 |
| Temas de FC |
B. Ochoa Fernández, B. González Martínez
Servicio de Hematología Pediátrica. Hospital Universitario de la Paz. Madrid
| Resumen
Los síndromes mielodisplásicos (SMD) son un grupo heterogéneo de trastornos clonales hematopoyéticos que, en la edad pediátrica, ocurren frecuentemente asociados a trastornos genéticos predisponentes. Son enfermedades raras en la edad pediátrica y presentan características clínicas y biológicas distintivas con respecto a esta enfermedad en otros grupos etarios. |
| Abstract
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic disorders, which in the pediatric age frequently occur in association with genetic predisposition disorders. MDS are rare in the pedi-atric age and present distinctive clinical and biological characteristics compared with this disease in other age groups. |
Palabras clave: Síndrome mielodisplásico en la infancia; Citopenia refractaria; Fallo medular congénito; Trasplante de progenitores hematopoyéticos.
Key words: Childhood myelodysplastic syndrome; Refractory cytopenia; Inherited bone marrow failure syndrome; Stem cell transplantation.
Pediatr Integral 2021; XXV (6): 320 – 325
Síndromes mielodisplásicos en Pediatría
Introducción
Los síndromes mielodisplásicos son un grupo heterogéneo de trastornos clonales hematopoyéticos con predisposición variable a evolucionar a leucemia aguda mieloblástica.
Los síndromes mielodisplásicos (SMD) son un conjunto de trastornos clonales de las células progenitoras hematopoyéticas, caracterizados por la presencia de hematopoyesis ineficaz (lo que condiciona citopenias periféricas) y alteraciones morfológicas celulares (dishemopoyesis)(1).
Este grupo de desórdenes hematopoyéticos son una entidad típica de personas de edad avanzada, con una mediana de presentación en adultos de 70 años(2), a diferencia de la edad pediátrica, donde presentan una baja frecuencia y se consideran una enfermedad rara.
Los SMD pediátricos presentan características diferentes a los de la edad adulta y su diagnóstico involucra un escenario diferente, tanto a nivel pronóstico como terapéutico(3).
El SMD en la edad pediátrica puede ocurrir de novo sin una aparente causa subyacente, ser secundario a otros factores (como la exposición previa a agentes citotóxicos) o desarrollarse en asociación a síndromes de fallo medular congénitos o adquiridos. Además, los niños con SMD tienen un riesgo aumentado de progresión a leucemia mieloide aguda (LMA)(4).
Es importante destacar que, el hallazgo de citopenias y/o rasgos displásicos, tanto en el hemograma como en medula ósea (MO), no son sinónimos de SMD; por lo que la sospecha de SMD requiere siempre poner en marcha estudios específicos, tanto en sangre periférica como en MO, que permitan excluir la existencia de otras enfermedades que presentan algunas características comunes.
En esta revisión, resumiremos brevemente los aspectos particulares del SMD pediátrico desde el diagnóstico hasta las opciones de tratamiento actuales.
Epidemiologia
El SMD es una patología muy común en adultos mayores de 70 años, pero muy infrecuente en niños, constituye menos del 5% de todas las hemopatías malignas.
Los SMD en la población pediátrica representan el 4% de la patología hemato-oncológica. Su incidencia anual es de 1 a 4 casos por millón, con una distribución similar en Europa y EE.UU.(1,5).
La edad media en el momento del diagnóstico suele ser entre 7 y 8 años, aunque la enfermedad se puede diagnosticar en todas las edades. Ambos sexos se ven igualmente afectados(6).
Etiopatogenia
Los SMD son enfermedades clonales que surgen en una célula progenitora hematopoyética independientemente de su estirpe, pudiendo afectar a la serie eritroide, mieloide o megacariocítica, causando alteraciones en la maduración normal de estas células.
Varios factores exógenos se han relacionado con el origen de los SMD, entre ellos, la exposición previa a agentes tóxicos y fármacos citostáticos, incluyendo agentes alquilantes e inhibidores de la topoisomerasa, usados como: quimioterápicos, tratamiento previo con radioterapia, ciertos antibióticos (p. ej., cotrimoxazol), algunos inmunosupresores y los factores estimulantes de eritropoyesis, granulocitos o análogos de trombopoyetina(2). Además, como hemos mencionado antes, en la infancia presenta ciertas peculiaridades y, típicamente, se asocia a ciertos trastornos genéticos predisponentes y enfermedades hematológicas congénitas (Tabla I).

Aproximadamente, el 50% de los niños con SMD tienen un trastorno constitucional conocido. Muchos otros, también, son portadores de alteraciones genéticas no conocidas previamente y que les confieren un riesgo mayor de desarrollar este tipo de enfermedad(7).
Comparado con los SMD en adultos, el conocimiento de las alteraciones genéticas somáticas en la población pediátrica es aún limitado. Estudios recientes han descrito que genes típicamente afectados en el SMD del adulto como: TET2, TP53, DNMT3A y SF3B1, no están relacionados con la patogenia del SMD pediátrico(8). Igualmente, la alteración citogenética típica del SMD del adulto, la deleción del 5q, no es frecuente en niños(9) (Tabla II).
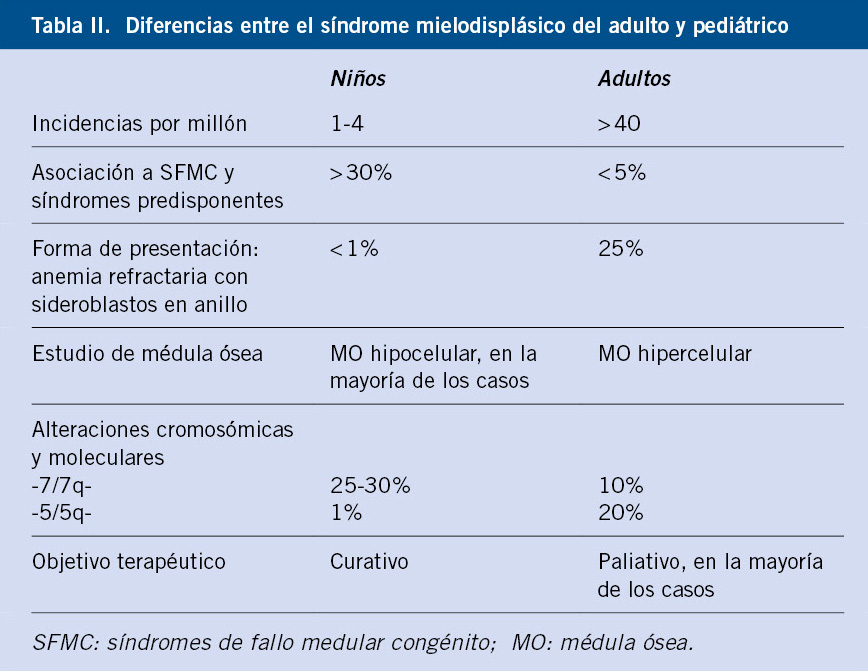
Dentro de las aberraciones citogenéticas relacionadas con los SMD pediátricos, la más frecuente es la monosomía del cromosoma 7, con una frecuencia del 30%(10). Otros genes relacionados descritos son: SETBP1 y los oncogenes ASXL1, RUNX1 y RAS, siendo en ellos las principales alteraciones somáticas encontradas en pacientes pediátricos con SMD(1).
También, se han identificado mutaciones en línea germinal en el gen GATA2, que representan el 15% de los SMD primarios avanzados (SMD con exceso de blastos, SMD-EB)(11,12). Actualmente, se ha incrementado la evidencia que apoya que las variantes de la línea germinal en diferentes factores de transcripción como: GATA2, RUNX1, ETV6 o CEBPA, están relacionados con SMD familiar(10).
Diagnóstico y clasificación
El hallazgo de citopenias y rasgos mielodisplásicos, tanto en el hemograma como en medula ósea, no son sinónimos de SMD. En todos los casos, deben excluirse las causas de citopenias y displasias transitorias.
El hallazgo de citopenias asociadas a disminución de la celularidad de la MO, es una combinación que puede ser causada por muchos trastornos diferentes; por lo tanto, es importante un adecuado diagnóstico diferencial para llegar a un juicio clínico correcto (Tabla III).

El diagnóstico de los SMD requiere siempre poner en marcha una serie de estudios específicos llevados a cabo por un hematólogo pediátrico, que permita excluir la existencia de otras enfermedades que presentan algunas características comunes y que incluya, tanto estudios en sangre periférica como en médula ósea. Los estudios realizados ante sospecha de SMD se describen en la tabla IV.

Es imprescindible tener presente que mielodisplasia no es sinónimo de SMD. Al no disponer de un dato patognomónico de SMD en todos los casos, se debe excluir toda causa de citopenia y displasia transitoria.
En cuanto a la clasificación de los SMD, existe cierta controversia, ya que las clasificaciones generales dirigidas a adultos no se ajustan a las características del paciente pediátrico(13). En el año 2008, la OMS publicó una clasificación recomendada para el paciente pediátrico, que es la más utilizada en nuestro medio(14). Posteriormente, en el año 2016, se ha publicado una actualización de la clasificación general, reflejada en la tabla V(15), que no introduce cambios a la subclasificación pediátrica (Tabla VI).


Presentación clínica y tratamiento
En general, las manifestaciones clínicas vienen condicionadas por el grado de citopenias que presente el paciente y las series celulares afectadas. En ocasiones, son hallazgos incidentales en controles analíticos realizados por otras causas. El trasplante de progenitores hematopoyéticos (TPH) es la única opción curativa para estos pacientes.
Citopenia refractaria de la infancia (CRI)
CRI es el subtipo de SMD más frecuente en niños y adolescentes, representando la mitad de los casos; esta entidad como su nombre indica, no suele presentarse en adultos. Afecta a ambos sexos por igual. La sintomatología será la derivada de sus citopenias, pero hasta un 20% de los enfermos están asintomáticos al diagnóstico(4). Estos pacientes suelen debutar con trombocitopenia y/o neutropenia, pudiendo asociar también anemia, aunque con menos frecuencia(13). La citopenia refractaria se define por: citopenias persistentes con <5% de blastos en MO, <2% de blastos en sangre periférica (SP) y la presencia de rasgos displásicos, más frecuentemente observados en las línea eritroide y megacariocítica. El estudio de MO suele mostrar celularidad disminuida en un 80% de los pacientes(1).
Es importante destacar que los SFMC y la aplasia medular adquirida pueden presentar características similares y, a veces, indistinguibles de CRI, por lo que las muestras deben ser valoradas por un hematólogo experto y correlacionadas con otras alteraciones encontradas en los pacientes.
El cariotipo es el factor pronóstico más importante de la progresión del SMD a leucemia mieloblástica (LMA). Las anomalías citogenéticas están presentes en aproximadamente un 30% de los pacientes con CRI y la alteración más frecuente encontrada es la monosomía del cromosoma 7. Estos pacientes tienen mayores probabilidades de progresión a SMD avanzado o LMA que aquellos niños que tienen cariotipos normales u otras alteraciones(3).
El tratamiento del CRI dependerá de la gravedad de las citopenias, requerimientos transfusionales, la presencia de infecciones de repetición y de la existencia de un donante HLA idéntico para realización de TPH.
El TPH es la única opción curativa y es el tratamiento de elección en pacientes con CRI que presentan alteraciones genéticas, como la monosomía del cromosoma 7 o cariotipos complejos, dado que se asocian a un mayor riesgo de progresión a SMD avanzado o LMA (Fig. 1).
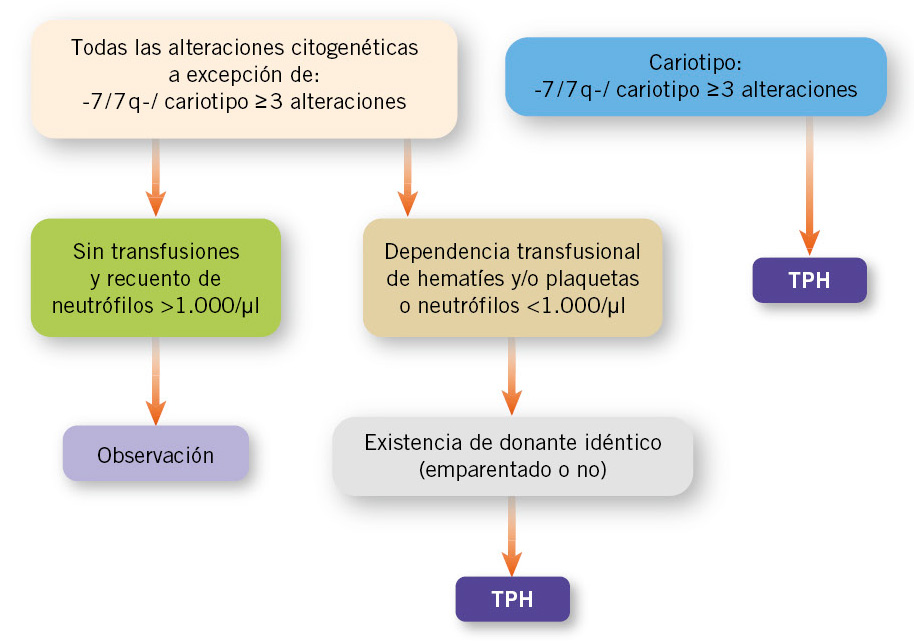
Figura 1. Algoritmo terapéutico de citopenia refractaria de la infancia. Algoritmo propuesto por Locatelli, et al. Adaptado y modificado(1). TPH: trasplante de progenitores hematopoyéticos.
SMD con exceso de blastos o SMD avanzado: (SMD-EB)
Se caracteriza por la presencia de entre 2 y 19% de blastos en SP o de entre 5 y 19% de blastos en MO.
Existe otra variante en la clasificación llamada SMD-EB en transformación (SMD-EB-t), que involucra un porcentaje de blastos de entre 20-29% en SP o MO(1). Es importante destacar que, el porcentaje de blastos por sí solo no es suficiente para diferenciar cualquiera de estos subtipos de SMD de una LMA de novo. Aspectos, como las alteraciones citogenéticas típicas de cada enfermedad y la ausencia de rápida progresión y organomegalias, nos orientarán a la existencia de un SMD.
El tratamiento de este tipo de pacientes con SMD-EB, con o sin transformación, representa un reto. La única terapia curativa es el TPH. Adicionalmente, este grupo de pacientes han sido relacionados con una gran toxicidad derivada del tratamiento y alto riesgo de recaída. La primera opción para realizar un TPH en este tipo de pacientes es el trasplante alogénico de donante HLA idéntico, ya sea emparentado o no. En caso de que el paciente posea mutaciones en línea germinal es importante tomar en cuenta el estudio genético de los posibles donantes familiares, quienes pueden ser portadores de las mismas mutaciones.
Actualmente, con las nuevas técnicas de manipulación del injerto de progenitores hematopoyéticos, que disminuyen los riesgos intrínsecos de un trasplante no idéntico, se está considerando el TPH haploidéntico como una alternativa adecuada en pacientes que no poseen un hermano idéntico y en quienes la posibilidad de encontrar un donante no emparentado representa un retraso en el tratamiento. Cabe destacar que el retraso en el TPH, se considera un factor de riesgo que disminuye la posibilidad de supervivencia.
Conclusiones
Los SMD en la infancia son enfermedades infrecuentes, pero dada su gravedad, hace que el diagnóstico de sospecha esté siempre presente ante citopenias o displasias morfológicas de las diferentes líneas celulares.
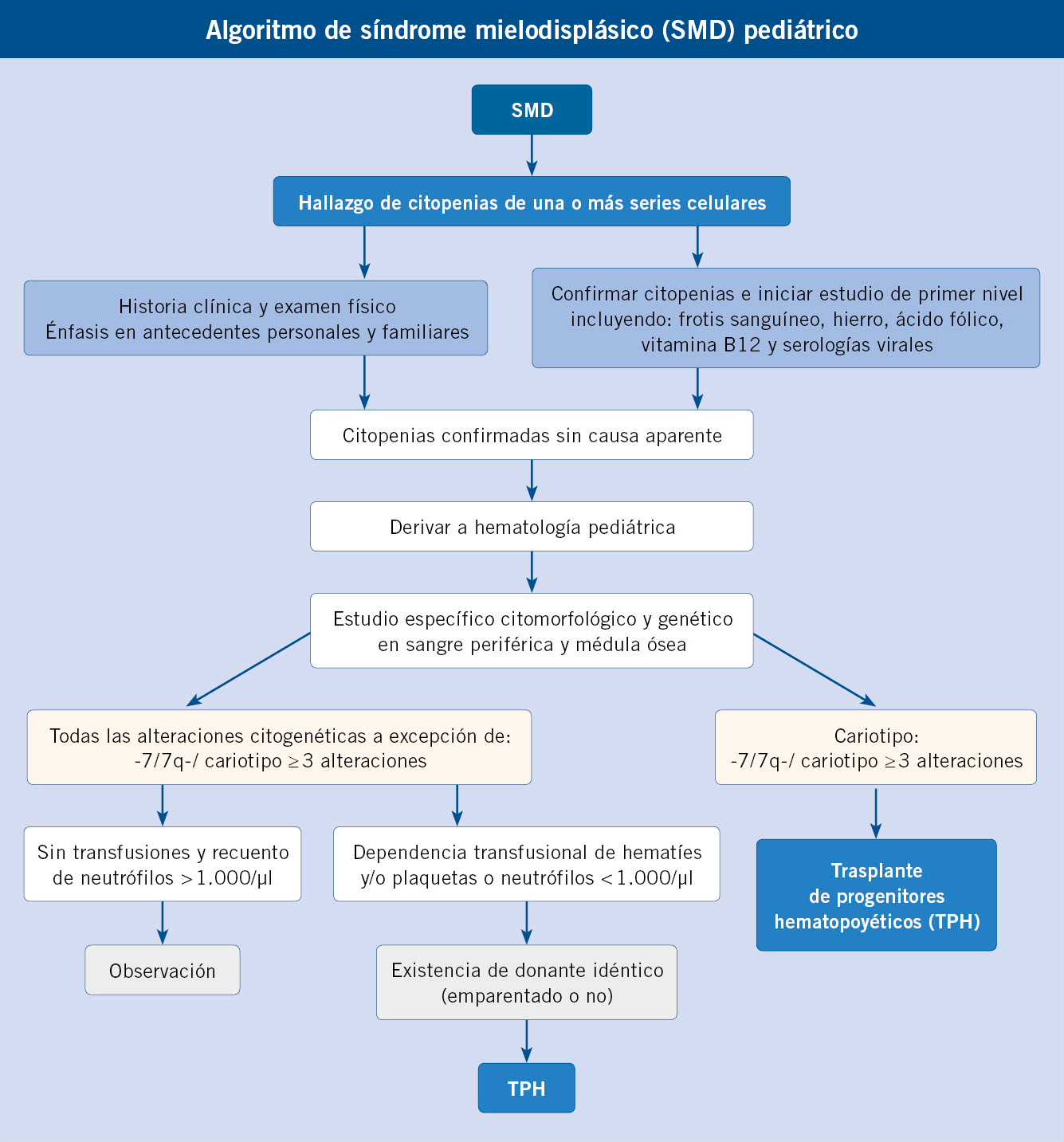
Los SMD pediátricos (ver algoritmo al final del artículo) son trastornos clonales heterogéneos de la hematopoyesis, con características específicas y diferencias significativas con los SMD de adultos.
Los síndromes de predisposición genética se detectan en una notable proporción de pacientes. El TPH está indicado en todos los pacientes con SMD-EB o SMD-EB-t, y en aquellos con CRI y características citogenéticas de alto riesgo, neutropenia grave o dependencia de transfusiones.
El retraso en el tratamiento es un importante factor de riesgo que empeora el pronóstico post-TPH. Actualmente, gracias a las nuevas técnicas de manipulación de los injertos de progenitores hematopoyéticos, la opción del TPH haploidéntico representa una opción viable cuando no existe un donante idéntico.
Función del pediatra de Atención Primaria
Ante el hallazgo de citopenia en analítica rutinaria, confirmar la determinación en un nuevo control, añadiendo frotis de sangre periférica y completar estudio con perfil férrico, vitamina B12 y fólico; es importante descartar causas secundarias (fundamentalmente, tóxicas e infecciosas). Si se comprueba la existencia de citopenia en 1 o más líneas celulares, sin causa justificada, derivar a hematología pediátrica.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1.*** Locatelli F, Strahm B. How I treat myelodysplastic syndromes of childhood. Blood. 2018; 131: 1406-1414. doi:10.1182/blood-2017-09-765214.
2.** Grupo español de síndromes mielodisplásicos (GESMD). Guía española de diagnóstico y tratamiento de los síndromes mielodisplásicos y leucemia mielomonocítica crónica. Haematologica. Edición 2020. http://www.gesmd.es/wpcontent/uploads/2021/09/GuiaSMDLMMC2020.pdf (acceso 14 septiembre 2021)
3. Hasle H. Myelodysplastic and myeloproliferative disorders of childhood. Hematol Am Soc Hematol Educ Progr. 2016; 2016: 598-604. doi:10.1182/asheducation-2016.1.598.
4.** Galaverna F, Ruggeri A, Locatelli F. Myelodysplastic syndromes in children. Curr Opin Oncol. 2018; 30: 402-8. doi:10.1097/CCO.0000000000000488.
5. Passmore SJ, Chessells JM, Kempski H, Hann IM, Brownbill PA, Stiller CA. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol. 2003; 121: 758-67. doi:10.1046/j.1365-2141.2003.04361.x.
6. Hofmann I. Pediatric myelodysplastic syndromes. J hemopathology. 2015; 8: 127-41. doi:10.1007/s12308-015-0253-4.
7. Rau ATK, Shreedhara AK, Kumar S. Myelodysplastic Syndromes in Children : Where Are We Today ? Ochser J. 2012; 12: 216-20.
8. Pastor V, Hirabayashi S, Karow A, Wehrle J, Kozyra EJ, Nienhold R, et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia. 2017; 31: 759-62. doi:10.1038/leu.2016.342.
9. Hasserjian RP. Myelodysplastic Syndrome Updated. pathobiology. 2019; 86: 7-13. doi:10.1159/000489702.
10.** Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017; 8: 1557. doi:10.1038/s41467-017-01590-5.
11. Göhring G, Michalova K, Beverloo HB, Betts D, Harbott J, Haas OA, et al. Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood. 2010; 116: 3766-9. doi:10.1182/blood-2010-04-280313.
12. Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016; 127: 1387-97; doi:10.1182/blood-2015-09-669937.
13. Escobar NF, Drelichman G, Viso M, Moreno Peñarrieta K, Daloi K. Realidad en nuestro país de una enfermedad infrecuente y grave. Hematología. 2016; 20: 90-102.
14. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114: 937-51. doi:10.1182/blood-2009-03-209262.
15. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127: 2391-405. doi:10.1182/blood-2016-03-643544.
Bibliografía recomendada
- Locatelli F, Strahm B. How I Treat How I treat myelodysplastic syndromes of childhood. Blood. 2018; 131: 1406-14. doi:10.1182/blood-2017-09-765214.
Completo artículo de revisión sobre los síndromes mielodisplásicos en la infancia, con descripción detallada del manejo y tratamiento según cada subtipo de SMD, incluyendo varios casos clínicos de ejemplo.
- Grupo español de síndromes mielodisplásicos (GESMD). Guía española de diagnóstico y tratamiento de los síndromes mielodisplásicos y leucemia mielomonocítica crónica. Haematologica. Edición 2020. http://www.gesmd.es/wpcontent/uploads/2021/09/GuiaSMDLMMC2020.pdf (acceso 14 septiembre 2021)
Guía amplia de la Sociedad Española del Síndrome Mielodisplásico, con orientación general tanto para adultos como en Pediatría, de los aspectos más importantes de la enfermedad, diagnóstico y tratamiento.
- Galaverna F, Ruggeri A, Locatelli F. Myelodysplastic syndromes in children. Curr Opin Oncol. 2018; 30: 402-8. doi:10.1097/CCO.0000000000000488.
Artículo reciente en el que se hace una amplia y magnífica revisión del estado actual del tratamiento del síndrome mielodisplásico en el paciente pediátrico, a su vez, actualizando revisiones anteriores.
- Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017; 8: 1557. doi:10.1038/s41467-017-01590-5.
Interesante artículo de revisión en el que se intenta ahondar sobre las cuestiones aún no resueltas sobre el síndrome mielodisplásico, profundizando en la etiología de estos síndromes.
| Caso clínico |
|
Niño de 10 años que refiere aparición de hematomas y petequias de manera recurrente, sin antecedentes de traumatismos importantes, durante los últimos 2 meses. No ha presentado sangrado a otros niveles ni otra sintomatología. Antecedentes familiares Antecedente de linfoma en abuela materna. Sin otros antecedentes familiares de interés. Antecedentes personales Sin antecedentes médicos ni quirúrgicos importantes. Vacunas según calendario. Exploración física Buen estado general. Bien hidratado, nutrido y perfundido. Numerosos hematomas dispersos en miembros superiores e inferiores. Petequias en región facial. Sin adenopatías palpables. Auscultación cardiopulmonar: normal. Abdomen: normal. Neurológico: normal. Exploración ORL: normal. Pruebas complementarias Hemograma: leucocitos: 1.380/μl (fórmula normal); neutrófilos: 750/μl; linfocitos: 540 /μl; monocitos: 80 /μl; eosinofilos: 10/μl. Hemoglobina: 9,5 g/dl; hematocrito: 28%; VCM: 98,9 fL; HCM: 33,6 pg; CHCM: 34 g/dl; plaquetas: 18.000/μl. Bioquímica básica, renal y hepática, amilasa: normales, proteínas totales 7,4 g/dl; ferritina: 46 ng/ml (7-140); hierro: 64 μg/dl (55-140); vitamina B12: 472 pg/ml; folato sérico: 8,6 ng/ml; coagulación: normal. PCR: <0,5 mg/L. EAB: normal. Orina: normal. Ecografía abdominal: sin hallazgos significativos. Radiografía de tórax: normal. Evolución: ante hallazgo de pancitopenia, se decide ingreso hospitalario para completar estudio a cargo del servicio de Hematología.
|

 How to suspect cancer in Primary Care
How to suspect cancer in Primary Care