 |
| Temas de FC |
P. Velasco Puyó*, L. Murillo Sanjuán**
*Servicio de Oncología y Hematología pediátricas. Profesor Asociado de la Universidad Autónoma de Barcelona. Vall d’Hebron Barcelona Hospital Campus. **Servicio de Oncología y Hematología pediátricas. Vall d’Hebron Barcelona Hospital Campus. Barcelona
| Resumen
La leucemia aguda es el cáncer infantil más frecuente. La leucemia aguda linfoblástica es más frecuente y de mejor pronóstico que la mieloblástica, aunque ambas han alcanzado una elevada supervivencia, de un 90% y 70%, respectivamente. El diagnóstico de precisión, añadiendo técnicas moleculares a las ya conocidas de morfología, citogenética y citometría de flujo, permiten una mejor clasificación de la enfermedad, seguimiento de la respuesta al tratamiento y uso de terapias dirigidas. La inmunoterapia ha demostrado buena respuesta en pacientes en recaída y refractariedad. |
| Abstract
Acute leukemia is the most common childhood cancer. Acute lymphoblastic leukemia is more common and has a better prognosis than myeloblastic leukemia, although both entities have achieved high survival rates of 90% and 70%, respectively. Precision diagnosis, incorporating molecular techniques to those already known for morphology, cytogenetics and flow cytometry, allow a better classification of the disease, monitoring of the response to treatment and the use of targeted therapies. Immunotherapy has shown a good response in patients with relapse and refractory to treatmen. |
Palabras clave: Leucemia linfoblástica aguda; Leucemia mieloide aguda; Niños.
Key words: Acute lymphoblastic leukemia; Acute myeloid leukemia; Children.
Pediatr Integral 2021; XXV (6): 296 – 307
Leucemia aguda en Pediatría
Introducción
La leucemia aguda es el resultado de la proliferación clonal de células inmaduras (blásticas) en la médula ósea. Estas células pierden su capacidad de diferenciación a células normales. Según la línea celular proliferante, se diferencian dos tipos básicos de leucemias agudas: mieloblástica (LAM) (línea mieloide, de la que en condiciones normales, derivan: glóbulos rojos, neutrófilos, monocitos, eosinófilos, basófilos y plaquetas) y linfoblástica (LAL) (línea linfoide, de la que derivan los linfocitos).
En ocasiones, la leucemia no tiene una diferenciación clara de un solo linaje, clasificándose como leucemia aguda de linaje ambiguo, suponiendo menos del 4% de todas las leucemias agudas. Pueden ser leucemias indiferenciadas (sin antígeno específico de linaje) o de fenotipo mixto (con expresión de antígenos de más de un linaje) en el mismo blasto (leucemia bifenotípica) o en distintos blastos del mismo paciente (leucemia bilineal). Suelen tener un mal pronóstico y se tratan generalmente con protocolos de leucemia linfoide aguda, a menos que la expresión de los marcadores o mutaciones más significativas sean mieloides, debiendo cambiar el tratamiento si el tipo de blasto predominante cambia durante el mismo.
Los primeros esquemas de tratamiento quimioterápico fueron diseñados para mejorar la supervivencia de la leucemia infantil, que era nula en los años 50. Como consecuencia de la estratificación de los pacientes en diferentes grupos de riesgo, la adaptación de la intensidad del tratamiento a cada grupo, los avances en el tratamiento de soporte, la introducción del trasplante de progenitores hematopoyéticos, la inmunoterapia y el tratamiento dirigido, en la actualidad se alcanzan tasas de supervivencia global a los 3 años cercanas al 90% en la LAL.
Epidemiología
La leucemia aguda linfoblástica es más frecuente y de mejor pronóstico que la leucemia mieloide aguda.
La leucemia aguda es el cáncer infantil más frecuente, diagnosticándose unos 300 nuevos casos al año en España, lo que supone el 27,9% de todos los tumores malignos pediátricos, según el registro español de tumores infantiles (RETI-SEHOP)(1). La LAL tiene un pico de incidencia entre 1 y 4 años de edad.
La LAL es más frecuente, supone el 80% de las leucemias agudas pediátricas y el 21,9% de las neoplasias malignas registradas en nuestro medio. La incidencia de LAL a nivel mundial es mayor en niños de etnia hispánica(2). La supervivencia a los 3 años del diagnóstico, ha aumentado de 66% en los años 80 a 91%, actualmente. Sin embargo, la supervivencia a la LAL disminuye con la edad, siendo de 65% en jóvenes adultos, 40% en adultos y 15% en ancianos.
En el caso de la LAM, la incidencia aproximada es de 7 casos por millón de niños anualmente, suponiendo aproximadamente el 20% de los casos de la leucemia aguda(3) y, aunque su pronóstico ha mejorado en las últimas décadas, su supervivencia global se encuentra en torno al 70-80% de los pacientes(1).
El esfuerzo en la colaboración de diferentes grupos cooperativos internacionales, está permitiendo un enfoque de manejo más homogéneo, intentando disminuir la toxicidad a corto y largo plazo, con tratamientos más dirigidos y personalizados.
Fisiopatología
Las alteraciones genéticas que llevan a la leucemia son resultados de alteraciones genéticas primarias que, en algunos casos, pueden tener una predisposición germinal.
La leucemia se origina en un precursor hematopoyético que, tras adquirir alteraciones en genes implicados en los procesos de diferenciación, maduración y/o regulación de la apoptosis, se transforma en una célula maligna inmadura.
En algunos casos, se ha evidenciado una predisposición genética a la leucemia aguda, como en el síndrome de Down (con más riesgo de LAM y LAL B con reordenamiento CRLF2), mutaciones en genes supresores de tumores (Li-Fraumeni), síndromes de fallo medular congénito (anemia de Fanconi, síndrome de Shwachman-Diamond, anemia de Blackfan-Diamond o disqueratosis congénita) e inmunodeficiencias (Wiskott-Aldrich, Agammglobulinemia de Bruton).
La predisposición familiar es rara pero, en estos casos, se han identificado variantes genéticas que predisponen a la leucemia y que también se observan en casos esporádicos (sin predisposición familiar) de leucemia, como las mutaciones en TP53 germinal en el síndrome de Li Fraumeni, que conlleva alto riesgo familiar de desarrollar distintos tumores, y que también se encuentra con frecuencia como mutación somática (en los blastos) en los casos de LAL con baja hipodiploidía. Ejemplos similares de mutaciones o variantes germinales son: los genes de ETV6, PAX5 y IKZF1 en el caso de la LAL; deficiencia del GATA2 en la LAM; así como diversos polimorfimos que predisponen a la LAL con menor penetrancia (ARID5B, BAK1, CDKN2A/CDKN2B,BMI1-PIP4K2A, CEBPE, ELK3, ERG, GATA3, IGF2BP1,IKZF1, IKZF3, USP7, LHPP)(4).
Algunos de los reordenamientos frecuentes en LAL pueden detectarse al nacimiento, demostrando un posible origen prenatal de la leucemia, como en algunos casos de reordenamientos KMT2A y ETV6-RUNX1 y, anecdóticamente, en algunos casos de hiperdiploidia o reordenamiento de ZNF384.
El papel de las células madre en el desarrollo y resistencia de la LAL al tratamiento, se ha estudiado en los últimos años. A raíz de su capacidad de quiescencia y capacidad de desarrollar tumores en ratones inmunodeprimidos, se les ha denominado células iniciadoras de tumores (LICs). Probablemente, el tratamiento de mantenimiento prolongado y la capacitación del propio sistema inmune a controlar la proliferación tumoral son determinantes para evitar las recaídas a partir de las LICs.
Además de la influencia genética, hay otros factores, como los ambientales o las infecciones virales. Se ha observado que la incidencia de LAL infantil ha aumentado con la industrialización de los países, lo que probablemente sea consecuencia de la exposición a diferentes agentes químicos o radiación, aunque el grado de contribución al desarrollo de LAL es controvertido. El hecho de que el pico de incidencia de LAL se sitúe entre los 1 y 4 años, la época en la que madura el sistema inmune, ha generado la hipótesis de que algunos casos de LAL podrían ser consecuencia de una respuesta inmune aberrante a infecciones comunes. Además, algunos virus se han relacionado con el desarrollo de determinados cánceres, como es el caso de la infección por el virus de Epstein-Barr (VEB) en casos de linfoma de Burkitt y LAL B madura. Recientemente, algunos autores han relacionado una menor incidencia de LAL con la menor exposición a virus, derivada del confinamiento por la pandemia de SARS-CoV-2, aunque son necesarios más estudios para confirmar esta asociación(5).
Los propios tratamientos de quimioterapia o radioterapia pueden ser la causa de leucemia, siendo la LAM el tipo de neoplasia secundaria más frecuente en pacientes pediátricos tratados con estas terapias.
Clínica
Las citopenias y las organomegalias no asociadas a infecciones, deben de hacer sospechar un proceso linfoproliferativo.
La clínica de la LAL suele ser aguda, presentando un 60% de los casos hepatoesplenomegalia y adenopatías, consecuencia de la infiltración blástica en órganos hematopoyéticos extramedulares, que también se puede observar en tejidos blandos (testes, orbita, piel, etc.), así como anemia, sangrado y/o fiebre, consecuencia de las citopenias resultantes de la ocupación de blastos en la médula ósea(6).
En el caso de la LAM, los síntomas también pueden ser inespecíficos al debut, aunque es importante destacar que, alrededor de un 30% de pacientes, pueden tener afectación extramedular, siendo la piel/tejido celular subcutáneo, el sistema nervioso central (SNC) y el hueso, los órganos más habitualmente afectados. También puede ser habitual la presencia de sarcomas granulocíticos o sarcomas mieloides, que son masas tumorales mieloides que aparecen en huesos con gran actividad hematopoyética y poco periostio, como es el caso de los huesos periorbitarios y de la base del cráneo, aunque también pueden aparecer en: tejido cutáneo, riñones, testes, etc. A veces, los sarcomas granulocíticos pueden aparecer como única manifestación de la enfermedad (2-4% de las LAM).
El debut de una leucemia aguda puede presentarse con complicaciones que precisan de un tratamiento urgente por el riesgo vital que conllevan, como:
• Síndrome de lisis tumoral espontánea, más frecuente en pacientes con LAL y alta carga tumoral e infiltración extramedular, que llevan a elevación de urato, potasio y fósforo.
• Hiperleucocitosis (> 100.000 leucocitos/mcL), más frecuente en la LAM que en la LAL y, dentro de esta última, más habitual en adolescentes y LAL T. Puede llevar a una clínica de leucostasis por acúmulo de los blastos en los vasos de pequeño calibre del SNC y/o del pulmón, dando lugar a síntomas neurológicos, como confusión, cefalea o coma, o manifestaciones pulmonares, como dificultad respiratoria, edema pulmonar o hemorragia pulmonar.
• Obstrucción aérea por adenopatías o ensanchamiento mediastínico (más frecuente en las LAL) (Fig. 1).
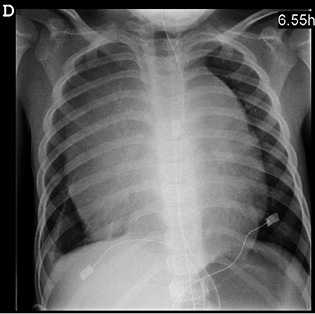
Figura 1. Radiografía de tórax antero posterior de un niño de 5 años al debut de una LAL T. Destaca la ocupación del mediastino anterior por una gran masa bilateral.
• Obstrucción de la vena cava superior por masa mediastínica, que lleva a edema y congestión facial, dificultad respiratoria y clínica neurológica.
• La infiltración blástica del sistema nervioso central, ocurre en menos del 5% de los pacientes.
• La hemorragia o la trombosis también pueden ser manifestaciones al debut, más habituales en el caso de la LAM. Pueden ocurrir incluso fenómenos de coagulación intravascular diseminada en el 5% de pacientes, especialmente en la leucemia promielocítica aguda (subtipo M3 de la clasificación franco-americano-británica o FAB), como consecuencia de la liberación de factores procoagulantes por parte de los blastos, lo que determina una auténtica emergencia médica.
Diagnóstico diferencial
La clínica de dolor osteoarticular con la que se presentan un 23% de los pacientes, puede llevar al diagnóstico erróneo de enfermedad reumatológica.
Las adenopatías y organomegalias pueden confundirse con infecciones víricas, como la mononucleosis (VEB).
Las citopenias que se encuentran habitualmente al debut de la leucemia aguda, pueden orientar el diagnóstico a enfermedades autoinmunes más frecuentes a esta edad, como la trombocitopenia inmune primaria (PTI); pero en casos de PTI, no es frecuente encontrar otras citopenias u organomegalias en la exploración. La extensión de sangre periférica y la evaluación de los parámetros de lisis tumoral pueden ayudar a orientar el caso. Si existen dudas de que el paciente tenga una leucemia, siempre se deberá descartar a través de un aspirado de médula ósea (AMO).
Los pacientes con leucemia aguda pueden presentarse con pancitopenia al debut (más frecuente en algunos subtipos de LAM, como la leucemia aguda promielocítica y la leucemia aguda megacarioblástica), lo que puede llevar a un diagnóstico erróneo de aplasia o fallo medular. En estos casos, el aspirado y/o biopsia medular confirmará el diagnóstico definitivo entre estas entidades y otras que pueden tener la misma presentación, como son: los síndromes mieloproliferativos con mielofibrosis, la infiltración medular de tumores sólidos (neuroblastoma, linfoma, sarcoma de Ewing, osteosarcoma y rabdomiosarcoma), la linfohistiocitosis hemofagocítica y la aplasia postinfecciosa por VH6 o parvovirus, entre otros.
La proliferación extramedular, en forma de ensanchamiento mediastínico, organomegalias o adenopatías masivas, son signos habituales de los linfomas B y T no Hodgkin, pero pueden darse también en el caso de la LAL. Es necesaria la realización de un AMO, que mostrará más de un 25% de linfoblastos en el caso de las LAL. La afectación extramedular puede plantear problemas de diagnóstico con: timoma, teratoma, linfoma, neuroblastoma, malformaciones o causas infecciosas en caso de masas mediastínicas y linfoma, tumor ovárico, tumor hepático, rabdomiosarcoma, tumor de Wilms y neuroblastoma en el caso de masas abdominales.
Diagnóstico
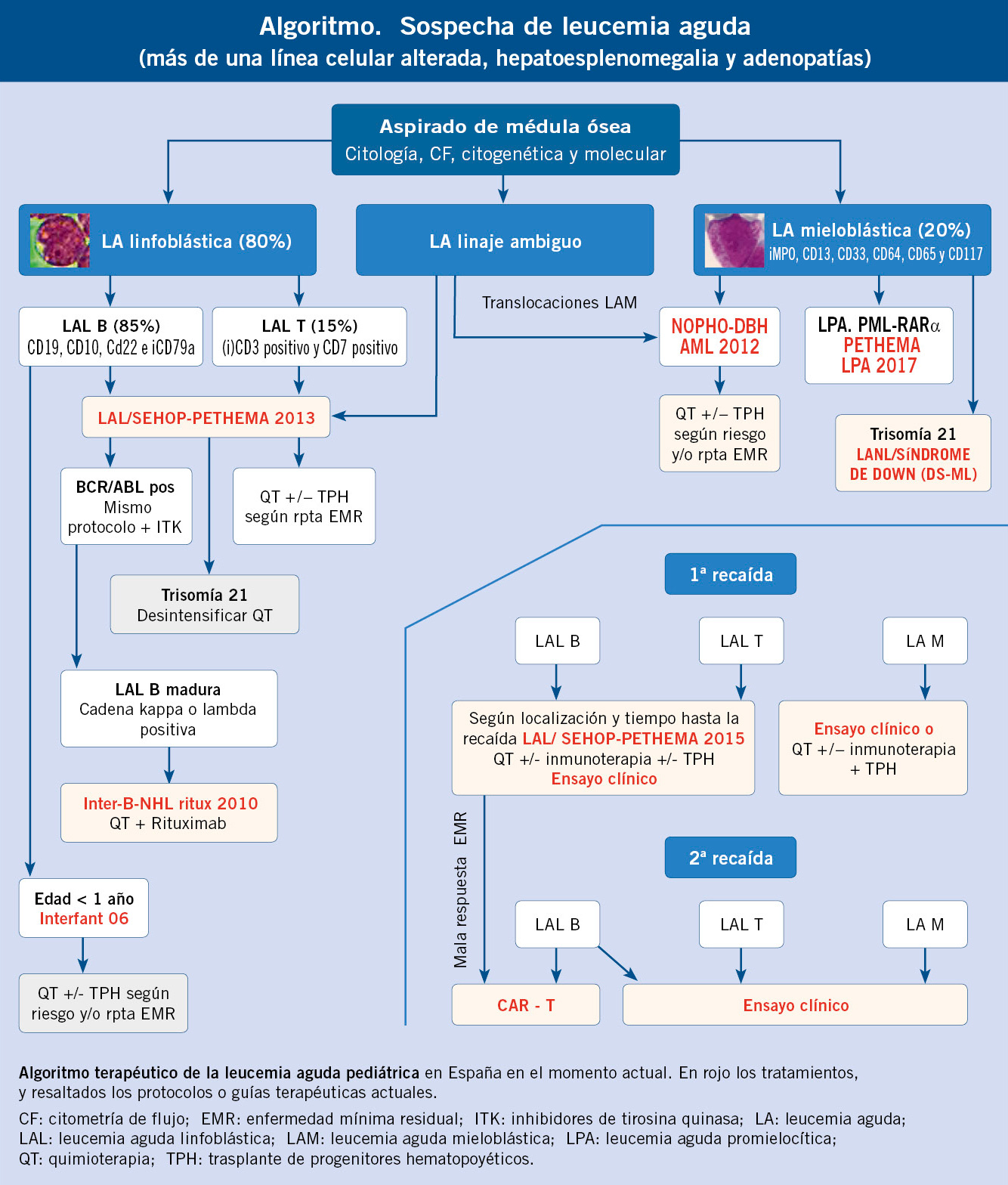
El diagnóstico de la leucemia aguda se basa en el estudio de la médula ósea mediante citomorfología, citometría de flujo y estudios citogenéticos y moleculares.
Para realizar el diagnóstico de leucemia aguda, es necesario demostrar la existencia de más de un 25% de blastos por citología en el AMO (Fig. 2).
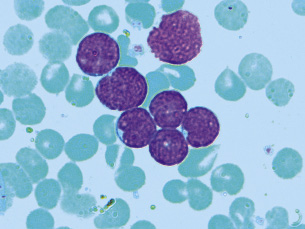
Figura 2. Frotis de médula ósea de una paciente con leucemia aguda linfoblástica (May-Grünwald-Giemsa), donde destaca: alta relación núcleo-citoplasmática, escaso citoplasma, núcleos de cromatina laxa, algunos de ellos con indentaciones y con nucléolos prominentes.
Los pilares diagnósticos de la leucemia aguda a partir de las muestras de AMO son:
• La citología, revisaremos por microscopía óptica la extensión de sangre periférica y de aspirado medular con tinción de May-Grünwald Giemsa, con el fin de identificar blastos de características de LAL L1 o L2 según la clasificación FAB (Franco-Americana-Británica), linfoblastos tipo Burkitt y mieloblastos. Si hay dudas en el diagnóstico de la estirpe mieloide de los blastos, se puede hacer la tinción de mieloperoxidasa característica del linaje de LAM.
• El inmunofenotipo por citometría de flujo, es la prueba gold estándar para la clasificación del linaje de la LAL. A través de las características de dispersión de la luz, según su tamaño y granulación, y a la fluorescencia que emiten las células gracias a la utilización de anticuerpos unidos a los antígenos propios, esta técnica sirve para la clasificación y caracterización de la enfermedad (Tabla I)(7).
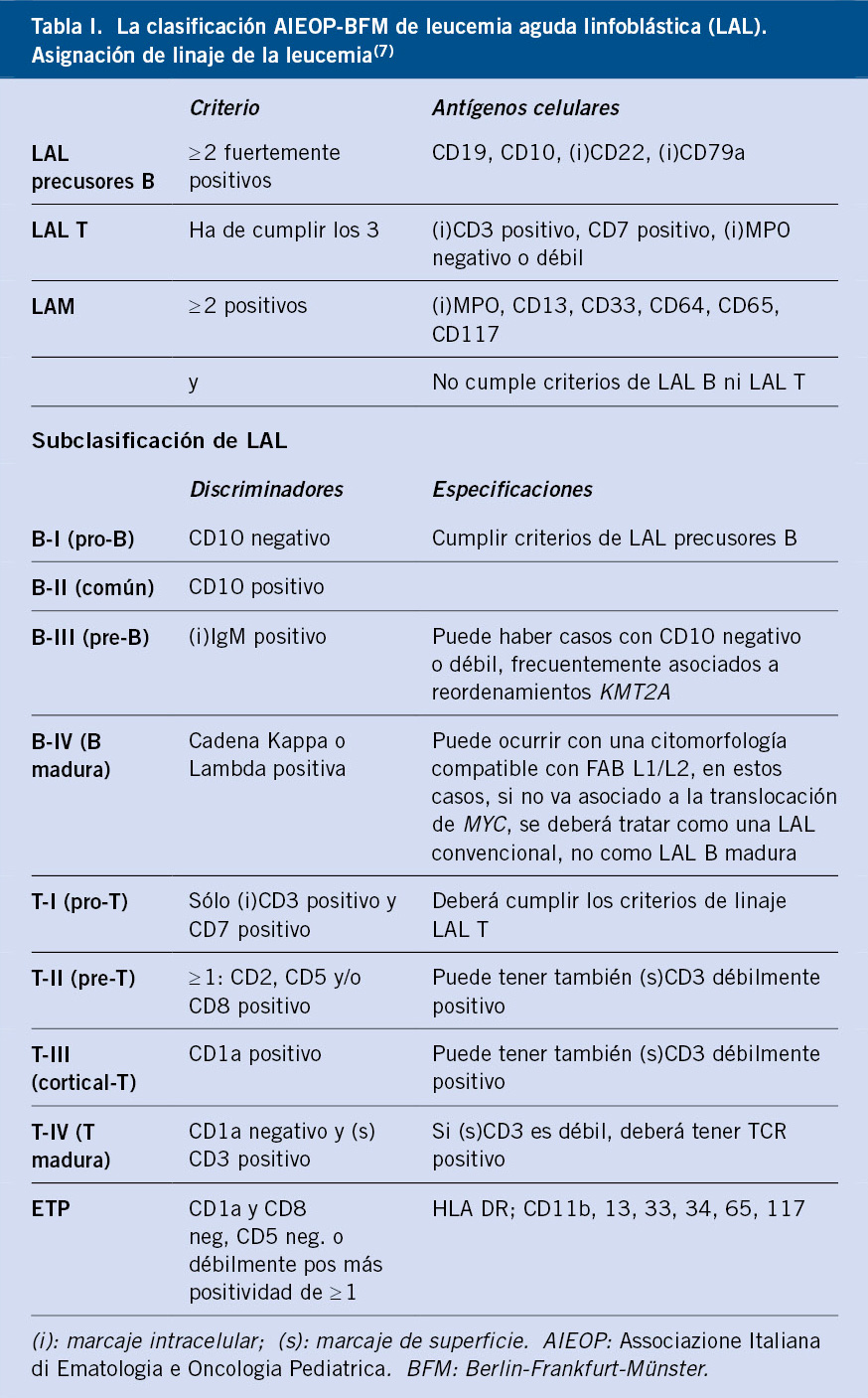
La caracterización inicial de la leucemia permite el seguimiento de la respuesta al tratamiento, tras alcanzar la remisión completa evaluando lo que conocemos como enfermedad mínima residual (EMR).
• La citogenética con la realización del cariotipo de los blastos, la hibridación in situ fluorescente (FISH), y el estudio molecular por RT-PCR y técnicas de secuenciación masiva (NGS), permiten detectar alteraciones genéticas primarias y secundarias específicas, con valor pronóstico y terapéutico.
Será necesaria la realización de una punción lumbar diagnóstica para poder establecer la afectación del SNC. En el caso de la LAM, dado el alto riesgo de sangrado que presentan estos pacientes al diagnóstico, la mayoría de los grupos recomiendan posponer este estudio hasta que el riesgo de hemorragia haya pasado.
Con todos estos estudios al debut, no solo confirmaremos el diagnóstico de leucemia aguda, sino que definiremos el riesgo de la enfermedad y con ello la intensidad del tratamiento, así como posibles dianas terapéuticas en caso de refractariedad.
Leucemia aguda linfoblástica
Los subgrupos de LAL se clasifican según criterios de la OMS en: LAL B (85%), LAL T (15%), y con muy escasa incidencia, LAL NK. La clasificación de la OMS define las translocaciones recurrentes de la LAL B, pero fuera de estos grupos hay un 30% de LAL B (que suponen un 50% de las recaídas) que clasificamos gracias a estudios de NGS como B others, siendo la mayor parte de mal pronóstico, especialmente las Philadelphia (Ph) like, que suponen un 50% de las B others y un 15% de las LAL pediátricas. Tienen un perfil genético similar a las LAL philadelphia positivo, pero sin reordenamiento BCR/ABL.
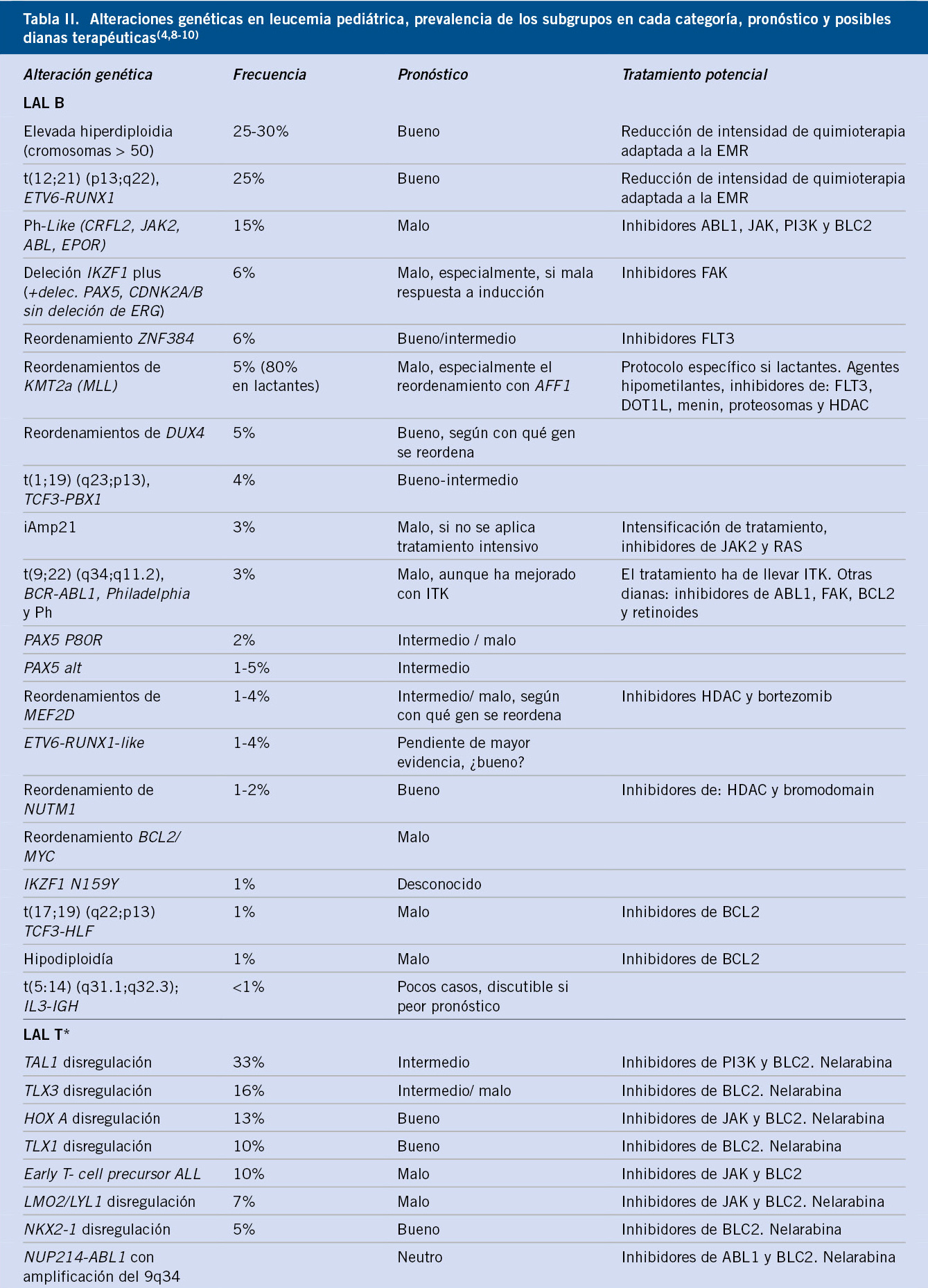
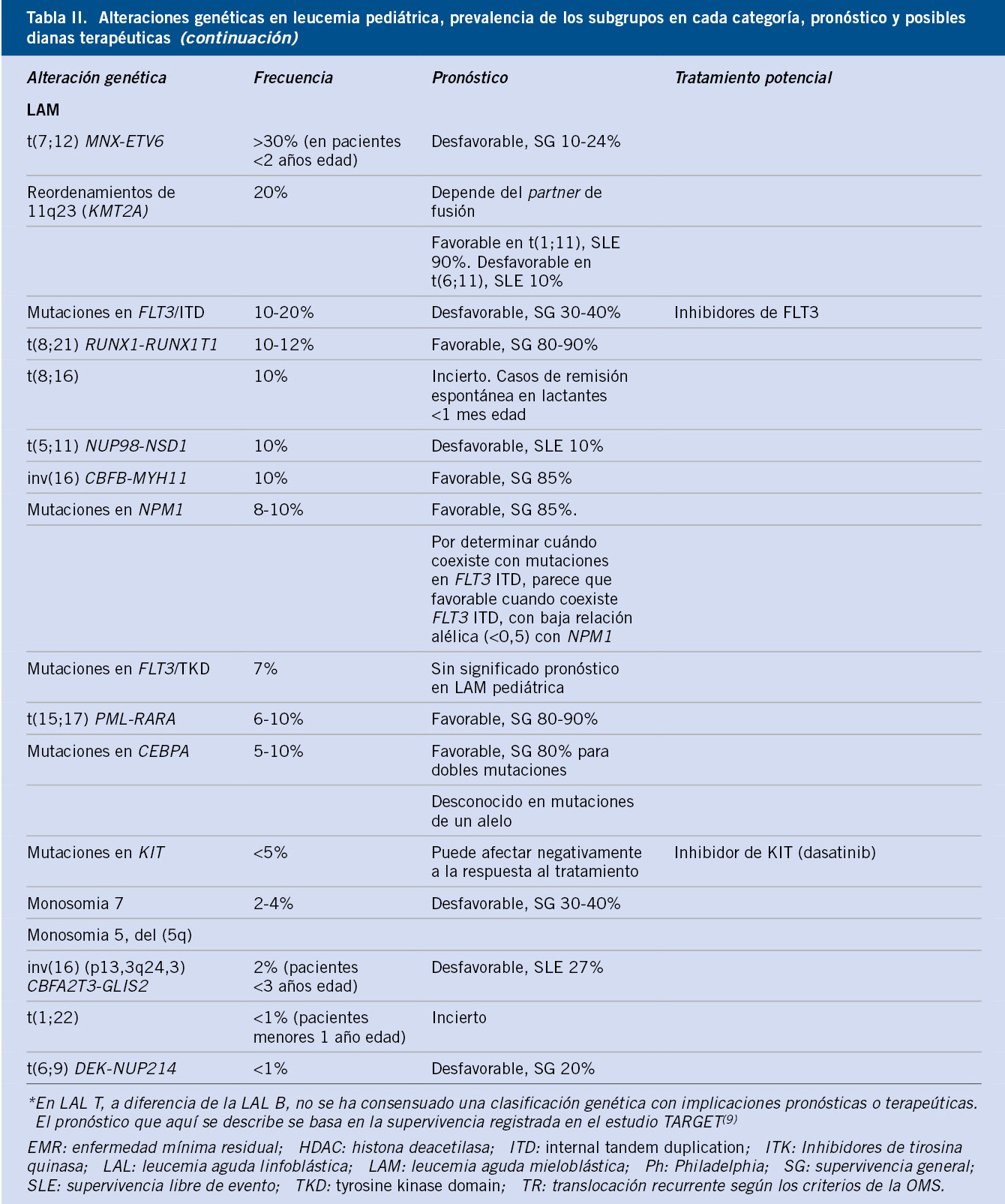
En la tabla II, resumimos las alteraciones genéticas, características y posibles dianas terapéuticas potenciales de las leucemias pediátricas(4,8-10).
Factores pronósticos y estratificación del riesgo
La presentación al diagnóstico y la respuesta al tratamiento, marcan el riesgo de la enfermedad.
Los factores de riesgo que permitirán la clasificación de los pacientes en grupos de riesgo y la intensidad del tratamiento a administrar, se detallan en la tabla III.

La respuesta al tratamiento, se basa en el seguimiento de la EMR, que se realiza principalmente por técnicas de citometría de flujo, así como por técnicas moleculares en determinados protocolos y subtipos de leucemia (Tabla IV).
La cinética de la EMR emerge como uno de los factores pronósticos más potentes, demostrándose en los últimos análisis del grupo COG (Children’s Oncology Group), que valores de EMR <0,01% al final de la inducción, se relacionan con una supervivencia libre de enfermedad un 17% mayor(1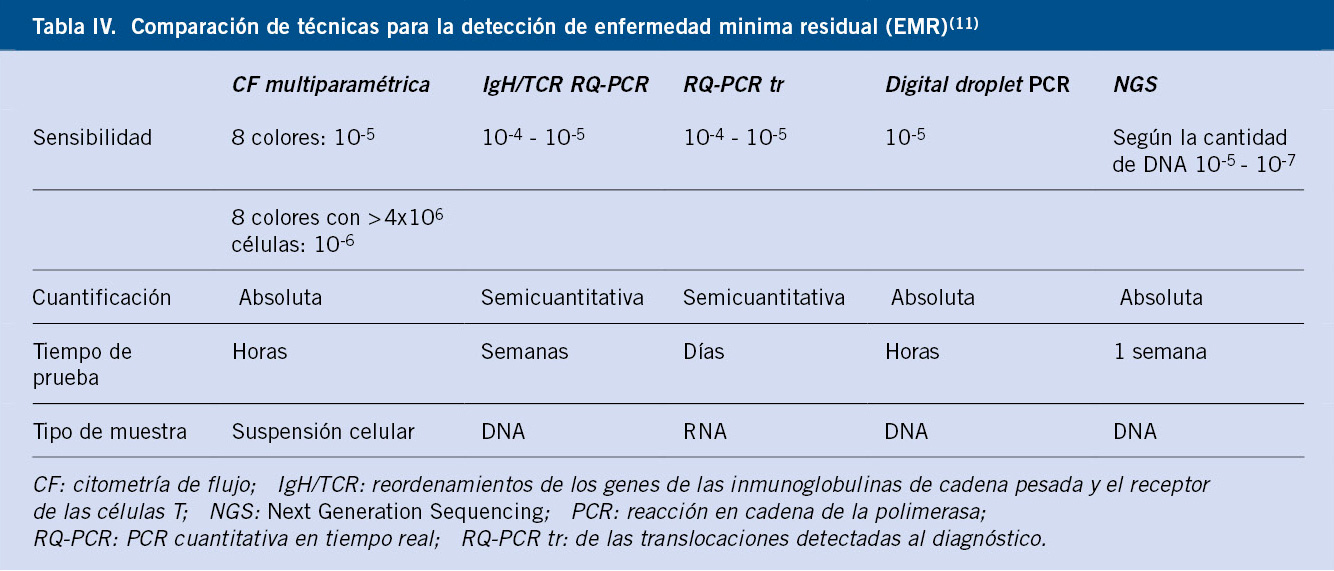 1). Los tiempos de evaluación y niveles de EMR exigidos en los futuros protocolos, se diferenciarán según el subtipo de LAL.
1). Los tiempos de evaluación y niveles de EMR exigidos en los futuros protocolos, se diferenciarán según el subtipo de LAL.
Tratamiento
El tratamiento de la LAL se basa en la poliquimioterapia durante 2 años. La inmunoterapia y el tratamiento diana han demostrado su efecto en pacientes en recaída o refractariedad.
El tratamiento de LAL en primera línea en nuestro país actualmente sigue la guía de recomendación terapéutica LAL/SEHOP-PETHEMA 2013, tiene una duración de 2 años en la mayoría de los casos, y comprende 3 fases:
• Inducción: con la combinación de prednisona, vincristina, daunorrubicina y asparaginasa, en la primera fase, y mercaptopurina, ciclofosfamida y citarabina, en la segunda fase de la inducción, se busca inducir a la remisión completa de la enfermedad. Para tratar o prevenir la enfermedad en SNC, se realizarán punciones lumbares con instilación de quimioterapia intratecal (IT), lo que ha conseguido resultados similares a la radioterapia con menor toxicidad.
• Consolidación: con la administración de metotrexate a altas dosis, mercaptopurina e IT, se consigue consolidar la respuesta, especialmente en tejidos extrahematopoyéticos, como el SNC.
• Reinducción: se repite un esquema similar a la inducción, con dexametasona en lugar de prednisona, para disminuir el riesgo de recaída.
• Mantenimiento: tras 6-10 meses de poliquimioterapia con los ciclos anteriores (en función del grupo de riesgo del paciente, la duración y la intensidad es mayor), los pacientes comienzan un tratamiento con quimioterapia de baja intensidad hasta completar 2 años de tratamiento. El tratamiento diario con mercaptopurina oral y metotrexate oral semanal, junto a un periodo inicial en el que se añaden IT y asparaginasa, según el grupo de riesgo, ha demostrado tener un gran impacto en la incidencia de la recaída, aumentando esta en 2,7 veces en casos en los que la adherencia sea menor de un 95%.
La monitorización de la actividad de la asparaginasa durante el tratamiento, permite detectar inactivaciones silentes de la misma, lo que tiene un gran impacto en la respuesta al tratamiento.
En el caso de pacientes con LAL y síndrome de Down, es necesario adaptar la intensidad del tratamiento a la elevada toxicidad del mismo en estos pacientes.
En el caso de lactantes con LAL menores de 1 año de vida, se tratan según el protocolo Interfant-06, que incluye fármacos dirigidos a LAM por la inmadurez propia de los blastos leucémicos de esta enfermedad con características de diferenciación linfoide y mieloide. Pese a ello, el pronóstico en estos pacientes continúa siendo malo, con una supervivencia libre de enfermedad a los 6 años del 46%.
En el caso de pacientes con LAL T, la supervivencia suele ser un 5-10% menor que en LAL B(9). Actualmente, siguen el mismo protocolo, aunque probablemente, en los próximos ensayos se beneficiaran de tiempos de evaluación distintos y de la incorporación de nuevos fármacos, como la nelarabina.
A los pacientes en los que no se consiga una correcta respuesta al tratamiento, se les someterá a un trasplante de progenitores hematopoyéticos (TPH) previo tratamiento de acondicionamiento. La estandarización de la selección del donante, el acondicionamiento, el tratamiento de soporte y la profilaxis de la enfermedad injerto contra huésped han mejorado mucho los resultados del trasplante(12).
Con los esquemas actuales de tratamiento, en torno a un 15% de pacientes pediátricos con leucemia aguda linfoblástica sufren una recaída y, tras la recaída, las posibilidades de curación se reducen a menos del 50%.
Con el fin de mejorar estas cifras, se incorporarán en los próximos protocolos de tratamiento de LAL en primera línea y en recaída, las moléculas diana y la inmunoterapia, que actualmente están demostrando mayor eficacia (y, en muchos casos, menor toxicidad) en ensayos clínicos, siendo los más relevantes:
• Distintas generaciones de inhibidores de tirosina quinasa junto a la quimioterapia de base en pacientes con LAL Philadelphia positivo.
• ABL1 inhibidores como dasatinib y JAK inhibidores como ruxolitinib para pacientes con LAL Ph like.
• Inhibidores de BCL-2 como venetoclax en LAL de alto riesgo como: LAL ETP, reordenamientos de KMT2a, TCF3-HLF o hipodiploidía.
• Blinatumomab es un anticuerpo biespecifico que, al unirse al CD3 de los linfocitos T y a los CD19 de los blastos, activa la destrucción de los mismos por los linfocitos T activados. La sustitución de parte de la quimioterapia previa al TPH, en casos de LAL B en recaída, por blinatumomab, ha demostrado conseguir un 26% más de supervivencia y menor toxicidad(13).
• Inotuzumab-ozogamicina es un anticuerpo monoclonal que se une al CD22 de los linfoblastos, que ha demostrado también buenos resultados, al incorporarse en tratamientos de LAL B en recaída o refractariedad (85% de respuesta completa en pacientes pediátricos con esta indicación), como tratamiento puente al trasplante(14).
• La terapia de células T con receptores quiméricos de antígenos (CAR-T) modifica los linfocitos T del paciente, añadiéndole un receptor sintético que les permite reconocer a los linfoblástos. Su administración en pacientes pediátricos con LAL B en segunda recaída o refractariedad, ha conseguido mejorar los resultados de supervivencia en esta población, alcanzando una supervivencia a los 2 años de 66%, con buenos resultados también en enfermedad extramedular. Actualmente, el ensayo clínico AALL1721/Cassiopeia está investigando la terapia CAR-T en primera línea de tratamiento para LAL B de muy alto riesgo con EMR persistente, tras la inducción(15).
• El daratumumab (anticuerpo monoclonal anti CD 38), dasatinib, venetoclax y los inhibidores de proteosomas, son algunos de los tratamientos que se están evaluando en pacientes con LAL T en recaída o refractariedad. También, se están abriendo ensayos clínicos para evaluar constructos CAR-T contra LAL T.
Con la inclusión de los nuevos tratamientos, se espera no solo mejorar la supervivencia, sino también reducir los efectos tóxicos a largo plazo de la quimioterapia, que son principalmente: osteonecrosis (incidencia de 6%), osteoporosis (10-30%), obesidad, hipogonadismo, oligozoospermia, cardiopatía por antraciclinicos y segundas neoplasias (1-2%).
Leucemia mieloblástica aguda
Factores pronósticos y estratificación del riesgo
La estratificación del riesgo en los pacientes pediátricos con LAM, se basa principalmente en características citogenéticas y moleculares de la enfermedad, así como en la respuesta obtenida tras el tratamiento de inducción.
Citogenética y genética molecular
La tabla II recoge las principales alteraciones genéticas observadas en la LAM pediátrica, su frecuencia y su pronóstico, según la mayoría de grupos cooperativos.
Respuesta al tratamiento
La respuesta al primer ciclo de tratamiento, junto con las características citogenéticas y moleculares, constituyen los marcadores pronósticos principales.
La enfermedad mínima residual (EMR) mediante citometría de flujo multidimensional tras la quimioterapia de inducción, es un factor pronóstico y predice el riesgo de recaída(16,17). Existen otros métodos para la medida de la EMR, como el análisis de reacción en cadena de la polimerasa (PCR por sus siglas en inglés), basada en ARN de los transcritos específicos de la leucemia, y la evaluación genómica del aclaramiento de la mutación, aunque estos últimos están siendo estandarizados en Europa.
Los pacientes con EMR negativa tras un ciclo de inducción, tuvieron una EFS y OS del 65 y 77%, respectivamente, comparado con el 22 y 51% en aquellos que tuvieron EMR positiva(18). Además, la EMR al inicio de la consolidación, parece ser el predictor más fuerte de supervivencia.
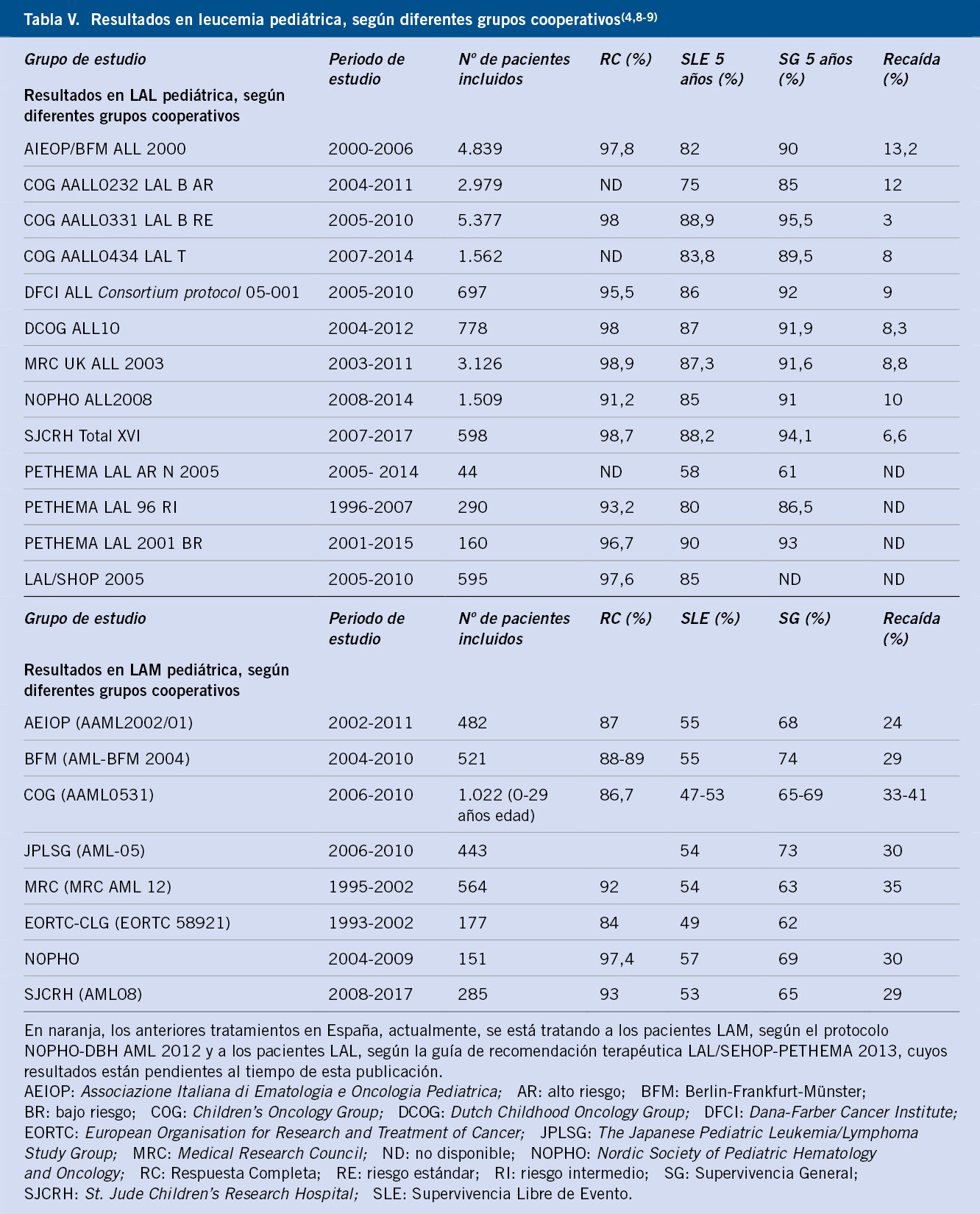
En la tabla V, se pueden observar los resultados de los principales grupos cooperativos a nivel internacional.
Tratamiento
El tratamiento de los pacientes pediátricos con LAM debería realizarse en el marco de ensayos clínicos randomizados. Este tratamiento se fundamenta en, al menos, 4-5 ciclos de quimioterapia intensiva basada en antraciclinas y citarabina.
Inducción
Todos los esquemas incluyen uno o dos ciclos de inducción. El régimen estándar de tratamiento de inducción incluye lo que se denomina “3 + 7” o “3 + 10” (3 días de un antraciclínico: daunorrubicina, idarrubicina o mitoxantrona; y 7-10 días de citarabina), con lo que se consiguen tasas de remisión >85%.
Varios estudios de los diferentes grupos cooperativos han tratado de determinar el tipo de antraciclina óptimo durante la inducción, la intensificación en las dosis de citarabina o la incorporación de otros agentes, como el etopósido, con resultados bastante similares en cuanto a supervivencia general entre las distintas ramas de tratamiento(17). La dosis acumulada de antraciclinas debe tenerse en consideración por el riesgo de cardiotoxicidad aguda y a largo plazo.
Consolidación
No está claro el número de ciclos necesario durante la consolidación, pero la mayoría de grupos administran entre 2 y 3 ciclos de consolidación. Estos ciclos consisten en combinaciones de citarabina a altas dosis con otros agentes.
Trasplante de progenitores hematopoyéticos (TPH)
El papel del TPH alogénico, como tratamiento de consolidación tras la adquisición de la 1ª remisión completa, es controvertido. Aunque varios estudios aportan evidencia sobre su eficacia anti-leucemia, se debe sopesar el beneficio con la mortalidad relacionada con el procedimiento y el beneficio sobre la supervivencia general. El beneficio del TPH alogénico parece mayor en los pacientes pediátricos con LAM de alto riesgo por sus características citogenéticas/moleculares y en aquellos que tienen EMR positiva tras la inducción(3), así como hay consenso en ofrecer el TPH a los pacientes pediátricos con LAM en 2ª remisión completa.
Profilaxis y tratamiento dirigido al SNC
La terapia dirigida al SNC se administra a todos los pacientes pediátricos con LAM, incluyendo aquellos sin afectación de SNC al debut, ya que la terapia sistémica tiene una eficacia limitada para erradicar los blastos ocultos en el compartimento del SNC. Se utiliza quimioterapia intratecal con un fármaco (citarabina o metotrexate) o triple terapia (citarabina, metotrexate e hidrocortisona) en función de los diferentes protocolos.
Nuevos agentes
Los efectos secundarios y la toxicidad de la quimioterapia son una preocupación importante en el tratamiento de la LAM pediátrica. Nuevos fármacos se están desarrollando en el marco de ensayos clínicos, enfocados a dianas moleculares.
Un agente se ha incorporado en primera línea en algunos ensayos fase 3: el gemtuzumab-ozogamicina, un anticuerpo monoclonal conjugado humanizado anti-CD33 expresado en la mayoría de casos de LAM pediátrica(19), así como otros, como los inhibidores de tirosín kinasa (sorafenib, midostaurin, gilteritinib) en los casos de mutaciones de FLT3, se están estudiando en ensayos clínicos de fases precoces.
Tratamiento de soporte
El tratamiento de soporte en estos pacientes ha contribuido, en gran medida, a mejorar la supervivencia en la última década.
Debido a la quimioterapia intensiva en el tratamiento de la LAM, es esperable el desarrollo de una aplasia profunda y prolongada (<500 neutrófilos/mcL durante >7 días). Es por ello, que estos pacientes se encuentran particularmente en riesgo de desarrollo de bacteriemias, principalmente por bacterias Gram-negativas y estreptococos del grupo viridans, así como infecciones fúngicas invasivas(20). El uso de tratamiento antibiótico profiláctico es controvertido por el riesgo de resistencias bacterianas, sin embargo, la profilaxis antifúngica está recomendada, así como la profilaxis frente al Pneumocystis jirovecii con cotrimoxazol. El uso profiláctico de G-CSF (factor estimulante de colonias de granulocitos) no está recomendado, ya que no ha demostrado influir en la supervivencia general.
Subgrupos especiales
Aunque el tratamiento de subgrupos especiales de LAM pediátrica no es el objeto de esta revisión, cabe destacar que existen protocolos específicos para algunos determinados subtipos de LAM.
Tal es el caso de la leucemia promielocítica aguda (LPA), que se caracteriza por reordenamientos de 17q21, involucrando al gen RARA. La incorporación al tratamiento del ácido transretinoico (ATRA) y el trióxido de arsénico (ATO) ha modificado sustancialmente el pronóstico y el tratamiento de estos pacientes.
Asimismo, los niños/as con LAM asociada a síndrome de Down, principalmente por mutaciones en GATA1, tienen un pronóstico muy favorable con esquemas de quimioterapia de intensidad reducida, alcanzando supervivencias > 80%.
Atención integral
El trabajo interdisciplinar de enfermería, auxiliares, celadores y profesionales de los servicios de: Cirugía oncológica, Anestesiología, Infectología, Medicina preventiva, Cuidados intensivos, Urgencias, Radiodiagnóstico, Diagnóstico integrado, Banco de sangre, Farmacología, Radioterapia y distintas especialidades pediátricas, es una pieza clave en la mejoría de la supervivencia de los pacientes.
En el tratamiento de la leucemia, es fundamental una atención integral del paciente que cuide también del bienestar emocional, educativo y económico del paciente y su familia. Para ello, el papel de: psicooncólogos, trabajadores sociales, voluntarios, educadores sociales y asociaciones de pacientes, es fundamental y es necesario potenciarlos y empoderarlos.
Para el cuidado continuado de los pacientes, es necesario mantener una comunicación fluida con su pediatra de Atención Primaria de referencia, que será el referente de proximidad del paciente durante el mantenimiento y tras finalizar el tratamiento.
Si durante el tratamiento el paciente presenta fiebre, sangrado o signos de anemia, deberá derivarse inmediatamente al centro de referencia para evaluar un tratamiento urgente.
En el seguimiento a largo plazo de los pacientes es necesaria la creación de unidades de largo seguimiento que evalúen junto al pediatra de atención primaria la presencia de las toxicidades a largo plazo y la reaparición de los síntomas de leucemia, con una anamnesis que evalúe su desarrollo físico, social y emocional, y una exploración minuciosa, incluyendo palpación de adenopatías, hígado, bazo y testículos.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.*** Pardo Romaguera E, Muñoz López A, Valero Poveda S, Porta Cebolla S, Cañete Nieto A, Barreda Reines MS, et al. Cáncer infantil en España. Estadísticas 1980-2019. Registro Español de Tumores Infantiles (RETI-SEHOP). Valencia Univ València. 2020 Edición preliminar.
2.*** Pizzo PA, Poplack DG, Adamson PC, Blaney SM, Helman L. Principles and Practice of Pediatric Oncology. 5th ed. Wolters Kluwer; 2016. p. 463-97.
3.*** Zwaan CM, Kolb EA, Reinhardt D, Abrahamsson J, Adachi S, Aplenc R, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015; 33: 2949-62.
4.*** Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020; 105: 2524-39.
5.** Schmiegelow K. Have COVID-19 affected ALL epidemiology? Acta Paediatr Oslo Nor 1992. 2021; 110: 387-8.
6.** Lassaletta Atienza, A. Leucemias. Leucemia linfoblástica aguda. Pediatr Integral. 2016; XX(6): 380-9.
7.*** Dworzak MN, Buldini B, Gaipa G, Ratei R, Hrusak O, Luria D, et al. AIEOP-BFM consensus guidelines 2016 for flow cytometric immunophenotyping of Pediatric acute lymphoblastic leukemia. Cytometry B Clin Cytom. 2018; 94: 82-93.
8.*** Pui C-H. Precision medicine in acute lymphoblastic leukemia. Front Med. 2020; 14: 689-700.
9.** Teachey DT, Pui C-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019; 20: e142-54.
10.*** Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet Lond Engl. 2020; 395: 1146-62.
11.** Contreras Yametti GP, Ostrow TH, Jasinski S, Raetz EA, Carroll WL, Evensen NA. Minimal Residual Disease in Acute Lymphoblastic Leukemia: Current Practice and Future Directions. Cancers. 2021; 13: 1847.
12.*** Peters C, Dalle J-H, Locatelli F, Poetschger U, Sedlacek P, Buechner J, et al. Total Body Irradiation or Chemotherapy Conditioning in Childhood ALL: A Multinational, Randomized, Noninferiority Phase III Study. J Clin Oncol Off J Am Soc Clin Oncol. 2021; 39: 295-307.
13.*** Locatelli F, Zugmaier G, Rizzari C, Morris JD, Gruhn B, Klingebiel T, et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA. 2021; 325: 843-54.
14.*** Brivio E, Locatelli F, López-Yurda M, Malone A, Díaz de Heredia C, Bielorai B, et al. A Phase I study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood. 2021; 137: 1582-90.
15.** Diorio C, Maude SL. CAR T cells vs allogeneic HSCT for poor-risk ALL. Hematology Am Soc Hematol Educ Program. 2020; 2020: 501-7.
16.** Loken MR, Alonzo TA, Pardo L, Gerbing RB, Raimondi SC, Hirsch BA, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: A report from Children’s Oncology Group. Blood. 2012; 120: 1581-8.
17.** Creutzig U, Van Den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, De Bont E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood. 2012; 120: 3167-205.
18.** Tierens A, Bjørklund E, Siitonen S, Marquart HV, Wulff-Juergensen G, Pelliniemi TT, et al. Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia; results of the NOPHO-AML 2004 study. Br J Haematol. 2016; 174: 600-9.
19.** Moore AS, Kearns PR, Knapper S, Pearson ADJ, Zwaan CM. Novel therapies for children with acute myeloid leukaemia. Leukemia. 2013; 27: 1451-60.
20.** Rubnitz JE. Current Management of Childhood Acute Myeloid Leukemia. Pediatr Drugs. 2017; 19: 1-10.
Bibliografía recomendada
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th edition. International Agency for Research on Cancer. 2017.
La clasificación de los tumores hematopoyéticos de la OMS es “la biblia” que clínicos y citólogos tienen siempre cerca, porque la clasificación es el lenguaje de la medicina a partir del cual las enfermedades son estudiadas y tratadas.
- Pizzo PA, Poplack DG, Adamson PC, Blaney SM, Helman L. Principles and Practice of Pediatric Oncology. 5th ed. Wolters Kluwer; 2016. p. 463-97.
Este libro desde su primera edición, en 1989, establece las bases de la Oncología pediátrica.
- Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020; 105: 2524-39.
Los 2 referentes mundiales, desde el punto de vista clínico y biológico, en la leucemia aguda linfoblástica, se juntan para ofrecernos una foto actual del “state of the art” de la misma. Cualquier lectura de ambos autores es recomendable para estar actualizado en las últimas novedades de la leucemia aguda pediátrica.
- Mukherjee S. El emperador de todos los males: Una biografía del cáncer (Spanish Edition). Penguin Random House Grupo Editorial España. 2014.
En este libro, el autor nos guía con una narrativa excelente por el viaje al pasado del cáncer, lo que nos servirá para entender mejor los actuales tratamientos y asimilar la necesaria evolución en el tratamiento de la leucemia.
| Caso clínico |
|
Anamnesis Varón de 3 años que acude a su pediatra por fiebre de 5 días de evolución y hematomas en extremidades inferiores y tronco. Se encuentra más cansado y, con frecuencia, le duelen las piernas desde hace 2 semanas. Ambiente epidemiológico negativo, no hay casos similares en el domicilio ni en la escuela. No ha tenido viajes recientes. No tiene antecedentes personales ni familiares de interés. Tiene 2 hermanas de 6 y 10 años, sanas. Exploración física Triángulo de evaluación pediátrica: estable. Frecuencia cardiaca: 150 lpm; tensión arterial: 100/60 mmHg; satHbO2: 99%. Activo y reactivo, normohidratado, pálido, con petequias generalizadas y hematomas en fase purpúrica en extremidades inferiores y tronco. Adenopatías laterocervicales y supraclaviculares, destaca una adenopatía dura de 3 cm a nivel laterocervical derecho. Auscultación cardiorrespiratoria dentro de la normalidad, salvo taquicardia. Abdomen blando y depresible, con hepatomegalia de 2 cm y esplenomegalia de 4 cm. Pruebas complementarias Analítica general: Hb 7 g/dl; VCM: 100fl; leucocitos: 2.500/mcl; neutrófilos: 200/mcl; 1.500 linfocitos/mcl; 800 monocitos/mcl; 5.000 plaquetas/mcl. Bioquímica: normal salvo urato de 7 mg/dl; potasio de 6 mmol/L y LDH 700 UI/L. PCR urgente de SARS-CoV-2 negativa.
|

 How to suspect cancer in Primary Care
How to suspect cancer in Primary Care