno (APC) activan en el proceso denominado sinapsis inmunológica a las CTL y células NK. Tras su activación, se produce la degranulación del contenido de las vesículas de las células CTL, NK y linfocitos T colaboradores (CD4 positivos). Estos gránulos contienen perforinas (perforan la membrana para que las otras enzimas puedan alcanzar el interior celular y activan la vía de las caspasas), granzimas (enzimas proteolíticas), así como otras sustancias como serglicanos y calreticulina. Este proceso es regulado mediante retroalimentación negativa a través de la apoptosis inducida por receptor, autolimitando así el fenómeno inflamatorio(1,11).
El complejo procedimiento de exocitosis requiere de la implicación de múltiples proteínas de membrana como: Lyst, Rab 27, AP3B1, Stx11 o Munc13-4(12) (Fig. 1).
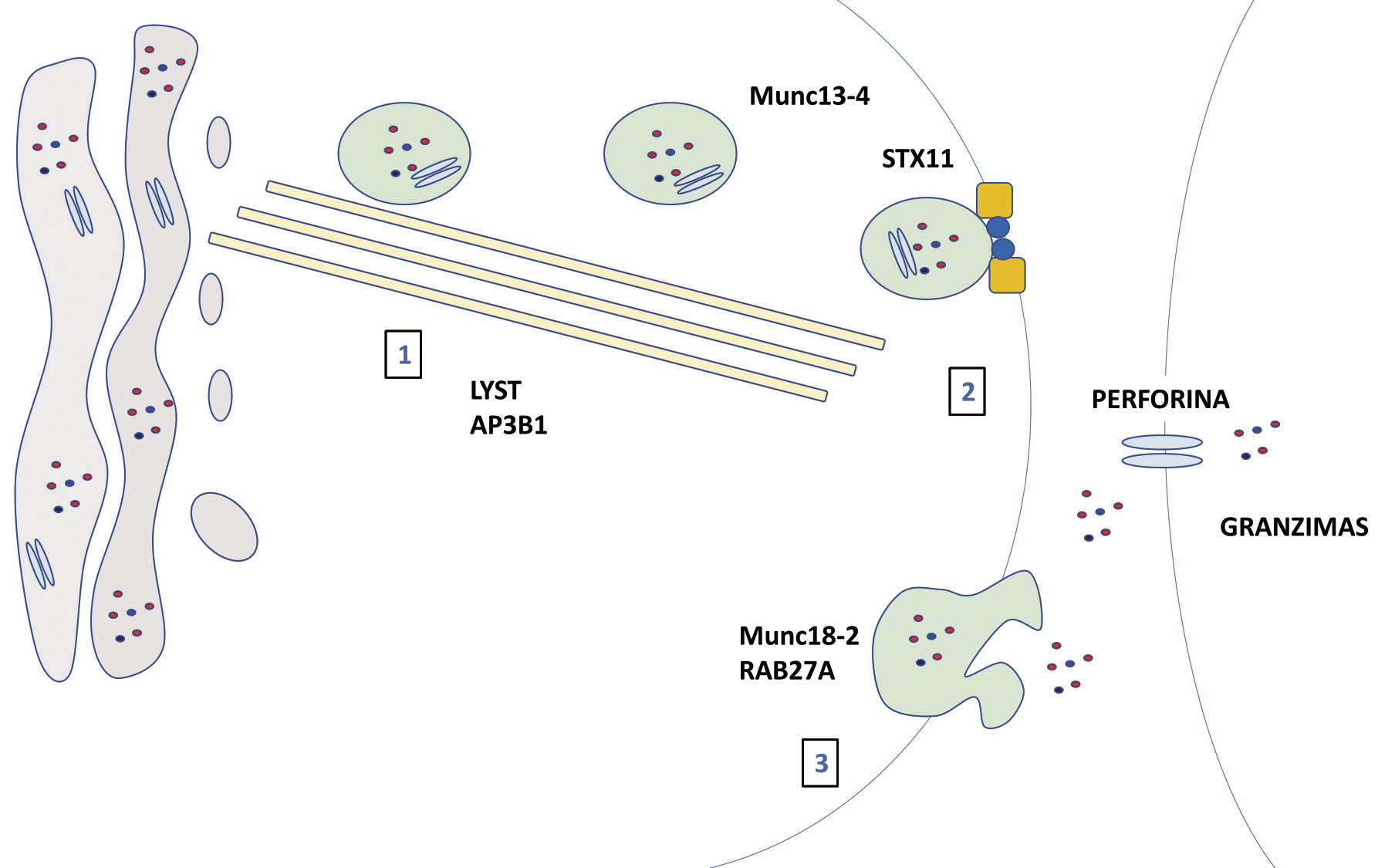
Figura 1. Esquema en el que se muestra el mecanismo fisiológico de la sinapsis inmunológica. Se representa el lugar de acción de diferentes proteínas en el proceso de secreción de las vesículas de la célula efectora, al realizar su actividad sobre la célula diana.
De esta manera, alteraciones en este proceso que anulen o produzcan un enlentecimiento en la sinapsis inmunológica, perpetuarán en el tiempo el estado inflamatorio. Las células presentadoras de antígenos continuarán activando a los linfocitos T, que continuarán, a su vez, secretando citoquinas inflamatorias y proliferando, generando de este modo un círculo vicioso inflamatorio (Fig. 2).
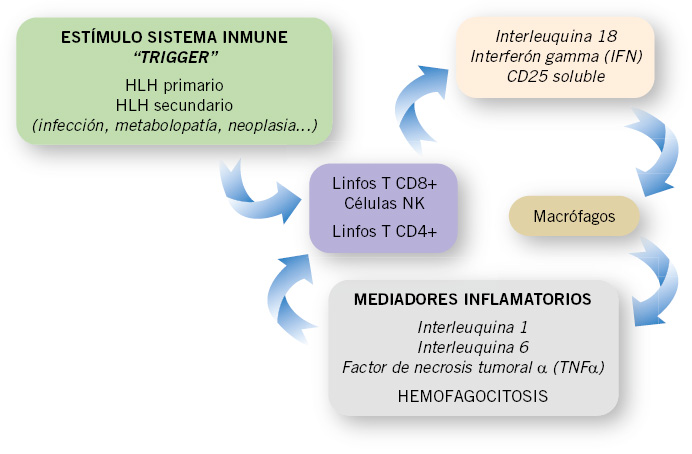
Figura 2. Esquema en el que se representan las bases fisiopatológicas de la linfohistiocitosis hemogafocítica (HLH), mediante la generación del círculo vicioso inflamatorio.
En el HLH se produce, por lo tanto, una hiperrespuesta inflamatoria favorecida por el estímulo constante de las APC y por la ausencia de retroalimentación negativa.
CTL, NK y macrófagos activados proliferan en los tejidos de manera incontrolada, favoreciendo el entorno inflamatorio mediante la secreción de mediadores inflamatorios (IL-1, IL-6, IFN gamma, TNF alfa, IL-8, IL-10, IL-18)(12). Esta infiltración es la causa de las megalias encontradas en estos pacientes.
La tabla I resume las bases fisiopatológicas de la enfermedad(11).

Formas primarias
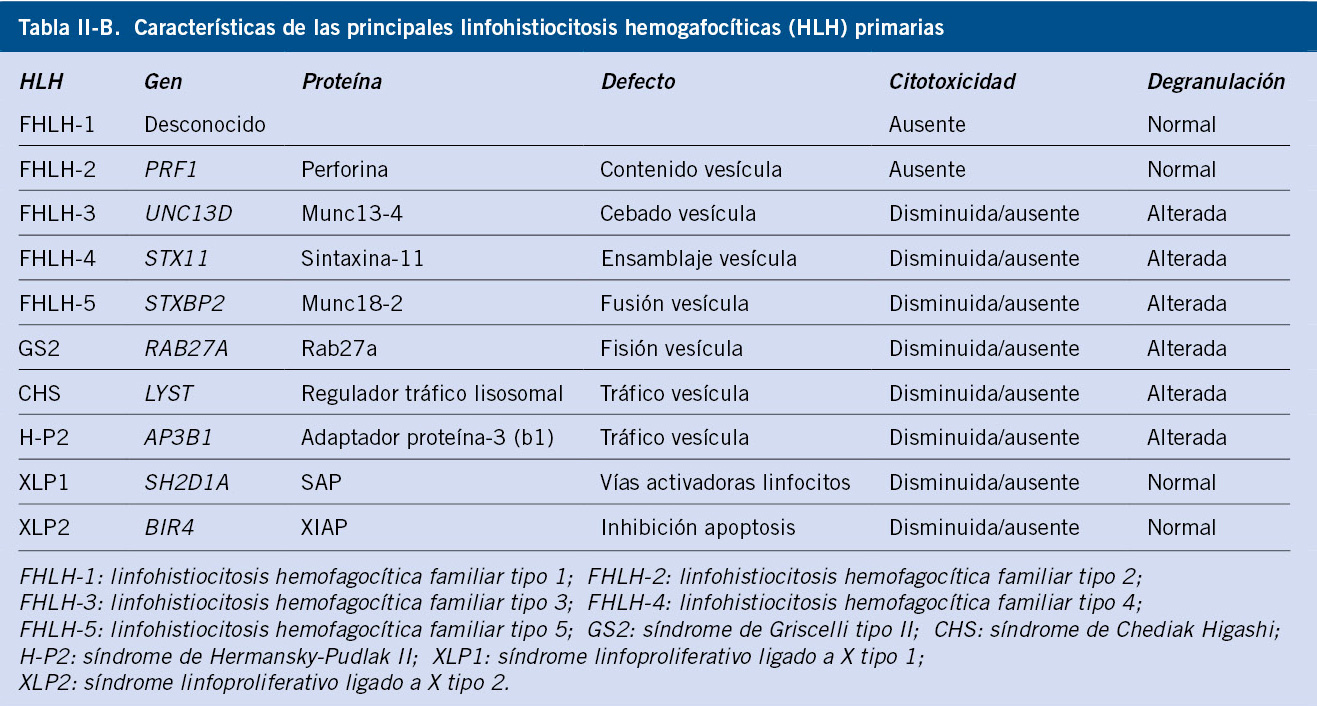
Conocemos causa genética subyacente en aproximadamente un 30-70% de las formas primarias de la enfermedad. Pertenecen a este grupo las HLH familiares (FHLH). Hasta el momento se han descrito 5 formas familiares (FHLH 1-5). Ejemplo de ello, son las formas que presentan mutaciones en el gen PRF1 (perforina) o en UNC13D (codificante para la proteína Munc13-14, necesaria para la fusión y liberación de los gránulos sinápticos).
Del mismo modo, pertenecen a las formas primarias inmunodeficiencias como: síndrome de Griscelli tipo 2, Chediak-Higashi, Hermansky-Pudlak tipo 2 o síndrome linfoproliferativo ligado al cromosoma X (XLP). En ellas, se han descrito anormalidades en las proteínas implicadas en el transporte de los gránulos citolíticos, así como en los melanosomas, por lo que estos pacientes presentan un grado variable de albinismo.
Formas secundarias
Estas formas, más frecuentes que las anteriormente citadas, se han descrito como: complicación de inmunodeficiencias primarias, errores innatos del metabolismo, algunas infecciones (generalmente víricas, típica VEB e importante por su auge en los últimos meses el SARS-CoV-2(13)), procesos neoplásicos (especialmente hematológicos, aunque no exclusivos), así como de origen autoinmune.
Centrándonos en el campo de la terapia celular, concretamente en el TPH, se han descrito fenómenos hemofagocíticos en pacientes trasplantados. Este hecho, inicialmente descrito en trasplantes autólogos, se ha estudiado con mayor detalle con los trasplantes alogénicos, especialmente en aquellos con disparidad HLA. La inmunosupresión derivada del acondicionamiento, las técnicas de manipulación del injerto y la profilaxis de la enfermedad de injerto contra receptor, genera una mayor susceptibilidad infecciosa, que como previamente se ha descrito, puede actuar como desencadenante (importante en este punto, la infección por VH8). Asimismo, la alorreactividad, tanto en dirección donante-receptor como receptor-donante, genera la liberación de citoquinas como resultado de la interacción entre las APC del receptor y los CTL del donante, activando, de este modo, a los macrófagos tisulares y amplificando así la respuesta inflamatoria(14).
Mención aparte merece una entidad de relativa reciente aparición, denominada síndrome de liberación de citoquinas, que mimetiza tanto la fisiopatología como la clínica del síndrome hemofagocítico, y que se observa en el contexto de la inmunoterapia, especialmente en el tratamiento con anticuerpos, como el blinatumomab o la terapia celular CAR-T.
Los mecanismos tras los que subyace el origen del cuadro clínico en estos casos secundarios no son del todo conocidos(3). En algunos casos, se postula la alteración en el equilibrio entre CTL/NK y APC.
Por lo general, estas formas secundarias tienen un curso menos agresivo, se presentan a edades más tardías y tienen un menor riesgo de recurrencia en comparación con las formas primarias.
En la tabla II, se recoge de manera resumida, la clasificación de los distintos tipos de HLH (Tabla IIA) y se resumen las principales características genéticas de las formas primarias (Tabla IIB)(1,15,16).

Clínica
La clínica puede simular inicialmente patologías banales con una evolución tórpida.
El espectro clínico de esta entidad es variable. A menudo, la distinta sintomatología irá sucediéndose en el tiempo, por lo que es importante una rápida sospecha clínica inicial. En algunas ocasiones, la forma de presentación será aguda, en el contexto de un paciente crítico, especialmente en las formas primarias.
Como norma general, los principales síntomas característicos de esta entidad son la fiebre persistente y la presencia de hepatoesplenomegalia. Con frecuencia, hasta en un tercio de los casos, puede existir afectación del sistema nervioso central, en cuyo caso, la clínica puede simular una encefalitis. Otros hallazgos clínicos menos frecuentes pueden ser: presencia de linfadenopatías, rash cutáneo, ictericia en casos con afectación hepatobiliar o edemas. A continuación, se detallan las entidades clínicas más frecuentes(11,16):
Fiebre persistente
Pese a tratarse de un motivo de ingreso relativamente frecuente, la fiebre persistente (mediana 19 días en algunos estudios publicados) junto con la elevación de parámetros inflamatorios, deben justificar el inicio del estudio del síndrome hemofagocítico.
La fiebre es el síntoma guía en la mayoría de las ocasiones, apareciendo de manera constante, prácticamente, en más del 90% de los casos(4,10,15).
Lactante febril
Generalmente, en cuadros primarios. El paciente, habitualmente un lactante menor de un año, se presenta con fiebre, citopenias y megalias. En ocasiones, estas formas pueden tener un debut neonatal (formas graves). A menudo, el aspirado de médula ósea se realiza para descartar malignidad en este contexto. Sin un diagnóstico precoz y tratamiento agresivo, estas formas pueden tener desenlace fatal de forma rápida.
Fallo hepático
En pacientes con presencia de fallo hepático agudo, debe plantearse esta posibilidad diagnóstica como opción. En ocasiones, la gravedad de esta entidad clínica es tal, que el paciente puede resultar candidato a trasplante hepático.
Es característica, la infiltración tisular linfocitaria (evidenciada en necropsias). En periodo neonatal, puede estar acompañado de hidrops fetal. Puede existir concomitantemente síndrome de oclusión sinusoidal, existiendo un mayor riesgo de presentación de esta complicación en pacientes que reciban trasplante de progenitores hematopoyéticos (TPH).
Junto a los datos clínico-analíticos de fallo hepático, aparecen el resto de parámetros que definen el síndrome hemofagocítico. En este grupo de pacientes, parámetros como la ferritina o el CD25 soluble, no son adecuados criterios diagnósticos, debido a que en situaciones de fallo hepático, pueden estar elevados.
Alteraciones en la coagulación
Alrededor de un 95% de los pacientes presentarán parámetros sugerentes de coagulación intravascular diseminada. En los casos de defecto de granulación de células NK, puede existir disfunción plaquetar.
Citopenias
Anemia y trombopenia suelen ser los hallazgos más frecuentes. Los estudios en médula ósea pueden ser normales, así como reflejar hipo o hipercelularidad. La presencia de hemofagocitosis oscila entre el 25-100% de los casos; si bien, es necesario destacar que este fenómeno no es exclusivo de esta entidad, pudiendo aparecer en pacientes críticos, politransfundidos, en infecciones o en enfermedades autoinmunes.
Alteraciones cutáneas
El espectro de afectación cutánea es variado, aunque frecuente (hasta el 65% de los casos), y se desencadena por la infiltración cutánea de linfocitos. Las manifestaciones más frecuentes varían desde un rash máculopapuloso generalizado (en ocasiones, morbiliforme), eritrodermia, edema o paniculitis. Puede simular un cuadro Kawasaki-like y aparecer junto con otros criterios clínicos, como conjuntivitis y alteraciones en mucosas.
Afectación pulmonar
Pueden aparecer infiltrados alveolares e intersticiales en el contexto de fracaso respiratorio agudo. En estos pacientes, la evolución a un cuadro fatal puede ocurrir hasta en un 88% de los casos.
Afectación neurológica
Aproximadamente, en un tercio de los casos, puede aparecer inicialmente afectación del sistema nervioso central. En estos casos, la clínica neurológica puede ser variada, desde: crisis convulsivas, fluctuación del nivel de consciencia similar a una encefalitis, meningismo, alteraciones en la marcha, disartria o alteraciones en los pares craneales. Es infrecuente la clínica neurológica aislada, coexistiendo, en la mayoría de los casos, con otras manifestaciones sistémicas. En cualquier caso, es importante una rápida sospecha.
Aunque la edad de aparición es variable, a menudo, aparece en niños mayores de un año.
Niño con infección
Como se muestra en la tabla IIA, agentes infecciosos, especialmente típico por su frecuencia, el VEB pueden actuar a modo de trigger del síndrome hemofagocítico.
Paciente crítico con cuadro “sepsis-like”
A menudo, este paciente aparece en unidades de críticos con un cuadro prácticamente indistinguible de una sepsis grave (llegando incluso a producirse fallo multiorgánico), en el que la clave diagnóstica es la gravedad del espectro inflamatorio. En estos pacientes, los fenómenos hemofagocíticos en la anatomía patológica son de escasa rentabilidad diagnóstica, debido a estar presentes en un alto porcentaje de pacientes críticos(16).
Síndrome hemofagocítico en paciente con patología autoinmune (síndrome de activación macrofágica)
Tradicionalmente, se ha denominado síndrome de activación macrofágica (SAM) al síndrome hemofagocítico, que aparece en el paciente con diagnóstico establecido de patología autoinmune. El ejemplo más característico, la artritis idiopática juvenil (AIJ), en la que aproximadamente un 10% pueden desarrollar este cuadro clínico. Ocasionalmente, puede ser la forma de debut de la enfermedad o ser desencadenado por el tratamiento inmunosupresor.
Aunque, tanto la clínica como los mecanismos fisiopatológicos son indistinguibles, existen una serie de peculiaridades en este tipo de presentación. El perfil de paciente es de mayor edad que en las formas primarias, y suele cursar con citopenias no tan marcadas, así como con cifras más elevadas de fibrinógeno.
Síndrome hemofagocítico asociado a neoplasia
En ocasiones, el síndrome hemofagocítico puede aparecer en pacientes con proceso oncológico concomitante. La frecuencia con la que esto aparece se incrementa con la edad, y los tumores hematológicos, especialmente los linfomas, son las patologías que con más frecuencia pueden asociar este síndrome (aunque no de forma exclusiva).
Síndrome hemofagocítico en paciente inmunocomprometido
Puede aparecer en pacientes con diagnóstico de inmunodeficiencia primaria o en pacientes con estado de inmunosupresión secundario a tratamiento. En este grupo, generalmente, un agente infeccioso (típicamente VEB o CMV) actúa a modo de desencadenante.
Terapias inmunológicas
En los últimos años, los avances en inmunoterapia, con el cada vez mayor empleo de anticuerpos monoclonales y células CAR-T, han creado la necesidad de estudiar una entidad fisiopatológicamente similar al HLH y que, en este contexto, recibe el nombre de síndrome de liberación de citoquinas, del inglés Cytokine Release Syndrome (CRS). El abordaje de esta complicación difiere del manejo estándar del HLH.
En definitiva, ante la evolución tórpida de cualquier enfermedad considerada a priori como común, el síndrome hemofagocítico debería ser descartado.
Además de la forma de presentación, es primordial también considerar el rango etario del paciente, debido a que las formas primarias, más agresivas, suelen debutar en los primeros meses de vida, incluso durante el periodo neonatal. En estos casos, es fundamental una correcta anamnesis que haga hincapié en los antecedentes familiares del paciente, especialmente importante si existe consanguinidad.
Diagnóstico
Las herramientas fundamentales para el diagnóstico son los criterios clínicos y analíticos. La presencia de una mutación conocida es suficiente para llegar al mismo.
El diagnóstico de la HLH es un diagnóstico genético, clínico y analítico. En la actualidad, se realiza según los criterios del protocolo HLH-2004(17) que se detallan en la tabla III.

Desde el punto de vista molecular, la presencia de mutaciones conocidas en genes implicados en la degranulación de las células NK, la homeostasis de la sinapsis inmunológica o de inmunodeficiencias, bastarían para diagnosticar al paciente. Actualmente, se conocen hasta un 70% de las alteraciones causantes de las formas familiares. El tipo de herencia, el fenotipo y la penetrancia son variables en función del tipo de trastorno del que se trate. Habitualmente, las formas que se presentan en homocigosis suelen ser de mayor gravedad.
En las formas primarias es obligado el estudio de los hermanos del paciente en caso de tenerlos, no solo por el riesgo de enfermedad que presentan en función de la herencia del trastorno, sino por su potencial elección como donante de precursores hematopoyéticos.
Se sospechará una forma primaria en los casos en que exista historia familiar compatible, en formas graves (especialmente de presentación temprana), recurrentes, o en casos de persistencia de disminución de actividad NK.
Desde un punto de vista clínico, son criterios diagnósticos la fiebre persistente y la hepatoesplenomegalia.
Analíticamente, los principales hallazgos son: citopenias (con afectación variable de las tres series, mínimo dos), hipertrigliceridemia, hipofibrinogenemia y la hiperferritinemia. Aunque no sea considerado criterio diagnóstico, apoyan el diagnóstico parámetros analíticos como: pleocitosis en el LCR, hiperproteinorraquia, disfunción hepatobiliar (hipertransaminasemia, hiperbilirrubinemia, coagulopatía con elevación de D-dímero), hipoalbuminemia, elevación de LDH en suero o hiponatremia. Estas alteraciones pueden aparecer especialmente en casos de gravedad(3,4,16).
Los estudios inmunológicos son de utilidad en el diagnóstico de la HLH, siendo característica la elevación de CD25 soluble (subunidad alfa del receptor de IL-2) o la ausencia/disminución de degranulación in vitro de células NK.
En estos estudios funcionales tienen especial relevancia los estudios inmunofenotípicos. Mediante citometría de flujo, puede estudiarse la presencia de determinadas proteínas de poblaciones celulares seleccionadas (p. ej., perforina en NK), así como la funcionalidad de su degranulación. De este modo, pueden complementar el diagnóstico de esta patología y apoyarlo hasta su confirmación definitiva.
Otro parámetro característico, aunque es importante conocer que no debe aparecer necesariamente, es la presencia de hemofagocitosis en médula ósea, muestra de tejido linfoide o LCR.
En la tabla IV, se relacionan los principales hallazgos clínico-analíticos con el mecanismo fisiopatológico subyacente(15,18).

Diagnóstico diferencial
Es necesario mantener un alto nivel de sospecha en cuadros persistentes o refractarios a tratamientos convencionales.
Desde el punto de vista clínico, el planteamiento de esta entidad se presenta habitualmente como un reto para el médico tratante.
A menudo, la mayor parte de estos cuadros son diagnosticados inicialmente (y no de forma equivocada) como infecciones. Patologías hematológicas como la histiocitosis de células de Langerhans o la enfermedad de Castleman, deberían ser tenidas en cuenta en el diagnóstico diferencial, debido a que pueden simular la forma de presentación. Del mismo modo, deben ser incluidas reacciones adversas a fármacos (estas últimas suelen cursar con rash cutáneo y eosinofilia), así como metabolopatías y enfermedades de depósito.
Es esencial considerar, no solo la forma de presentación clínica, sino el momento de aparición de la misma, especialmente relevante en los neonatos. Entidades como la hepatitis aloinmune gestacional o GALD (del inglés Gestational Alloinmune Liver Disease) debe ser considerada en neonatos críticos con fallo hepático agudo.
En la tabla V, se recoge el diagnóstico diferencial del HLH(3,16).

Tratamiento y pronóstico
La piedra angular del tratamiento de esta entidad es la inmunosupresión. El TPH estará reservado para formas primarias o secundarias persistentes o recidivantes.
Sin tratamiento, el pronóstico de la linfohistiocitosis hemofagocítica es fatal, por lo que una rápida sospecha clínica y su instauración precoz, mejorará notablemente el pronóstico del paciente. En las formas primarias, recurrentes y refractarias, la gravedad del cuadro clínico puede suponer el fallecimiento del paciente. Las formas secundarias, sin embargo, suelen presentar un pronóstico favorable una vez controlado el factor desencadenante.
El objetivo principal del tratamiento es poner fin al círculo vicioso inflamatorio característico de la enfermedad (independientemente de la causa subyacente), mediante el empleo de potentes fármacos inmunosupresores y citostáticos que bloqueen o eliminen la elevada población de células activadas(3).
Una vez conseguido, se actuará sobre la causa desencadenante mediante el tratamiento etiológico específico, espectro que puede abarcar desde la resolución de una infección hasta el TPH, en el caso de las formas primarias.
Los datos reportados a través de dos grandes ensayos clínicos promovidos por la Sociedad Internacional del Histiocito, HLH-94 y HLH-2004, apoyan de manera general el tratamiento de esta entidad mediante el empleo de: dexametasona, ciclosporina, citostáticos como el etopósido y el trasplante de progenitores hematopoyéticos. La terapia intratecal estará indicada en casos de afectación del sistema nervioso central(11,17,19).
En los últimos años y, en especial, en las formas secundarias, se han empleado fármacos biológicos que han resultado de utilidad. Ejemplo de ello es el empleo de: inmunoglobulina antitimocítica (ATG), rituximab (anticuerpo anti CD20), anakinra (anticuerpo anti IL-1), etanercept (anticuerpo anti TNFa), tocilizumab (anticuerpo anti receptor de IL-6) o anticuerpos anti IFN gamma como el emapalumab(20).
Este último anticuerpo neutralizador de IFN gamma ha sido protagonista de numerosos ensayos clínicos, tanto en formas primarias como en HLH secundario, en adultos y en pacientes pediátricos. En combinación con los fármacos estándares ha demostrado buenas tasas de respuesta, si bien, los resultados publicados hasta el momento son controvertidos.
El alemtuzumab, otro anticuerpo monoclonal (anti CD52), ha sido empleado en casos refractarios seguido en la mayoría de los casos de TPH con buenos resultados, si bien la efectividad de este fármaco en monoterapia es discutida y el riesgo de infecciones, especialmente víricas, debe ser individualizado en cada paciente(21,22).
Asimismo, inhibidores de las JAK/STAT kinasas como el ruxolitinib, se han empleado en el tratamiento de esta entidad(15).
El tratamiento debe iniciarse siempre guiados por la gravedad de los síntomas, pese a no llegar a cumplir en ese momento todos los criterios diagnósticos, debido al compromiso vital que vincula el cuadro(15,19).
El TPH es el único tratamiento curativo en las formas primarias, y se recomienda en formas secundarias recidivantes o persistentes. Se recomienda la realización de un acondicionamiento de intensidad reducida.
En torno al 70% de las formas primarias son trasplantadas, alcanzando supervivencias a largo plazo del 50-60%. El TPH en las formas secundarias, alcanza supervivencias a los 5 años del 86% cuando el desencadenante es infeccioso, e inferiores al 15% cuando la causa es tumoral.
En la tabla VI, se resumen las principales estrategias terapéuticas del síndrome hemofagocítico.

Prevención
En las formas primarias con alteraciones conocidas, está indicado el consejo genético.
No existe ninguna medida preventiva hasta el momento conocida. Como se detalló en un inicio, es de gran importancia la sospecha clínica precoz, ya que de ello depende el inicio temprano del tratamiento y el pronóstico del paciente.
En pacientes con formas primarias, donde sea conocida la alteración genética subyacente, estará indicado el consejo genético de los progenitores a cargo de un experto en genética clínica.
Función del pediatra de Atención Primaria
Es fundamental considerar este cuadro clínico ante cualquier paciente con una evolución tórpida de su enfermedad, a pesar de la administración de un tratamiento adecuado.
Además, la correcta historia clínica haciendo especial hincapié en antecedentes familiares y consanguinidad, así como una detallada exploración física, son claves en el diagnóstico de esta enfermedad.
Ante un paciente con sospecha de HLH, se recomienda su derivación a un centro con experiencia en el manejo de este tipo de pacientes y con posibilidad de realización de TPH.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.*** Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020; 135: 1332-43.
2. Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatr Int. 2016; 58: 817-25.
3. Janka GE, Lehmberg K. Hemophagocytic syndromes–an update. Blood Rev. 2014; 28: 135-42.
4. Sen ES, Steward CG, Ramanan AV. Diagnosing haemophagocytic syndrome. Arch Dis Child. 2017; 102: 279-84.
5. Allen CE, McClain KL. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2015; 2015: 177-82.
6. Yanagisawa R, Nakazawa Y, Matsuda K, Yasumi T, Kanegane H, Ohga S, et al. Outcomes in children with hemophagocytic lymphohistiocytosis treated using HLH-2004 protocol in Japan. Int J Hematol. 2019; 109: 206-13.
7. Simon AC, Delhi Kumar CG, Basu D, Ramesh Kumar R. Hemophagocytic Lymphohistiocytosis in Children: Clinical Profile and Outcome. J Pediatr Hematol Oncol. 2020; 42: e281-5.
8. Meeths M, Horne A, Sabel M, Bryceson YT, Henter J-I. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015; 62: 346-52.
9. Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008; 50: 1227-35.
10. Dapena Díaz JL, Díaz de Heredia Rubio C, Bastida Vila P, Llort Sales A, Elorza Álvarez I, Olivé Oliveras T, et al. Haemophagocytic syndrome: A common pathogenic mechanism of various aetiologies. An Pediatr (Barc). 2009; 71: 110-6.
11.*** Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011; 118: 4041-52.
12. Ishii E. Hemophagocytic Lymphohistiocytosis in Children: Pathogenesis and Treatment. Front Pediatr. 2016; 4: 47.
13. Soy M, Atagündüz P, Atagündüz I, Sucak GT. Hemophagocytic lymphohistiocytosis: a review inspired by the COVID-19 pandemic. Rheumatol Int. 2021; 41: 7-18.
14. Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001; 27: 893-8.
15.*** Astigarraga I, González-Granado LI, Allende LM, Alsina L. Haemophagocytic syndromes: The importance of early diagnosis and treatment. An Pediatr (Barc). 2018; 89: 124.e1-e8.
16. Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019; 66: e27929.
17. Henter J-I, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007; 48: 124-31.
18. Brisse E, Wouters CH, Matthys P. Hemophagocytic lymphohistiocytosis (HLH): A heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev. 2015; 26: 263-80.
19. Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the Use of Etoposide-Based Therapy and Bone Marrow Transplantation for the Treatment of HLH: Consensus Statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018; 6: 1508-17.
20. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N Engl J Med. 2020; 382: 1811-22.
21. Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013; 60: 101-9.
22. Marsh RA, Jordan MB, Talano J-A, Nichols KE, Kumar A, Naqvi A, et al. Salvage therapy for refractory hemophagocytic lymphohistiocytosis: A review of the published experience. Pediatr Blood Cancer; 2017. p. 64.
Bibliografía recomendada
- Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020; 135: 1332-43.
Artículo reciente sobre el HLH pediátrico publicado en la revista de la Asociación Americana de Hematología, donde se hace una revisión de la fisiopatología, formas de presentación, diagnóstico y tratamiento.
- Astigarraga I, González-Granado LI, Allende LM, Alsina L. Haemophagocytic syndromes: The importance of early diagnosis and treatment. An Pediatr (Barc). 2018; 89: 124.e1-e8.
Artículo especial publicado en la revista de la Asociación Española de Pediatría cuya primera autora es una de las mayores expertas nacionales de esta patología, donde se revisa el HLH de manera completa y sencilla.
- Dapena Díaz JL, Díaz de Heredia Rubio C, Bastida Vila P, Llort Sales A, Elorza Álvarez I, Olivé Oliveras T, et al. Haemophagocytic syndrome: A common pathogenic mechanism of various aetiologies. An Pediatr (Barc). 2009; 71: 110-6.
Artículo original publicado en la revista de la Asociación Española de Pediatría por el grupo del Hospital Vall d’Hebron, donde se analizan las características de 22 pacientes con diagnóstico de HLH.
- Pérez-Martínez A. Síndromes hemofagocíticos (I): concepto, clasificación, fisiopatología y clínica. Anales de Pediatría Continuada (Internet). 2013. Citado el 11 de mayo de 2021; 11: 237-44. Disponible en: https://linkinghub.elsevier.com/retrieve/pii/S1696281813701446.
– Pérez-Martínez A. Síndromes hemofagocíticos (II): diagnóstico y tratamiento. Anales de Pediatría Continuada (Internet). 2013. Citado el 11 de mayo de 2021; 11: 245-53. Disponible en: https://linkinghub.elsevier.com/retrieve/pii/S1696281813701458.
Actualización de uno de los autores de este trabajo, donde se revisa ampliamente el HLH. De interés, los apartados de fisiopatología y diagnóstico, especialmente el papel de la citometría de flujo en esta entidad.
| Caso clínico |
|
Niña de 8 años procedente de Bangladesh, con diagnóstico de leucemia linfoblástica aguda de precursores B, de riesgo intermedio, refractaria a dos líneas de tratamiento. Se presenta candidata a receptora de terapia celular CAR-T CD19 (tisagenlecleucel). El producto es infundido tras recibir quimioterapia de linfodepleción. En el momento actual, la paciente se encuentra ingresada en la Unidad de Cuidados Intensivos (UCIP), 5 días después de la infusión, por presentar un síndrome de liberación de citoquinas (CRS), con: fiebre, hipoxemia que requiere la administración de oxigenoterapia de alto flujo y taquicardia con hipotensión, que requiere la administración de soporte vasoactivo. Debido a la intensidad de la respuesta inflamatoria, se administran tres dosis de tocilizumab y se decide iniciar tratamiento esteroideo con metilprednisolona a 2 mg/kg/día. A las 48 horas de su ingreso en UCIP, se solicitan las siguientes pruebas complementarias: • Hemograma: hemoglobina: 9 g/dL (9,4-14,6); leucocitos: 1,02x10e3/µL (6,6-16,2); neutrófilos: 0,3x10e3/µL (1,3-8,7); linfocitos: 0,6x10e3/µL (2,7-12,6); monocitos 0,1x10e3/µL (0,2-1,9); plaquetas: 32x10e3/µL (240-550); frotis de sangre periférica sin evidencia de formas inmaduras. • Coagulación: tiempo de protrombina: 10,3 segundos; actividad de protrombina: 112% (70-120); INR: 1 (0,8-1,2); fibrinógeno: 120 mg/dl (150-450). • Bioquímica general: ASAT/GOT: 130 UI/L (0-95); ALAT/GPT: 236 UI/L (0-35); LDH: 116 UI/L (110-295); GGT: 77 UI/L (0-73); bilirrubina total: 2,72 mg/dL (0,3-1,2); bilirrubina directa: 1,9 mg/dL (0-0,3); colesterol total: 165 mg/dL (0-200); triglicéridos: 750 mg/dL (0-150); ferritina: 15.863 ng/mL (22-322). • Ecografía abdominal: esplenomegalia homogénea de 10,3 cm de diámetro longitudinal. • Radiografía de tórax: opacidad en lóbulo superior derecho. Ante la sospecha de síndrome hemofagocítico secundario (fiebre, esplenomegalia, citopenias, hipertrigliceridemia con hipofibrinogenemia e hiperferritinemia), se inició tratamiento con dexametasona a 10 mg/m2, suprimiendo metilprednisolona. Evolución posterior Ante la persistencia de la fiebre y la elevación de los marcadores inflamatorios 48 horas después, pese al tratamiento esteroideo, se decide administración de anakinra subcutáneo (tres dosis), tras lo cual desaparece la fiebre y comienzan a descender los parámetros hemofagocíticos. Debido a la mejoría progresiva de la paciente, el soporte inotrópico pudo retirarse en las 48 horas posteriores, así como la asistencia respiratoria. La dosis esteroidea de dexametasona fue reduciéndose de manera paulatina hasta su retirada en 10 días.
|


 How to suspect cancer in Primary Care
How to suspect cancer in Primary Care