 |
| Regreso a las bases |
M. González Vicent, B. Molina Angulo, I. López Torija, A. Peretó Moll, M.A. Díaz Pérez
Unidad de Trasplante Hematopoyético del Hospital del Niño Jesús. Madrid
| Resumen
En Pediatría, algunas enfermedades neoplásicas o congénitas no malignas, no tienen más tratamiento curativo que el trasplante alogénico de progenitores hematopoyéticos de un donante sano. |
| Abstract
In Pediatrics, several hematological malignancies or non-malignant congenital diseases have no curative treatment other than allogeneic hematopoietic stem cell transplantation from a healthy donor. |
Palabras clave: Pediatría; Trasplante hematopoyético.
Key words: Pediatric; Hematopoietic stem cell transplantation.
Pediatr Integral 2021; XXV (6): 328.e1 – 328.e6
Trasplante alogénico de progenitores hematopoyéticos en Pediatría
Introducción
Hacer de la necesidad virtud es sacar provecho de la desventaja, y la historia del trasplante alogénico de progenitores hematopoyéticos (TPH) está llena de dichos ejemplos. Dicha historia se inicia con la necesidad de proporcionar un tratamiento curativo para aquellos pacientes con fallo en la hematopoyesis (parcial o total).
El primer trasplante fue realizado sin éxito por Osgood en 1939, en un paciente con aplasia medular(1). En la segunda mitad de la década de los años 40, hubo un gran interés por el estudio de las patologías derivadas de las explosiones nucleares a raíz de la Segunda Guerra Mundial. Entre otros, se comprobó el efecto destructor de la irradiación sobre la médula ósea. Lorenz(2), en 1951, comprobó que la administración parenteral de médula ósea singénica protegía a los ratones de una irradiación supraletal. Aunque, inicialmente, el efecto protector de la médula ósea (MO) fue atribuido a un factor humoral, en 1956 se demostró, mediante diversos marcadores genéticos, que el efecto protector era debido a las células hematopoyéticas del donante(3).
Estos primeros avances clínicos sirvieron para llevar este procedimiento a aquellos pacientes con hemopatías malignas, incurables de otro modo. Entre 1957 y 1959, el Dr. Thomas comienza a realizar trasplantes singénicos en pacientes con leucemia linfoblástica aguda (LLA) en fases avanzadas de la enfermedad, utilizando como acondicionamiento la irradiación corporal total (ICT). En estos enfermos, se puso de manifiesto que la médula ósea, en determinadas condiciones, podía ser infundida por vía intravenosa y que esta era capaz de conseguir una reconstitución hematopoyética; sin embargo, estos pacientes fallecieron posteriormente por recaída de su enfermedad(4).
En los años posteriores, los investigadores orientaron sus estudios a localizar y utilizar en la clínica donantes HLA idénticos, tratando de aminorar una enfermedad que fue descrita por Van Bekkum(5) en animales de experimentación, como “wasting disease” o “enfermedad secundaria” y conocida ahora, como enfermedad del injerto contra huésped (EICH). Durante la década de los 1960, Dausset & Van Rood demuestran que en el ser humano, existe un sistema de histocompatibilidad denominado antígeno leucocitario humano (HLA), localizado en el cromosoma 6 y que da explicación al desarrollo de EICH en los trasplantes hematopoyéticos (Fig. 1).

Figura 1. Complejo mayor de histocompatibilidad (adaptada de www.cancer.gov).
El primer trasplante con éxito fue realizado en 1968 por Gatti y cols. en un niño con inmunodeficiencia combinada severa (IDCS), al que se le infundió médula ósea de su hermana sana HLA-idéntica(6). Entre 1969 y 1980, el trasplante hematopoyético se fue consolidando como una opción terapéutica para pacientes con leucemia que no habían respondido a la quimioterapia convencional, así como para alguna enfermedad metabólica, comprobándose que era posible la supervivencia a largo plazo en algunos de ellos.
Desde 1980 hasta nuestros días, se han ido definiendo diferentes grupos de riesgo en cada patología, que han permitido un uso más racional del trasplante, alcanzando así unos mejores resultados.
La creación de registros internacionales de donantes altruistas ha proporcionado una nueva perspectiva al trasplante hematopoyético, permitiendo su utilización en pacientes que carecían de un donante familiar adecuado. Inicialmente, los resultados no fueron totalmente satisfactorios, pero con posterioridad, se han publicado diferentes estudios multicéntricos en los que los resultados son similares a los que se obtienen en el trasplante de médula ósea con donante familiar HLA idéntico.
En estos últimos años, se ha demostrado la utilidad de los progenitores hematopoyéticos de otras fuentes diferentes a la médula ósea, como son la sangre periférica y la sangre de cordón umbilical (Fig. 2).
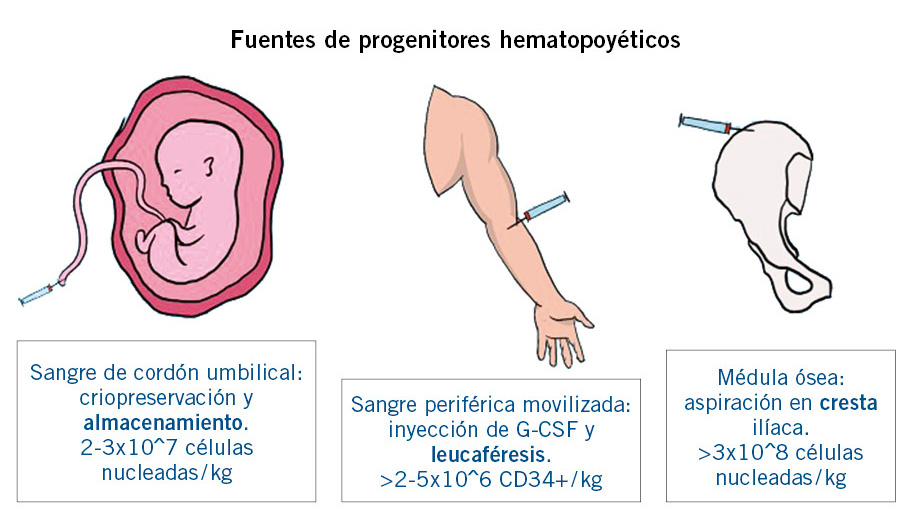
Figura 2. Fuentes de progenitores hematopoyéticos.
La utilización de estas fuentes de progenitores fue inicialmente considerada como un procedimiento experimental, pero, actualmente, se consideran una alternativa más para los trasplantes de progenitores hematopoyéticos en Pediatría(7).
Durante todos esos años, el TPH ha sido considerado como un tratamiento definitivo para los pacientes con hemopatías malignas de alto riesgo o aquellos con fallo primario o secundario de la hematopoyesis.
Para realizar el trasplante alogénico, el paciente era sometido a un acondicionamiento con dosis mieloablativas de quimioterapia y/o radioterapia y, posteriormente, se infundían las células progenitoras hematopoyéticas procedentes de la médula ósea o de la sangre periférica del donante sano. El paciente recibía tratamiento inmunosupresor por un tiempo indeterminado para evitar las complicaciones inmunes asociadas y, posteriormente, una vez obtenida la tolerancia inmune, se suspendía dicho tratamiento.
La hipótesis sobre la que se sustentaba su uso, consideraba la sustitución de una hematopoyesis enferma por una de un donante sano que, una vez injertada, funcionaría de forma normal en el cuerpo del paciente al modo como lo haría el riñón o el hígado de un donante sano en un paciente con fallo de dichos órganos. Su base biológica era la compatibilidad entre donante y receptor en el sistema HLA.
Sin embargo, y a pesar de la compatibilidad HLA entre donante y receptor, el procedimiento se veía y se ve aún, frecuentemente complicado por la presencia de la denominada EICH, tanto en su forma aguda como en su forma crónica, más parecida esta última al desarrollo de una enfermedad autoinmune. Esta grave complicación está mediada por la respuesta alo-inmune (alorreactividad) de los linfocitos T del donante contra los tejidos no hematopoyéticos del receptor (Fig. 3).

Figura 3. Fisiopatología de la enfermedad injerto contra huésped (adaptada de Ferrara y cols.). CPA: células presentadoras de antígenos; FNT: factor de necrosis tumoral; IL1: interleuquina 1; LPS: lipopolisacáridos; Th1: T helper 1; LTC: linfocitos T citotóxicos.
Sin embargo, estudios iniciales demostraron que la EICH reducía la probabilidad de recaída de los enfermos con leucemia. A este efecto, se le denomina efecto injerto contra leucemia (EICL) y está mediado por la alorreactividad de los linfocitos T del donante sobre los tejidos hematopoyéticos malignos y no malignos del receptor(8).
Necesidades no resueltas del trasplante hematopoyético
La separación de la EICH del EICL
Como se puso de manifiesto, más pronto que tarde, en los primeros estudios clínicos, existía una estrecha relación entre la EICH y el EICL(9). Desafortunadamente, la EICH es la causa de morbilidad y mortalidad más importante asociada con el TPH, si descartamos la propia recaída leucémica, y, hasta la fecha, no han existido métodos de prevención y tratamiento lo suficientemente exitosos.
Durante muchos años, la prevención de la EICH ha estado basada en el uso de donantes HLA compatibles y en la profilaxis farmacológica con ciclosporina A ± metotrexato. Con todo, un 30% de los pacientes desarrollan EICH aguda grave y casi un 40% de estos fallecen como consecuencia de las complicaciones sistémicas (fallo orgánico) de la EICH o de su tratamiento inmunosupresor prolongado (infecciones oportunistas).
Es bien conocido, actualmente, que los linfocitos T “naive” son los responsables de la EICH aguda, especialmente de sus formas clínicas más graves. Desafortunadamente, también esta subpoblación de linfocitos T es la principal responsable de la reacción antileucémica, generando una población memoria responsable de la inmunovigilancia tras el TPH.
Por tanto, EICH y EICL han sido considerados como las dos caras de una misma moneda denominada “alorreactividad”. Así, una necesidad no resuelta del TPH alogénico en el contexto de donante/receptor HLA compatible, es la separación de la EICH del EICL(10).
La reconstitución inmune postrasplante juega un papel determinante en el resultado del mismo. En los días inmediatos tras el acondicionamiento, en el paciente existe una profunda linfopenia y un exceso de citocinas (especialmente IL-7 e IL-15), que actúan como “combustible” para la reconstitución inmune. Los linfocitos T-naive y los CD4+ tienen bajos niveles del receptor de la IL-15, y dependen más de la IL-7 para su expansión y mantenimiento.
En ausencia del control ejercido por la inmunidad innata (especialmente por las células “natural killer”, NK), la expansión de los linfocitos T CD8+ incrementa los niveles séricos de interferón γ (IFN-γ) y factor de necrosis tumoral α (FNT-α), acelerando la alorreactividad y generando EICH grave. Dado que un inóculo sin manipular contiene linfocitos “naive” y memoria del donante, estos compiten por dicho combustible para su proliferación y diferenciación(9).
Tras el TPH, la reconstitución inmune puede ser dirigida por antígenos o bien ser homeostática, es decir, fisiológica. La reconstitución homeostática ocurre a partir del conjunto de linfocitos T memoria del donante y protege al paciente de las infecciones. Sin embargo, estos linfocitos T memoria compiten con aquellas poblaciones que reconocen antígenos tisulares del paciente presentados por el complejo principal de histocompatibilidad (MHC) al receptor de la célula T (TCR)(9). Estos antígenos tisulares dominan la escena post-TPH, debido a los daños producidos por el acondicionamiento y reconducen la reconstitución inmune, favoreciendo la proliferación y expansión de los linfocitos T alorreactivos, dando como resultado la EICH. Para resolver este dilema se han propuesto algunas aproximaciones.
Una alternativa propone la manipulación del inóculo eliminando las poblaciones responsables de la EICH y apostando por la reconstrucción del sistema inmune post-trasplante, como modo de asegurar la vigilancia inmune anti-infecciosa y anti-leucémica(10). Así, la eliminación de los linfocitos T “naive” del inóculo ha mostrado resultados prometedores en el TPH HLA idéntico en adultos con hemopatías malignas(11). Sin embargo, aún no hay datos con suficiente seguimiento para saber si afectará a la probabilidad de recaída post-TPH.
Otra aproximación, es la creación de un ambiente inmunológico post-TPH libre de EICH y de inmunosupresión que permita la reconstrucción del mismo mediante la infusión de linfocitos del donante (ILD). Esta alternativa contempla la inducción de tolerancia inmunológica entre donante y receptor, mediante la generación de un estado de quimerismo mixto transitorio(12).
La tolerancia inmune en el paciente es conseguida a través de la presencia de las células T, tanto del donante como del receptor, por un periodo más o menos prolongado de tiempo post-TPH, y está sustentada en los efectos beneficiosos de una subpoblación de linfocitos T denominada T-reguladores (Tregs)(13).
Ambas plataformas de trasplante generan un estado post-TPH con muy bajo riesgo de EICH agudo grave, lo que permite la rápida retirada de los fármacos inmusupresores y el uso de ILD postrasplante(14,15). Dicha ILD se ha mostrado clínicamente útil en el tratamiento de la recaída post-TPH, especialmente en los pacientes con leucemia mieloide crónica y leucemia mieloblástica aguda. Sin embargo, aún no hay datos suficientemente maduros como profilaxis de la recaída post-TPH.
Dado que todos los eventos inmunológicos ocurren muy precozmente post-TPH, las estrategias clínicas de ILD deberían realizarse en los primeros días post-TPH, dependiendo del tipo de linfocitos que vayan a infundirse.
Actualmente, existe la posibilidad de realizar ILD inespecíficas, o bien solo con subpoblaciones seleccionadas (CD4+, Tregs, Tmemoria)(16). Aunque aún es prematuro sacar conclusiones sólidas de los estudios realizados basados en estas estrategias, en general la ILD post-TPH mejora la reconstitución inmune, pero no hay datos maduros respecto al riesgo de recaída. No obstante, los estudios clínicos con manipulación del inóculo en pacientes con leucemia aguda mieloblástica en 1ª remisión completa, no han mostrado un aumento de la probabilidad de recaída(17).
Nuevas fuentes de progenitores hematopoyéticos: oportunidades y desafíos
La necesidad de obtener una mayor cantidad de células progenitoras para el TPH, llevó al uso de los factores de crecimiento hematopoyético (G-CSF) en la movilización. A mediados de los 90, se generalizó el uso de G-CSF para movilizar y cosechar células progenitoras hematopoyéticas de la sangre periférica (PBSC). Esto hizo que la sangre periférica se convirtiera en la fuente preferida para TPH autólogo(18). Era cuestión de tiempo que semejante estrategia se trasladara al TPH alogénico.
Tras más de dos décadas de experiencia clínica, se puede concluir que el uso de la sangre periférica movilizada como fuente de progenitores, se asocia a una más rápida cinética de injerto hematopoyético especialmente plaquetar, similar incidencia de EICH aguda, pero mayor incidencia de EICH crónica(16). Esto ha llevado a considerar a la médula ósea como la fuente preferida de progenitores en el TPH alogénico de patologías no malignas y, en general, en los pacientes pediátricos. Sin embargo, en adultos, la sangre periférica es la fuente de progenitores más usada en el TPH con donantes no familiares, debido fundamentalmente a ser la fuente preferida por los donantes.
A finales de los 80, el cordón umbilical fue usado por primera vez como fuente de progenitores para TPH alogénico. Durante la década de los 90, su uso se extendió considerablemente para dar respuesta a la necesidad de tener donantes para aquellos pacientes que no tenían un donante HLA idéntico familiar o no emparentado(7).
Las células de sangre de cordón umbilical son inmunológicamente menos reactivas y permiten una mayor disparidad HLA entre donante y receptor. En general, puede afirmarse que el TPH, tanto de sangre periférica como de cordón umbilical, no han proporcionado resultados mejores que la medula ósea, aunque han proporcionado ventajas operativas innegables en los programas de TPH de los centros y han facilitado el acceso al TPH de muchos pacientes en las últimas dos décadas(16). Sin embargo, el TPH de cordón umbilical presenta desventajas en cuanto al escaso número de células para el TPH, su uso único y la necesidad de crear complejas estructuras para su adecuado almacenaje y gestión(7).
El donante familiar haploidéntico: el donante que siempre está ahí
A pesar de la disponibilidad de donantes no familiares y de unidades de cordón umbilical criopreservadas, todavía un gran número de pacientes no tienen acceso a un TPH por falta de un donante apropiado y con la premura necesaria.
Sin embargo, el donante haploidéntico (aquel que comparte un haplotipo con el receptor) permitiría que casi todos los pacientes que necesitan un TPH alogénico puedan tener acceso al mismo. Más aún, en el caso de los pacientes pediátricos, donde prácticamente todos tienen, al menos, un donante haploidéntico disponible.
El TPH haploidéntico rompe radicalmente con el principio de la compatibilidad HLA y superar dicha barrera ha sido posible mediante el uso de nuevas estrategias de prevención de la EICH(19). Estas estrategias no solo muestran que es posible realizar un TPH superando la barrera del HLA, sino que ponen de manifiesto la importancia de la inmunidad innata en el control de la enfermedad leucémica post-TPH. Dicho control se basa en que las células NK se activan al reconocer diferencias en los epítopos de HLA clase I.
Actualmente, existen dos plataformas clínicas de TPH haploidéntico. Una se basa en el uso de ciclofosfamida post-TPH junto con otros inmunosupresores, y la otra se basa en la eliminación total o parcial de los linfocitos T del donante en el inóculo, o de algunas subpoblaciones de los mismos (Fig. 4)(19,20).
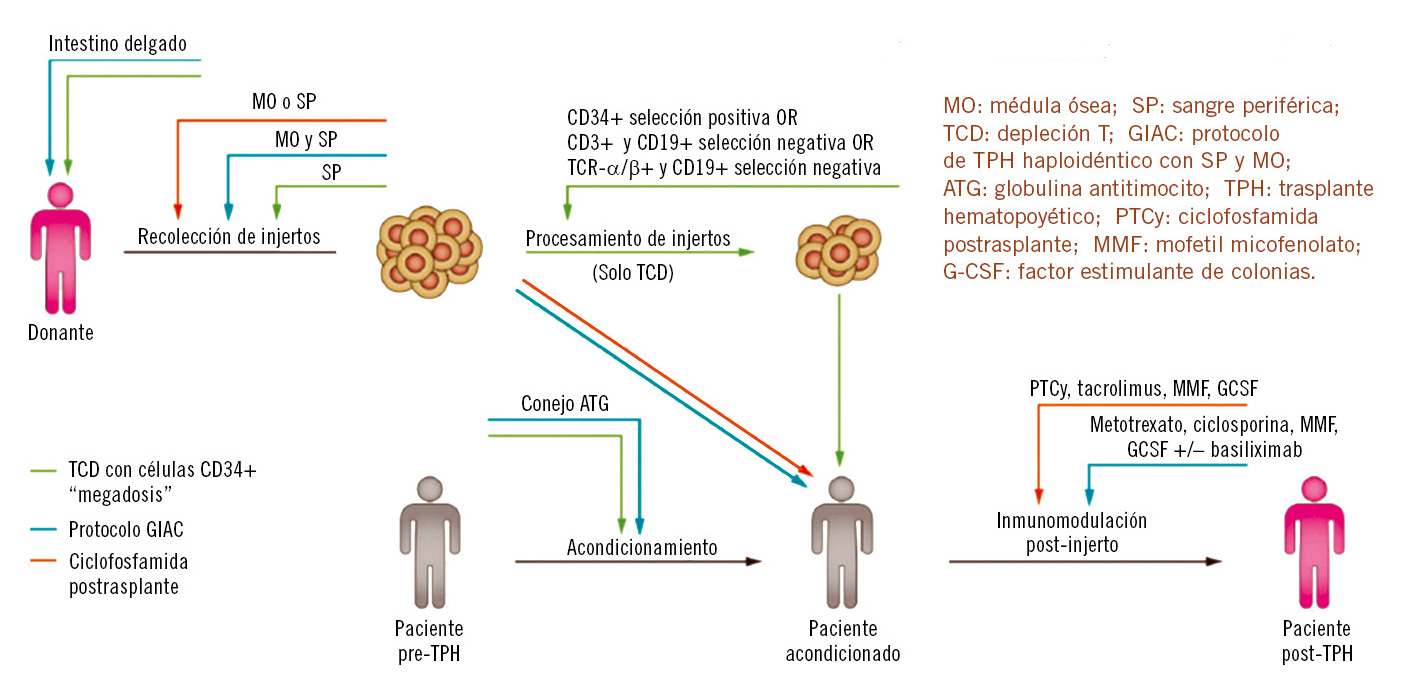
Figura 4. Plataformas de trasplante haploidéntico.
La eliminación de los linfocitos T o de algunas subpoblaciones permite suprimir el uso de fármacos inmunosupresores post-TPH, haciendo del TPH haploidéntico una plataforma para la terapia celular post-TPH.
Las células NK y los linfocitos T compiten por la IL-15, modulando la actividad inflamatoria post-TPH. Las células NK son potentes reguladoras de la inmunidad adaptada y lo hacen mediante la inhibición de la expansión clonal de los CD4+, estimulados antigénicamente y eliminando los CD8+ y las células dendríticas presentadoras de antígenos, además de produciendo citocinas inmunomoduladoras como la IL-10. En la tabla I, se ven las diferentes características de las dos subpoblaciones: linfocitos T y células NK.

Las células NK son la primera subpoblación de linfocitos en emerger tras el injerto hematopoyético y su rápida recuperación, tanto en número como en su perfil madurativo, se ha asociado con un mejor resultado post-TPH haploidéntico(21,22). Otros factores que influyen en el resultado del TPH haploidéntico son el genotipo KIR de las células NK (mejor con donante con genotipo B)(21) y la edad del donante (preferible usar donantes más jóvenes)(23). Ambos factores se asocian a una mejor supervivencia del TPH, debido a una menor probabilidad de recaída en el primer caso, y a una menor mortalidad del trasplante en el segundo.
Estudios clínicos recientes muestran superioridad en los resultados del trasplante haploidéntico frente al cordón umbilical(24) y resultados similares a los obtenidos con donantes convencionales, como el donante HLA compatible(25). Estos resultados han cambiado el modo de abordar la búsqueda de un donante “alternativo” al donante HLA familiar idéntico. De hecho, el TPH haploidéntico es el tipo de trasplante con mayor incremento en su actividad en los últimos años, en detrimento del trasplante de cordón umbilical.
Futuro del TPH alogénico
En la actualidad, y a pesar de las nuevas terapias emergentes contra algunas hemopatías malignas, el número de trasplantes realizados en los últimos años, no ha hecho más que aumentar, posiblemente debido a que algunas de estas terapias han rescatado pacientes que anteriormente no hubieran sido considerados como candidatos a TPH. Además de un aumento en la actividad, hemos asistido a una reducción importante de la mortalidad relacionada con el trasplante.
Sin embargo, el gran problema del TPH, especialmente en las enfermedades malignas, ha sido y continúa siendo, la recaída de la enfermedad leucémica. Por lo tanto, los esfuerzos van dirigidos a reducir las tasas de recaída post-TPH.
En primer lugar, reduciendo la carga tumoral pre-TPH. Es decir, reducir todo lo posible la enfermedad residual del paciente.
En segundo lugar, incrementando el efecto injerto contra leucemia, diseñando terapias celulares basadas en células NK y linfocitos T activados contra antígenos específicos de la hematopoyesis y que no generen EICH.
Por último, las terapias de mantenimiento post-TPH han mostrado una reducción significativa en la recaída en algunos tipos de leucemia, mejorando la supervivencia.
El futuro del TPH pasa por algunos hitos importantes que están en fase de ensayo clínico.
Uno sería el uso de fármacos no genotóxicos (anticuerpos monoclonales y radioinmunoterapia) como acondicionamiento que, además de facilitar el injerto de las células hematopoyéticas del donante, reduzca la enfermedad residual pre-TPH sin incrementar el riesgo de secuelas graves a largo plazo, muy importante en la población pediátrica.
Otros esfuerzos van dirigidos a considerar el TPH como una plataforma para la terapia celular específica. Esto permitiría reconstruir el sistema inmune tras el TPH con células específicas, para controlar las infecciones oportunistas y la recaída. Es esta última frontera, por ahora, la que abre más expectativas en la forma de terapia celular más antigua conocida, que es el TPH.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito.
Bibliografía
1. Osgood EE, Riddle MC, Matthews TJ. Aplastic anemia treated with daily transfusions and intravenous bone marrow; case report. Ann Intern Med. 1939; 13: 357-8.
2. Lorenz E, Uphoff D, Reid TR, Shelton E. Modifications of irradiation injury in mice and quinea pigs by bone marrow injections. J Nat Cancer Inst. 1951; 12: 197-201.
3. Santos GW. History of bone marrow transplantation. Clin Haematol. 1983; 12: 611-39.
4. Thomas ED, Lochte HL Jr, Lu W, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. 1957; 257: 491-7.
5. Van Bekkum DW. Biology of acute and chronic graft-versus-host reactions: predictive value of studies in experimental animals. Bone Marrow Transplant. 1994; 14: S51-5.
6. Gatti RA, Meawissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet. 1968; 2: 1366-9.
7. Broxmeyer HE, Douglas GW, Hangoc G, Cooper S, Bard J, English D, et al. Human umbilical cord blood as a potential source of transplantable haematopoietic stem/progenitor cells. Proc Natl Acad Sci USA. 1989; 82: 3828-32.
8. Gale RP, Fuchs EJ. Is there really a specific graft-versus-leukaemia effect? Bone Marrow Transplant. 2016; 51: 1413-5.
9. Thomas ED, Storb R, Clift RA, Fefer A, Johnson L, Neiman PE, et al. Bone marrow transplantation. N Engl J Med. 1975; 292: 832-43; 895-902.
10. Pierini A, Ruggeri L, Mancusi A, Carotti A, Falzetti F, Terenzi A, et al. T cell depletion and no post-transplant immune suppression allow separation of graft versus leukemia from graft versus host disease. Bone Marrow Transplantation. 2019; 54: 775-9.
11. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. 2015; 125: 2677-89.
12. Spitzer TR, McAfee S, Sackstein R, Colby C, Toh HC, Multani P, et al. Intentional induction of mixed chimerism and achievement of antitumor responses after non myeloablative conditioning therapy and HLA-matched donor bone marrow transplantation for refractory hematologic malignancies. Biol Blood Marrow Transplant. 2000; 6: 309-20.
13. Kinsella FAM, Zuo J, Inman CF, Pearce H, Maggs L, Eldershaw SE, et al. Mixed chimerism established by hematopoietic stem cell transplantation is maintained by host and donor T regulatory cells. Blood Adv. 2019; 3: 734-43.
14. Kolb H-J. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood. 2008; 112: 4371-83.
15. Pérez A, González-Vicent M, Ramírez M, Sevilla J, Madero L, Díaz MA. Intentional induction of mixed haematopoietic chimerism as platform for cellular therapy after HLA-matched allogeneic stem cell transplantation in childhood leukaemia patients. Br J Haematol. 2008; 140: 340-3.
16. Granot N & Storb R. History of hematopoietic cell transplantation: challenges and progress. Haematologica. 2020; 105: 2716-29.
17. Montoro J, Ceberio I, Hilden P, Maloy MA, Barker J, Castro-Malaspina H, et al. Ex Vivo T Cell-Depleted Hematopoietic Stem Cell Transplantation for Adult Patients with Acute Myelogenous Leukemia in First and Second Remission: Long-Term Disease-Free Survival with a Significantly Reduced Risk of Graft-versus-Host Disease. Biol Blood Marrow Transplant. 2020; 26: 323-32.
18. Díaz MA, Villa M, Alegre A, Lamana ML, de la Vega A, Granda A, et al. Collection and transplantation of peripheral blood progenitor cells mobilized by G-CSF alone in children with malignancies. Br J Haematol, 1996; 94: 148-54.
19. Kanarky CJ, Fuchs, EJ, Luznik L. Modern Approaches to HLA-Haploidentical Blood or Marrow Transplantation. Nat Rev Clin Oncol. 2016; 13: 10-24.
20. Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med. 1998; 339: 1186-93.
21. Díaz MA, Pérez-Martínez A, Herrero B, Deltoro N, Martínez I, Ramírez M, et al. Prognostic factors and outcomes for pediatric patients receiving an haploidentical relative allogeneic transplant using CD3/CD19-depleted grafts. Bone Marrow Transplant. 2016; 51: 1211-6.
22. Díaz MA, Zubicaray J, Molina B, Abad L, Castillo A, Sebastián E, et al. Haploidentical Stem Cell Transplantation in Children With Hematological Malignancies Using alphabeta(+) T-Cell Receptor and CD19(+) Cell Depleted Grafts: High CD56(dim)/CD56(bright) NK Cell Ratio Early Following Transplantation Is Associated With Lower Relapse Incidence and Better Outcome. Front Immunol. 2019; 10: 2504.
23. González-Vicent M, Molina B, Deltoro N, Sevilla J, Vicario JL, Castillo A, et al. Donor age matters in T-cell depleted haploidentical hematopoietic stem cell transplantation in pediatric patients: Faster immune reconstitution using younger donors. Leuk Res. 2017; 57: 60-4.
24. Sanz J, Montoro J, Solano C, Valcarcel D, Sampol A, Ferra C, et al. Prospective randomized study comparing myeloablative unrelated umbilical cord blood transplantation versus HLA-haploidentical related stem cell transplantation for adults with hematologic malignancies. Biol Blood Marrow Transplant. 2020; 26: 358-66.
25. Nagler A, Labopin M, Houhou M, Aljurf M, Mousavi A, Hamladji RM, et al. Outcome of haploidentical versus matched sibling donors in hematopoietic stem cell transplantation for adult patients with acute lymphoblastic leukemia: a study from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. J Hematol Oncol. 2021; 14: 53.

 How to suspect cancer in Primary Care
How to suspect cancer in Primary Care