 |
| Temas de FC |
P. Guerra García, D. Plaza López de Sabando
Hospital Universitario La Paz, Servicio de Hemato-Oncología Pediátrica
| Resumen
Los linfomas son el tercer cáncer más frecuente en la infancia. Son tumores derivados de los linfocitos, en sus distintos estadios de maduración. Se dividen en dos grandes grupos: linfoma de Hodgkin (LH) y linfoma no Hodgkin (LNH). El principal objetivo es reducir los efectos secundarios a largo plazo del tratamiento. El LNH es un grupo heterogéneo de linfomas donde se incluyen a todos aquellos linfomas que no son Hodgkin. En su gran mayoría, son linfomas de alto grado y clínicamente agresivos. Sin embargo, gracias a la colaboración internacional en forma de grandes ensayos clínicos, los tratamientos actuales tienen buenos resultados, alcanzando supervivencias por encima del 80%. La toxicidad aguda de los tratamientos supone un reto, por lo que el tratamiento en unidades especializadas es primordial. |
| Abstract
Lymphomas are the third most common childhood cancer. They are malignant tumours of the lymphoid tissues. There are two main groups: Hodgkin (HL) and non-Hodgkin lymphomas (NHL). |
Palabras clave: Linfoma de Hodgkin; Linfoma no Hodgkin; Linfadenopatía; Masa mediastínica.
Key words: Hodgkin lymphoma; Non-Hodgkin lymphoma; Lymphadenopathy; Mediastinal mass.
Pediatr Integral 2021; XXV (6): 308 – 319
Linfomas de Hodgkin y no Hodgkin
Introducción
Los linfomas son el tercer cáncer más frecuente en la infancia. Se dividen en dos grandes grupos: linfomas de Hodgkin (LH) y linfomas no Hodgkin (LNH). El LH es frecuente en los adolescentes, y se presenta como adenopatías de crecimiento progresivo. Con la quimioterapia y radioterapia, la supervivencia supera el 90%, y el objetivo de los protocolos de tratamiento actuales es disminuir las toxicidades a largo plazo. El LNH es un grupo heterogéneo de linfomas que tienen una presentación clínicamente agresiva, en forma de adenopatías/masas de rápido crecimiento, que pueden producir síntomas de compresión y lisis tumoral. El tratamiento ha mejorado en los últimos años, obteniendo supervivencias por encima del 80%. Las toxicidades agudas son la principal complicación.
Epidemiología
El linfoma es la tercera neoplasia más frecuente de la infancia y la más frecuente en adolescentes.
Los linfomas suponen el 12% de las neoplasias diagnosticadas en niños y adolescentes en nuestro medio; se registran 130-150 casos nuevos cada año según los datos del Registro Español de Tumores Infantiles (RETI-SEHOP). Son el tercer grupo más frecuente tras las leucemias y los tumores del sistema nervioso central; si bien, su incidencia se incrementa con la edad. En el grupo de adolescentes entre 15 y 19 años, se posicionan como la entidad más frecuente junto con los tumores óseos, alcanzando el 22% de los diagnósticos(1).
La mayor parte de los linfomas aparecen de novo; sin embargo, en una pequeña proporción de casos, es posible identificar factores predisponentes determinados: inmunodeficiencias primarias o secundarias (SIDA, tratamiento inmunosupresor, trasplante de órgano sólido o hematopoyético) y síndromes de predisposición familiar al cáncer.
La distribución de los diferentes subtipos de linfomas se muestra en la tabla I; a continuación, se describen las características específicas de cada grupo.

Linfoma de Hodgkin
El LH representa la tercera parte de los linfomas en la edad pediátrica (6% del total de los cánceres en la infancia). Muestra dos picos de incidencia en países económicamente desarrollados: uno en adolescentes y adultos jóvenes, y otro en mayores de cincuenta años. La incidencia aumenta con la edad durante las dos primeras décadas de la vida hasta alcanzar los 29 casos por millón en el grupo de edad entre 15 y 19 años, siendo una de las neoplasias más frecuentes del adolescente. La distribución por sexos difiere también en función de la edad: si en el niño menor de cinco años la incidencia es cinco veces mayor en varones, en adolescentes es superior en mujeres(2).
Linfoma no-Hodgkin
El LNH representa dos terceras partes de los casos de linfoma diagnosticados en la infancia y el 7% de las neoplasias pediátricas, constituyendo el grupo más frecuente de linfomas hasta los 15 años. La edad media al diagnóstico es de 10 años; son raros en el niño pequeño y la incidencia aumenta con la edad hasta alcanzar 25 casos por millón entre los 15-19 años. Es más frecuente en el sexo masculino y en individuos de raza blanca.
Se trata de un grupo heterogéneo que comprende distintos subtipos histológicos (Tabla I) con distribución e incidencia variables. Los linfomas de célula B madura son los más frecuentes (54%) y, entre ellos, el linfoma de Burkitt (LB) (42%), que es el predominante entre los 5 y 15 años. Los linfomas linfoblásticos (LL) suponen el 20% de los LNH y su incidencia es constante durante la infancia, mientras que la del linfoma difuso de célula B grande (LDCBG) y la del linfoma anaplásico de célula grande (LACG) (11% y <10% de los LNH) se incrementan con la edad.
Los LNH presentan también variabilidad geográfica, como es el caso del LB endémico.
Biología y Patología de los linfomas
Linfoma de Hodgkin
El LH es una neoplasia derivada del linfocito B caracterizada por la presencia de escasas células tumorales y un infiltrado heterogéneo.
La célula maligna en la variante clásica del LH es la célula de Reed-Sternberg (CRS). Derivan de linfocitos B del centro germinal y conforman el 1% de la celularidad tumoral en el LH. El resto está conformado por un infiltrado compuesto de: linfocitos, eosinófilos, macrófagos, células plasmáticas y fibroblastos, que crean un microambiente que favorece la supervivencia de la CRS. Es una célula grande, binucleada, con nucléolos prominentes. Existen variantes mononucleadas (célula de Hodgkin) o multinucleadas. El inmunofenotipo característico es CD15/CD30+ y CD20/CD45-; no forman inmunoglobulinas y presentan negatividad para otros marcadores B o T. La clasificación de la OMS reconoce dos tipos: el LH clásico (LHc) y el de predominio linfocítico nodular (LHPL)(3).
El LHc representa el 85-90% de los casos. Los subtipos son: esclerosis nodular (supone el 80% de los casos en el niño mayor y adolescente, y el 55% en el niño pequeño; en él, bandas densas de colágeno sectorizan el ganglio en nódulos), celularidad mixta (representa el 20% de los casos en niños menores de 10 años y el 10% en adolescentes, y se define por una eosinofilia prominente). Las variantes depleción linfoide y rico en linfocitos son muy infrecuentes en edad pediátrica.
El LHPL supone el 10-15% de los casos. Muestra un infiltrado de linfocitos pequeños, histiocitos y la célula tumoral característica en “palomitas de maíz”, morfológica, genética e inmunofenotípicamente distinta de la CRS (expresa CD20, CD45, BCL6 y PAX5, mientras que es negativa para CD30 y CD15).
La oncogénesis está relacionada principalmente con cuatro factores(4):
• Mutaciones durante la maduración del linfocito B.
• Generación por la CRS de sistemas aberrantes de señalización autocrina o paracrina, que reclutan el infiltrado inflamatorio.
• Infección latente por VEB (virus de Epstein-Barr).
• Desarrollo de sistemas de evasión inmune por pérdida de expresión de MHC y sobreexpresión de PD-L1 y PD-L2.
Linfoma no-Hodgkin
Los LNH son un grupo heterogéneo de neoplasias malignas derivadas de progenitores B o T, células B maduras o células T maduras. La mayoría de los LNH pediátricos presentan translocaciones cromosómicas que producen proteínas de fusión que alteran los mecanismos de control celular, favoreciendo la oncogénesis.
• LB: deriva de linfocitos B maduros. Histológicamente, es característico el patrón en cielo estrellado, producido por macrófagos (histiocitos) que han ingerido las células tumorales apoptóticas. Las células tumorales son monomórficas, de mediano tamaño y con un citoplasma basófilo. Tienen un índice de proliferación muy elevado (cercano al 100%). Expresan, entre otros, antígenos de células B (CD19, CD20, CD22, CD79a) e inmunoglobulina (Ig) de superficie. Es característica la translocación del oncogen MYC (cromosoma 8) con uno de los genes de las cadenas pesadas (cromosoma 14, lo más frecuente) o ligeras (cromosomas 2 y 22) de las Ig(5).
• LDCBG: neoplasias de células B maduras con un patrón difuso y un índice de proliferación alto (>40%). También expresan marcadores de células B y pueden expresar CD30 y MUM1/IRF4. Es frecuente la translocación de BCL2 y BCL6, aunque MYC también puede estar translocado(5).
• LL: son linfomas de precursores B o T (linfoblastos). Las mutaciones en NOTCH/FBXW7 confieren mejor pronóstico(5,6) y la pérdida de heterocigosidad en 6q (LOH6q) peor pronóstico(18).
• LACG: son linfomas derivados de células T maduras. Expresan CD30 y más del 95% son ALK positivos; la gran mayoría expresan la proteína de fusión NPM1-ALK(6) producida por la translocación t(2;5) (Tabla II)(3).

Clínica
Los LH se suelen presentar con linfadenopatías de crecimiento progresivo y masa mediastínica asintomática. Puede acompañarse de síntomas sistémicos (síntomas B). Los LNH son clínicamente agresivos, pudiendo provocar síntomas de compresión y lisis tumoral.
Los linfomas suelen presentarse con linfadenopatías generalizadas y pueden aparecer en cualquier localización. Las adenopatías son grandes, no dolorosas y duras a la palpación.
Estas masas pueden producir síntomas de compresión y ser una urgencia oncológica. Entre los síntomas de compresión, se encuentran: síndrome de vena cava superior, obstrucción de vía aérea, obstrucción intestinal/ invaginación intestinal, compresión espinal, taponamiento cardíaco y obstrucción ureteral/ hidronefrosis.
Puede haber afectación del sistema nervioso central (SNC), en forma de meningitis linfomatosa o masa cerebral, o afectación de la médula ósea (MO), produciendo citopenias (anemia, trombopenia, leucopenia) con la sintomatología característica asociada.
Además, es muy característica la presencia de síntomas sistémicos (los llamados síntomas B: fiebre, pérdida de peso (>= 10% en los últimos 6 meses) y sudoración nocturna).
A continuación, se exponen algunas particularidades de cada subtipo de linfoma.
Linfoma de Hodgkin
Las linfadenopatías están presentes en el 80% de los casos, y suelen localizarse a nivel cervical, supraclavicular, axilar e inguinal. Son masas de crecimiento progresivo e indoloras. En el 75% de los casos, se acompañan de masa mediastínica y es frecuente la presencia de síntomas B. Puede haber hepatoesplenomegalia y son característicos, aunque infrecuentes, los fenómenos de autoinmunidad (trombopenia inmune, anemia hemolítica, neutropenia inmune, alteraciones tiroideas…).
Linfomas no Hodgkin
A diferencia del LH, los LNH son tumores de crecimiento rápido y clínicamente agresivos, que suelen debutar con síndrome de lisis tumoral o con síntomas de compresión.
• Linfoma Burkitt: el LB se presenta como una masa de rápido crecimiento. Su tamaño puede duplicarse en 25 horas, por lo que el diagnóstico precoz y la instauración de un tratamiento rápido es primordial. Debido a la capacidad de multiplicarse, el LB se puede presentar con lisis tumoral ya establecida. Hay dos subtipos:
- Esporádico: suele tener localización abdominal (es muy frecuente la presentación en forma de invaginación intestinal), en cabeza y cuello o afectación amigdalar. Puede haber afectación de la MO en un 30% de los casos y del SNC en un 15%.
- Endémico: se presenta como un tumor en la mandíbula o en algún hueso de la cara, y se asocia a VEB. Son característicos de países africanos; no están presentes en nuestro medio.
• Linfoma difuso de células B grandes: se presenta como una masa de rápido crecimiento, generalmente linfadenopatías en cuello o abdomen. Un subtipo concreto es el linfoma B mediastínico primario, que se presenta como una masa en mediastino, generalmente en niñas adolescentes.
• Linfoma linfoblástico: el LL-T se presenta en forma de linfadenopatías generalizadas o en forma de masa mediastínica, con sintomatología de compresión asociada como: dificultad respiratoria, sibilancias, síndrome de vena cava superior… Suele acompañarse de derrame pleural o cardíaco. Hay afectación de MO en un 30% de los casos y del SNC en un 5%.
En el LL-B, es frecuente la afectación cutánea y ósea, junto con las adenopatías, y suele diagnosticarse en estadios localizados.
• Linfoma anaplásico de células grandes: hay dos formas clínicas:
- Afectación cutánea exclusiva.
- Afectación sistémica, en forma de linfadenopatías, masa en mediastino o adenopatías abdominales. Se acompaña muy frecuentemente de síntomas constitucionales, fiebre y afectación extranodal. En más del 90%, se detecta la translocación ALK.
Diagnóstico
Las exploraciones complementarias se deben orientar a conseguir el diagnóstico histológico y establecer una estadificación adecuada.
Pruebas de laboratorio
Es frecuente que los estudios analíticos sean normales; sin embargo, es necesario realizar estudios rutinarios que incluyan: hemograma completo con recuento diferencial y frotis de sangre periférica, velocidad de sedimentación eritrocitaria (VSE), estudio de función renal y hepática, e ionograma con inclusión de parámetros marcadores de síndrome de lisis tumoral. Las principales alteraciones que se pueden encontrar son las siguientes:
• Citopenias de una o varias series. Su presencia puede indicar afectación de médula ósea (más frecuente en los LNH). En los LH, la pancitopenia es rara; es más frecuente observar anemia moderada con patrón de trastornos crónicos. Asimismo, en el LH, la elevación de la VSE es un dato de pronóstico desfavorable.
• Signos de síndrome de lisis tumoral: hiperuricemia, hiperfosforemia, hiperpotasemia, hipocalcemia y elevación de LDH. Son excepcionales en pacientes con LH y frecuentes en los LNH, especialmente en los LB y LDCBG, que son las neoplasias con mayor tasa de proliferación. La LDH se utiliza en la estadificación de los LNH.
Obtención de tejido para estudio histológico
Con el objetivo de minimizar riesgos derivados de procedimientos, es recomendable seleccionar adenopatías periféricas y accesibles. En caso de que no fuera viable, se seleccionaría la región que implique menos riesgo: derrame pleural, abdominal, etc. Se recomienda evitar la obtención de muestras de masas mediastínicas; es preciso recordar que todo paciente con una masa mediastínica voluminosa tiene riesgo de obstrucción de vía respiratoria en los procedimientos que impliquen algún grado de sedación profunda.
Mientras que en los pacientes con LNH la obtención de tejido mediante punción aspirativa con aguja fina puede ser suficiente, no sucede así en los pacientes con LH debido a que la escasa cantidad de células tumorales y a que el alto grado de fibrosis pueden condicionar un aspirado seco. Por ello, en estos casos, es necesario realizar una biopsia escisional y, además, la subclasificación depende de la arquitectura ganglionar y la composición del infiltrado.
Estudios de extensión en el LH
La extensión en el LH se produce generalmente por contigüidad y en sentido craneocaudal. La afectación de médula ósea y del sistema nervioso central es mucho menos frecuente que en los LNH.
• Estudios de imagen: la realización de una radiografía simple de tórax inicial puede ser útil para evaluar el riesgo derivado de grandes masas mediastínicas. En el momento actual, está plenamente reconocido el papel de la PET/TC con 18-fluoro-2-desoxiglucosa (FDG) para realizar el estudio de extensión inicial (Fig. 1), de forma que los sistemas de estadificación actuales se basan en los hallazgos de esta prueba.
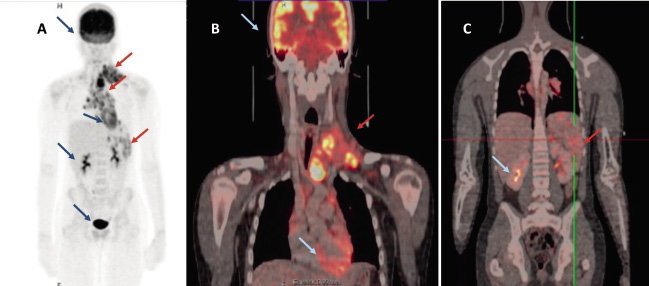
Figura 1. FDG-PET en paciente con diagnóstico de linfoma de Hodgkin. Flecha roja: lugares de afectación: cervical, supraclavicular, mediastínico, esplénico. Flecha azul: lugares de captación fisiológica: cerebro, corazón y vía urinaria.
Es más, es suficiente por sí misma para definir la afectación de hueso y de médula ósea, de forma que no es necesaria ninguna prueba adicional para el estudio de estas localizaciones. Además, la respuesta precoz a los tratamientos en la PET/TC tiene valor pronóstico y se emplea para modular la intensidad de la terapia(11) (Fig. 2).
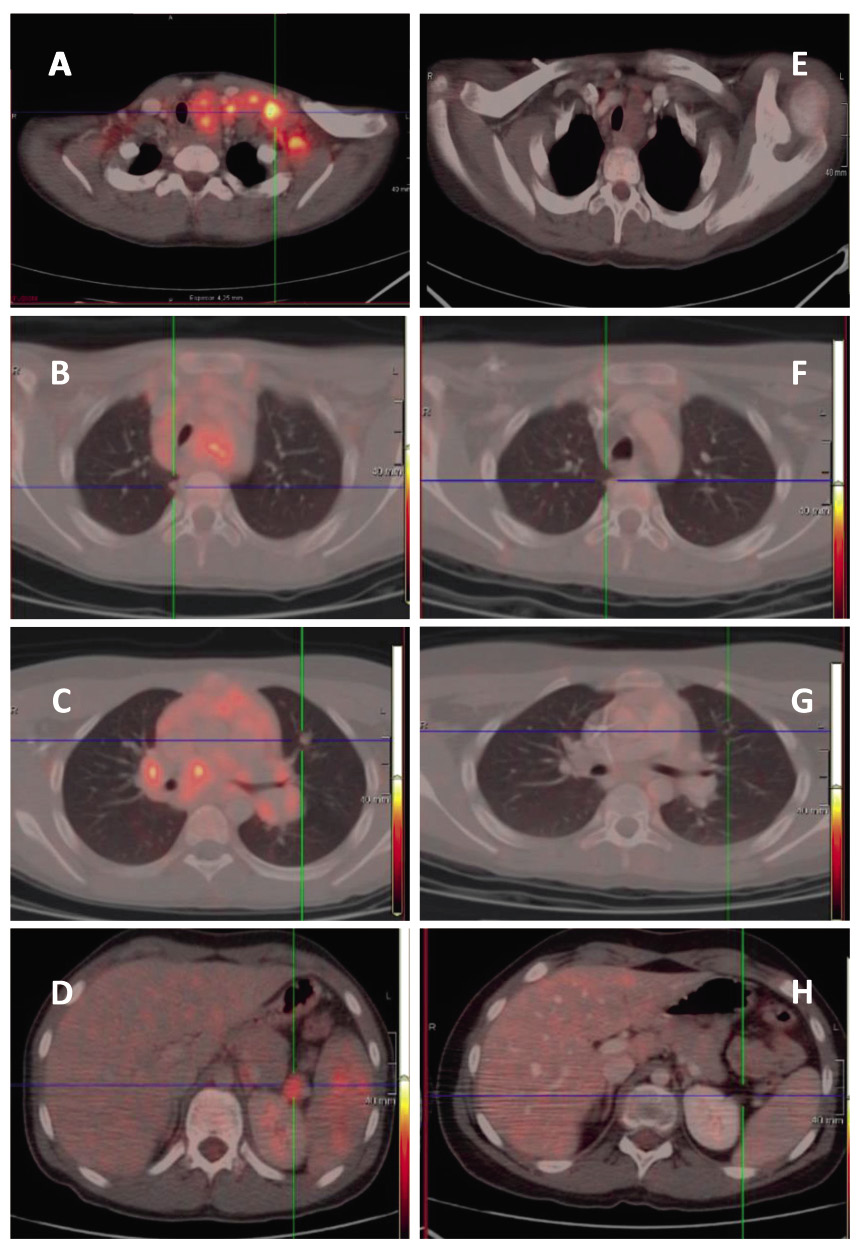
Figura 2. FDG-PET en paciente con diagnóstico de linfoma de Hodgkin. A, B, C y D: imágenes al diagnóstico. E, F, G y H: imágenes en la evaluación precoz tras dos ciclos de quimioterapia de inducción. Se puede observar la respuesta metabólica completa, con resolución de la captación de todas las regiones afectas (infraclavicular, mediastino, hilios pulmonares, parénquima pulmonar, hilio esplénico y bazo).
Se recomienda completar el estudio con una ecografía abdominal para detectar lesiones focales esplénicas no captantes y con visualización directa del anillo de Waldeyer, pues presenta una captación aumentada de forma fisiológica en la PET-FDG.
• En caso de sintomatología neurológica concomitante, se realizará estudio dirigido con RM.
Para ver los sistemas de estadificación del LH, consultar la tabla III.

Estudios de extensión en los LNH
Se deben rastrear los principales lugares de extensión (otras regiones ganglionares o parenquimatosas, médula ósea y sistema nervioso central).
• Estudios de imagen: los LNH pueden afectar regiones ganglionares no contiguas. Se deberían incluir las regiones cervicales, torácicas, axilares, abdominales y pélvicas. Esto se consigue con una tomografía computarizada (TC) con contraste. El papel de la tomografía por emisión de positrones integrada (PET/TC) no está tan bien definido como en el caso del LH, pero podría ser más sensible y específica que la TC en algunos subtipos histológicos, aunque con mayor tasa de falsos positivos y negativos, y, además, se desconoce si el tipo de respuesta se podría emplear para modular la terapia(7-9). En caso de sospecha de afectación ósea o neurológica, se realizaría estudio dirigido con resonancia magnética (RM).
• Estudio citológico de líquido cefalorraquídeo mediante punción lumbar.
• Biopsia/aspirado de médula ósea. El estudio debe realizarse mediante biopsia bilateral, ante la posibilidad de afectación dispersa en nidos aislados.
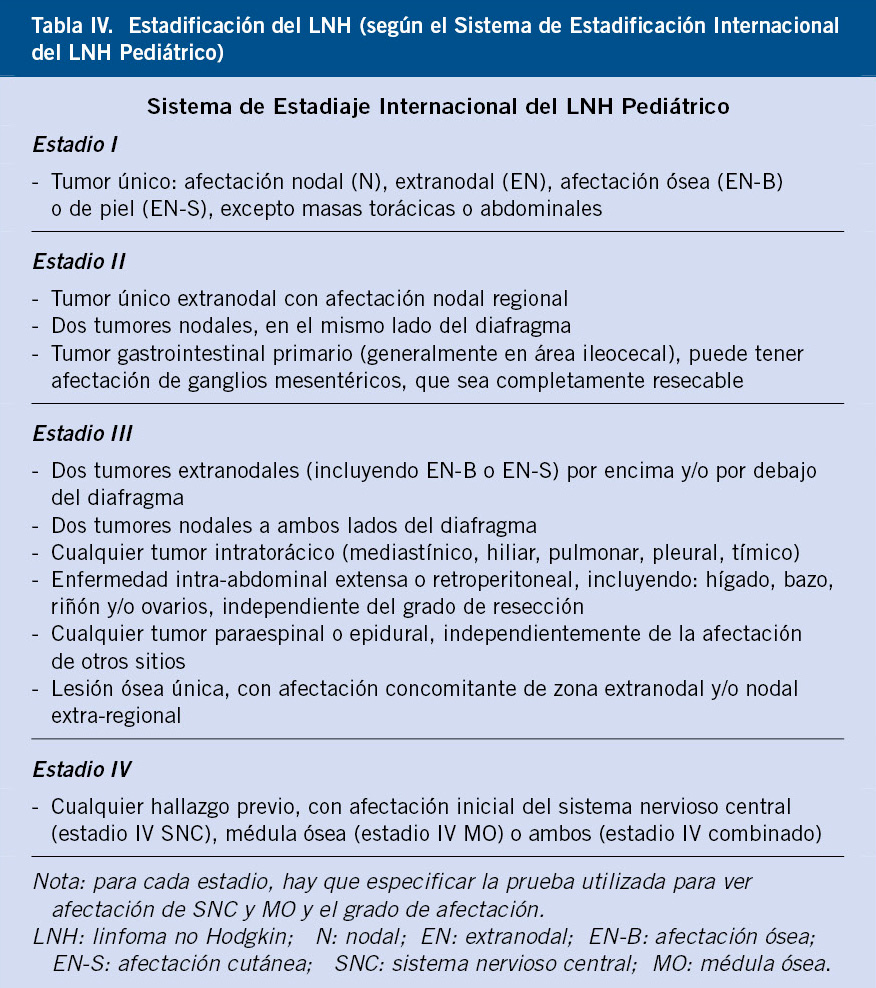
Actualmente, la estadificación se realiza mediante el Sistema de Estadificación Internacional del LNH Pediátrico (International Pediatric Non-Hodgkin Lymphoma Staging System- IPNHLSS)(10) (Tabla IV).
Tratamiento y pronóstico del linfoma de Hodgkin
El tratamiento del LH se basa en la quimioterapia; se consolida con radioterapia a los mal respondedores al tratamiento de inducción.
Linfoma de Hodgkin clásico
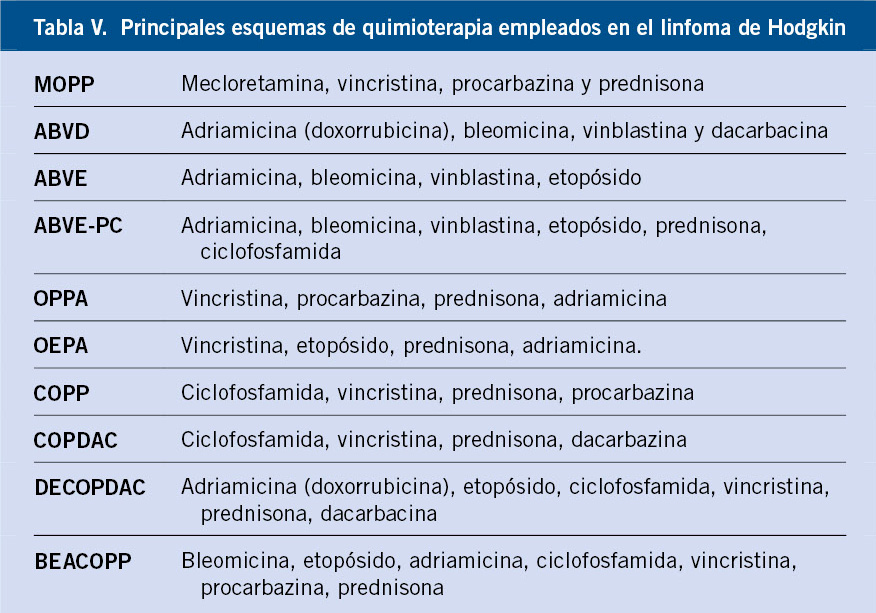
Tras la introducción de regímenes de poliquimioterapia (ciclos MOPP y ABVD) (Tabla V) y radioterapia (RT), es una de las neoplasias pediátricas con mejor tasa de curación, pero con alta incidencia de secuelas tardías.
Los esfuerzos para optimizar la terapia pretenden modular la intensidad para prevenir la ocurrencia de tales complicaciones, particularmente la infertilidad masculina (secundaria a la toxicidad gonadal de la procarbazina) y los efectos tardíos derivados de la RT (segundas neoplasias y complicaciones cardiovasculares secundarias a la combinación de antraciclinas con RT mediastínica). Dichas adaptaciones consisten principalmente en:
• Disminución de la intensidad de tratamiento en los estadios de menor riesgo.
• Sustitución de la procarbacina por etopósido en los ciclos de inducción (cambio de OPPA por OEPA) y por dacarbazina en los de consolidación (COPP por COPDAC)(12) (Tabla V).
• Reducción de la RT. El estudio colaborativo europeo EuroNet-PHL-C1 comprobó que era posible erradicarla si se cumplen criterios de remisión metabólica evaluada por FDG-PET tras dos ciclos de inducción sin compromiso en la supervivencia ni en la tasa de recidivas.
A efectos explicativos, se describe el esquema de tratamiento EuroNet-PHL.C2 (European Network Pediatric Hodgkin Lymphoma) (Fig. 3).
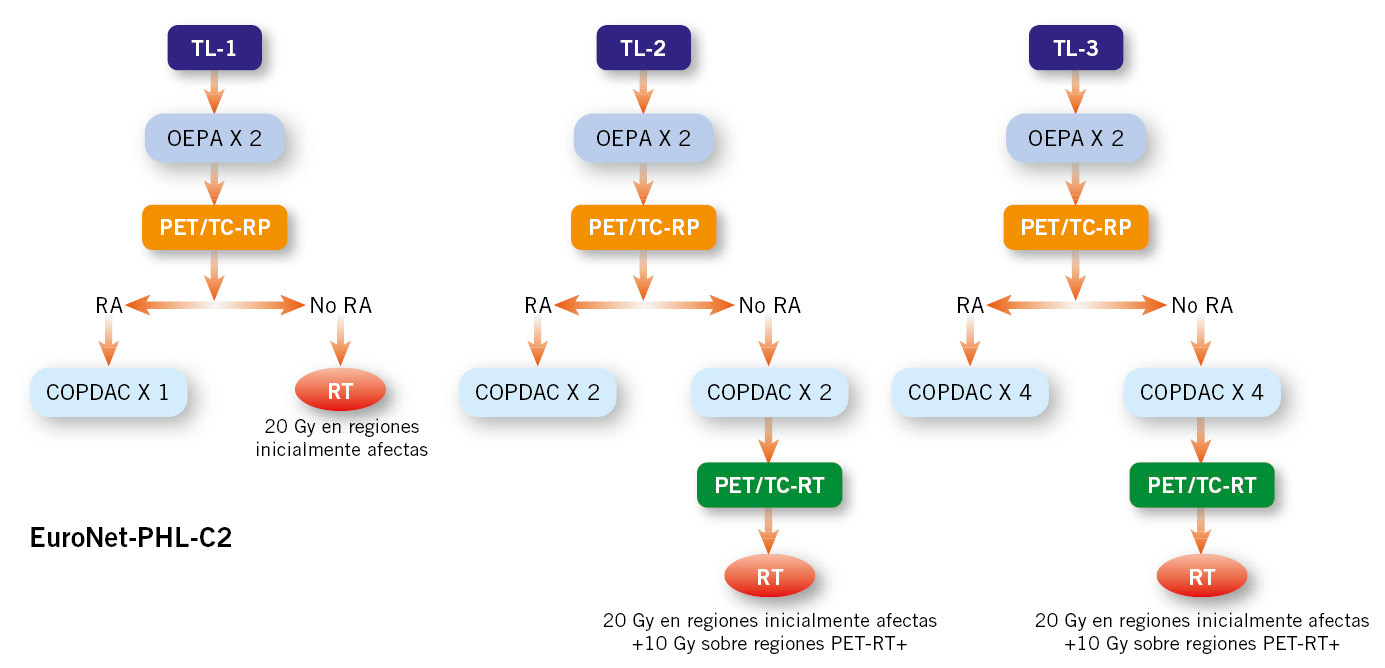
Figura 3. Esquema de tratamiento del Linfoma de Hodgkin clásico (LHc), según propuesta EuroNet-PHL-C2.
PET/TC-RP: PET/TC de evaluación de respuesta precoz; PET/TC-RT: PET/TC de respuesta tardía; RA: respuesta metabólica adecuada;
RT: radioterapia; TL: nivel de tratamiento.
Ciclos: OEPA: vincristina, etopósido, prednisona, adriamicina; COPDAC: ciclofosfamida, vincristina, prednisona, dacarbazina.
• Se administran dos ciclos de inducción tipo OEPA (Tabla V). Tras ellos, se realiza evaluación de respuesta precoz mediante PET, que tiene valor modulador de la terapia: los que alcancen buena respuesta no reciben RT.
• Los pacientes del grupo TL-1 con respuesta metabólica adecuada reciben un ciclo de consolidación COPDAC; los que tienen una respuesta inadecuada, RT.
• Los pacientes de los grupos TL-2 y TL-3 reciben tratamiento de consolidación con 2 y 4 ciclos COPDAC, respectivamente.
• La RT se administra al finalizar el tratamiento citostático a los pacientes con respuesta inadecuada en la evaluación precoz.
Los mayores esfuerzos se orientan a la reducción de las dosis e indicaciones de RT. Existen varias alternativas: intensificar la quimioterapia de consolidación (ciclos DECOPDAC) o asociar terapias biológicas o dirigidas: brentuximab-vedotín (anticuerpo monoclonal anti-CD30) o inhibidores de punto de control inmune (nivolumab, pembrolizumab, etc.)(13).
Tratamiento de la recidiva y enfermedad refractaria
Las tasas de recidiva son del 10% para estadios bajos y del 15-20% para estadios avanzados. Los principales factores de pronóstico favorable son: tiempo transcurrido hasta la recidiva superior a 12 meses, tratamiento previo sin RT (o recidiva en un lugar no radiado) o con intensidad baja de quimioterapia, ausencia de síntomas B y estadio I-II. Los pacientes de bajo riesgo serían rescatables con quimioterapia a dosis estándar y RT, mientras que para el resto, aparte de quimioterapia a dosis estándar, se propone consolidación con dosis altas de quimioterapia seguidas de rescate con trasplante autólogo de progenitores hematopoyéticos. Los pacientes con mala respuesta conforman un grupo de alto riesgo y son subsidiarios de recibir trasplante hematopoyético alogénico u otras terapias experimentales(13).
Linfoma de Hodgkin de predominio linfocítico nodular
Más indolente que el LHc, debe tratarse de forma menos intensiva. Los estadios de bajo riesgo (I, II) pueden tratarse con regímenes de muy baja intensidad, como CVP (ciclofosfamida, vincristina, prednisona) o con resección y observación en los estadios IA completamente resecados, mientras que estadios más avanzados (III, IV) se podrían tratar de forma similar al LHc. La supervivencia global es excelente, cercana al 100%. En enfermedad refractaria, al tratarse de neoplasias CD20+, es posible emplear esquemas con rituximab.
Pronóstico y secuelas tardías
La tasa de supervivencia global a los 5 años es del 97% (RETI)(1). Por grupos de riesgo, la supervivencia libre de eventos (SLE) a los 2 años es del 87%, 91% y 86% en los estadios tempranos, intermedios o avanzados, respectivamente(14).
Se asocia a una tasa importante de morbimortalidad tardía. La incidencia de hemopatías malignas es relativamente baja (1% a los 15 años). En cambio, el riesgo acumulado de desarrollo de un tumor sólido como segunda neoplasia se incrementa hasta el 25% a los 30 años (sobre todo, cáncer de mama y tiroides). Asimismo, es la neoplasia con mayor incidencia de cardiotoxicidad derivada de la terapia. Se trata de una población con una tasa de mortalidad estandarizada en los supervivientes, 12 veces superior a la de la población general (entre las más altas en Oncología pediátrica), así como con una mayor incidencia de condiciones crónicas de salud(15-17).
Tratamiento y pronóstico del linfoma no-Hodgkin
El tratamiento consiste en la poliquimioterapia intensiva. Requiere administrarse en centros oncológicos especializados que cuenten con unidades de cuidados intensivos, por las toxicidades muy frecuentes que conllevan.
• Linfoma Burkitt: se utilizan protocolos derivados de los grupos francés y alemán (FAB-LMB y BFM)(18), con regímenes que contienen: ciclofosfamida, citarabina, metotrexato, vincristina, doxorrubicina, etopósido y corticoides.
El tratamiento tiene una duración de entre 2 y 6 meses según el grupo de riesgo (Fig. 4), que depende de la estadificación, la elevación de la LDH y la respuesta al tratamiento.
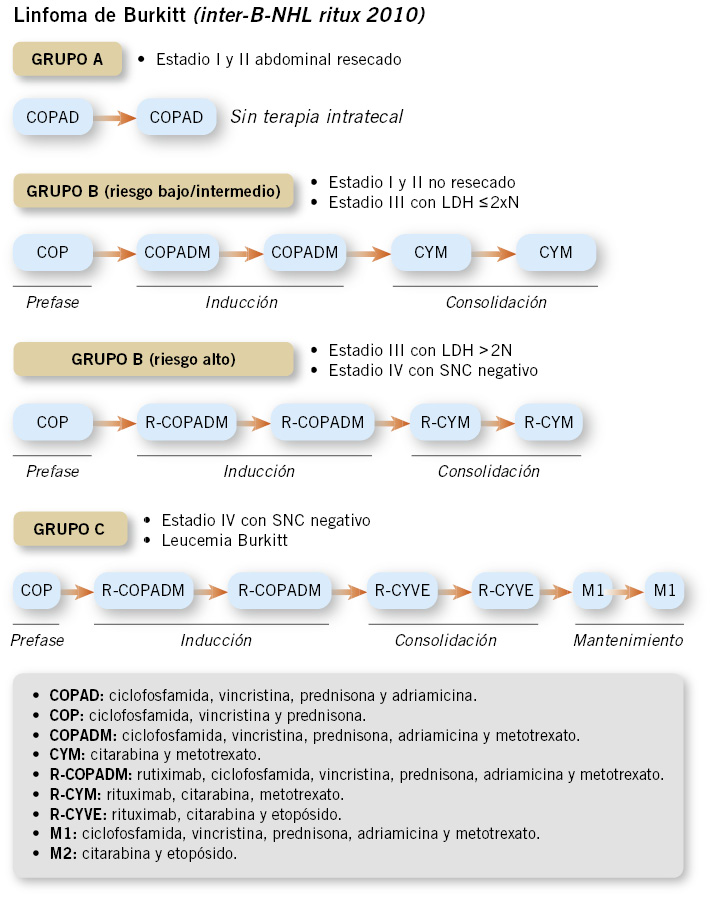
Figura 4. Esquema-resumen del tratamiento del linfoma de Burkitt en Pediatría. Se utilizan ciclos de quimioterapia intensiva, en un corto período de tiempo. El tratamiento incluye además terapia intratecal.
Cabe destacar que, en el grupo de alto riesgo, se administra, junto con la poliquimioterapia intensiva, rituximab (un anticuerpo quimérico monoclonal anti-CD20). Este tratamiento se basa en el protocolo internacional Inter-B-NHL ritux 2010, cuyos resultados han sido publicados recientemente(19).
• Linfoma difuso de células B grandes: en Pediatría, se tratan con los mismos protocolos que para el LB, con resultados similares.
• Linfoma linfoblástico: se utilizan esquemas de tratamiento muy similares a los de la leucemia linfoblástica aguda(18). Se utiliza la poliquimioterapia intensiva seguida de un tratamiento de mantenimiento prolongado, con una duración total de 2 años (Fig. 5).
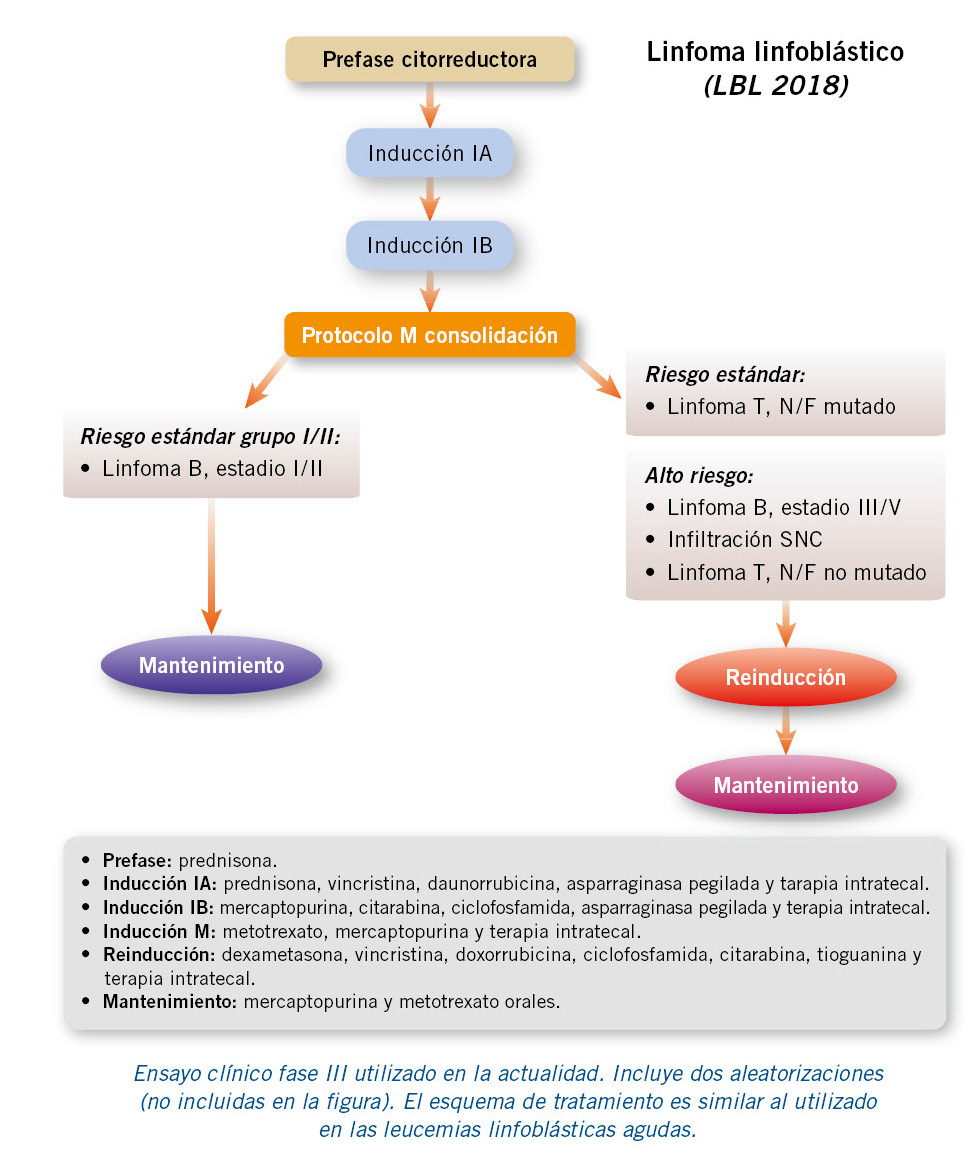
Figura 5. Esquema-resumen (adaptado) del tratamiento del linfoma linfoblástico pediátrico.
La quimioterapia incluye: corticoides, vincristina, antraciclinas, ciclofosfamida, citarabina, asparraginasa y metotrexate, además de terapia intratecal (profilaxis/ tratamiento del SNC).
En el momento actual, el estándar de tratamiento del LL es dentro de un ensayo clínico fase III europeo (LBL 2018- EudraCT number: 2017-001691-39), donde se estratifica la intensidad del tratamiento según el fenotipo, la estadificación, la presencia o no de mutaciones en NOTCH1/FBXW7 y la afectación o no del SNC (Fig. 5).
• Linfoma anaplásico de células grandes: se utiliza un esquema basado en el protocolo ALCL 99, derivado del grupo alemán BFM(18). Consiste en ciclos alternos (tres o seis según el grupo de riesgo) de quimioterapia, utilizando: dexametasona, metotrexato, ifosfamida/ciclofosfamida, etopósido, citarabina y doxorrubicina. No necesita tratamiento intratecal.
El grupo europeo de LNH está planificando un ensayo clínico internacional donde se incorpore, al tratamiento de primera línea, un inhibidor de ALK o vinblastina semanal.
Pronóstico y tratamiento de la recidiva del LNH
El pronóstico de los LNH ha mejorado de manera sustancial en los últimos 30 años. En la actualidad, la supervivencia global de los LNH es superior al 80% siendo >90% en los estadios I o II y del 80-90% para los estadios avanzados.
Sin embargo, el pronóstico de las recaídas en los LNH es generalmente infausto, con una curación global por debajo del 30%.
En cuanto a los efectos secundarios, la intensidad de los tratamientos se correlaciona con toxicidades agudas que pueden ser muy graves. Por otro lado, las toxicidades a largo plazo son escasas, pero incluyen: aumento de mortalidad asociada a segundas neoplasias y cardiomiopatía, además de aumento de morbilidad en forma de defectos neurocognitivos, peor calidad de vida y dificultades en las relaciones sociales, comparado con población sana(20).
• LB: en el LB, la SLE se encuentra en un 80-90%. Las recaídas son precoces (en los primeros 15 meses) y tienen muy mal pronóstico, con supervivencias globales por debajo del 25%, a pesar de regímenes intensivos de quimioterapia seguidos de trasplante de progenitores hematopoyéticos autólogo. Se está estudiando la terapia con células CAR-T en los linfomas B de alto grado en recaída/ refractarios, dentro de ensayos clínicos y del anticuerpo monoclonal anti CD-22 inotuzumab.
• LDCBG: las recaídas pueden ser más tardías, hasta 3 años desde el diagnóstico, y se tratan de manera similar a las recaídas del LB.
• LL: la supervivencia alcanza el 75-85%, por lo que un 10-20% de pacientes son refractarios o recaen. Las recaídas suelen ser locales y la supervivencia inferior al 30%. El tratamiento de segunda línea incluye el trasplante de progenitores hematopoyéticos alógenico, si se consigue una segunda remisión completa. Nuevas terapias, como la nelarabina o la clofarabina, se han mostrado poco eficaces. Se está investigando, dentro de un ensayo clínico, el uso del daratumumab (anticuerpo monoclonal contra el CD-38) para las recaídas del subtipo de LL-T.
• LACG: la SLE se sitúa en el 65-75%. Sin embargo, a diferencia del resto de LNH, este subtipo tiene muy buena respuesta al tratamiento tras la recaída, obteniendo supervivencias globales por encima del 90% con el uso del trasplante de progenitores hematopoyéticos alógenico. Se están utilizando inhibidores de ALK para alcanzar la remisión previo al TPH. Otras posibles terapias para alcanzar la remisión incluyen vinblastina o brentuximab vedotin (un anticuerpo monoclonal anti-CD30).
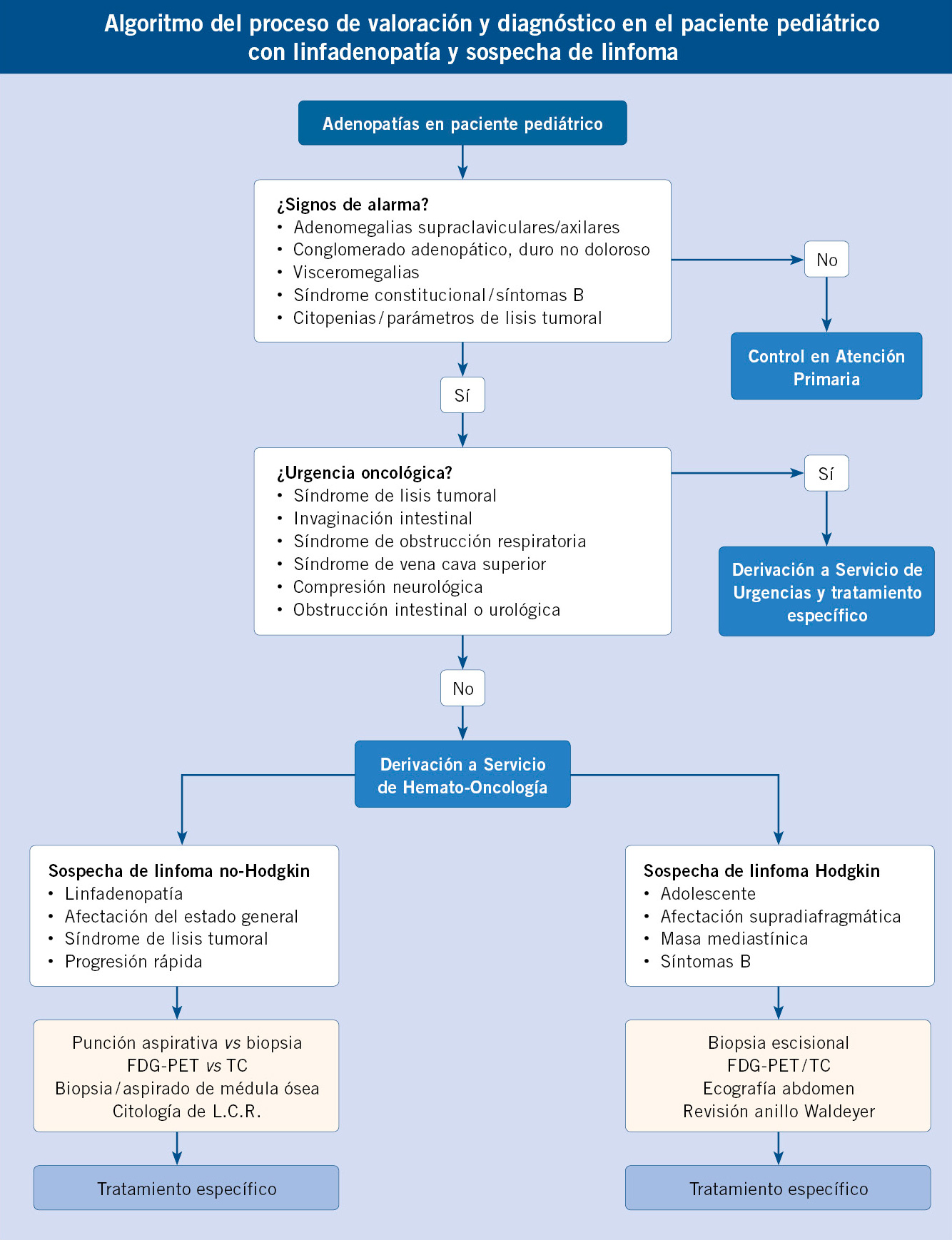
Función del pediatra de Atención Primaria
Es muy importante el papel del pediatra de Atención Primaria para la detección precoz de los linfomas. El pediatra, a menudo, va a ser el que reciba la primera consulta y debe sospechar esta entidad para poder derivar al paciente a un centro especializado.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.** Pardo E, Muñoz A, Valero S, Porta S, Cañete A, Barreda MS, et al. Cáncer infantil en España. Estadísticas 1980-2019. Registro Español de Tumores Infantiles (RETI-SEHOP). Valencia: Universitat de València. 2020.
2.** Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA: A Cancer Journal for Clinicians.
3.*** Swerdlow SH, Campo E, Pileri SA, Lee Harris N, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127: 2375-90.
4.*** Bräuninger A, Schmitz R, Bechtel D, Renné C, Hansmann ML, Küppers R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. International Journal of Cancer. 2006; 118: 1853-61.
5.** Miles RR, Shah RK, Frazer JK. Molecular genetics of childhood, adolescent and young adult non-Hodgkin lymphoma. British Journal of Haematology. 2016; 173: 582-96.
6.** Shiramizu B, Mussolin L, Woessmann W, Klapper W. Paediatric non-Hodgkin lymphoma – perspectives in translational biology. British Journal of Haematology. 2016; 173: 617-24.
7.** Juweid ME, Stroobants S, Hoekstra OS, Mottaghy FM, Dietlein M, Guermazi A, et al. Use of positron emission tomography for response assessment of lymphoma: Consensus of the imaging subcommittee of international harmonization project in lymphoma. Journal of Clinical Oncology. 2007; 25: 571-8.
8.** Allen CE, Kelly KM, Bollard CM. Pediatric lymphomas and histiocytic disorders of childhood. Pediatric Clinics of North America. 2015; 62: 139-65.
9.** Rahman HA, El Semary SF, Ahmed G, Kenaai N El, Omar W, Zaky I, et al. Can FDG-PET replace biopsy for the evaluation of residual tumor in pediatric mature B-cell non-Hodgkin lymphoma? Pediatric Blood and Cancer; 2020. p. 67.
10.*** Rosolen A, Perkins SL, Pinkerton CR, Guillerman RP, Sandlund JT, Patte C, et al. Revised international pediatric non-Hodgkin lymphoma staging system. Journal of Clinical Oncology. 2015; 33: 2112-8.
11.** Barrington SF, Qian W, Somer EJ, Franceschetto A, Bagni B, Brun E, et al. Concordance between four European centres of PET reporting criteria designed for use in multicentre trials in Hodgkin lymphoma. European Journal of Nuclear Medicine and Molecular Imaging. 2010; 37: 1824-33.
12.** Mauz-Körholz C, Hasenclever D, Dörffel W, Ruschke K, Pelz T, Voigt A, et al. Procarbazine-free OEPA-COPDAC chemotherapy in boys and standard OPPA-COPP in girls have comparable effectiveness in pediatric Hodgkin’s lymphoma: The GPOH-HD-2002 study. Journal of Clinical Oncology. 2010; 28: 3680-6.
13.*** Lo AC, Dieckmann K, Pelz T, Gallop-Evans E, Engenhart-Cabillic R, Vordermark D, et al. Pediatric classical Hodgkin lymphoma. Pediatric Blood and Cancer; 2021. p. 68.
14.** Landman-Parker J, Wallace H, Hasenclever D BW. First international inter-group study for classical Hodgkin lymphoma in children and adolescents: euroNet-PHL-C1. Report of the latest interim analysis. Haematologica. 2016; 191: 35.
15.** Bhatia S, Yasui Y, Robison LL, Birch JM, Bogue MK, Diller L, et al. High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin’s disease: Report from the Late Effects Study Group. Journal of Clinical Oncology. 2003; 21: 4386-94.
16.*** Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic Health Conditions in Adult Survivors of Childhood Cancer. New England Journal of Medicine. 2006; 355: 1572-82.
17.** Fidler MM, Reulen RC, Henson K, Kelly J, Cutter D, Levitt GA, et al. Population-based long-Term cardiac-specific mortality among 34 489 five-year survivors of childhood cancer in Great Britain. Circulation. 2017; 135: 951-63.
18.*** Minard-Colin V, Brugières L, Reiter A, Cairo MS, Gross TG, Woessmann W, et al. Non-Hodgkin lymphoma in children and adolescents: Progress through effective collaboration, current knowledge, and challenges ahead. Journal of Clinical Oncology. 2015; 33: 2963-74.
19.** Minard-Colin V, Aupérin A, Pillon M, Burke GAA, Barkauskas DA, Wheatley K, et al. Rituximab for High-Risk, Mature B-Cell Non-Hodgkin’s Lymphoma in Children. New England Journal of Medicine. 2020; 382: 2207-19.
20.** Cairo MS, Beishuizen A. Childhood, adolescent and young adult non-Hodgkin lymphoma: current perspectives. British Journal of Haematology. 2019; 185: 1021-42.
21. Sánchez de Toledo Codina J, Sábado Álvarez C. Linfomas de Hodgkin y no Hodgkin. Pediatr Integral. 2016; XX(6): 390-400.
Bibliografía recomendada
- Swerdlow SH, Campo E, Pileri SA, Lee Harris N, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127: 2375-90.
Clasificación de los linfomas actualizada. Interesante lectura que engloba las novedades histológicas, genéticas y moleculares.
- Bräuninger A, Schmitz R, Bechtel D, Renné C, Hansmann ML, Küppers R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. International Journal of Cancer. 2006; 118: 1853-61.
Artículo en el que se expone claramente la biología en la oncogénesis del linfoma de Hodgkin, útil para comprender el desarrollo de las nuevas terapias que se emplean en esta entidad.
- Rosolen A, Perkins SL, Pinkerton CR, Guillerman RP, Sandlund JT, Patte C, et al. Revised international pediatric non-Hodgkin lymphoma staging system. Journal of Clinical Oncology. 2015; 33: 2112-8.
Se actualiza el sistema de estadificación de Murphy/ St Jude en uso desde 1980, para poder incorporar nuevas entidades, avances moleculares, inmunofenotípicos y citogenéticos, el seguimiento de la enfermedad mínima residual o diseminada, y nuevas técnicas de imagen.
- Lo AC, Dieckmann K, Pelz T, Gallop-Evans E, Engenhart-Cabillic R, Vordermark D, et al. Pediatric classical Hodgkin lymphoma. Pediatric Blood and Cancer; 2021. p. 68.
Artículo que expone, clara y concisamente, la perspectiva de los grupos estadounidense y europeo en la estadificación y el tratamiento del linfoma de Hodgkin.
- Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic Health Conditions in Adult Survivors of Childhood Cancer. New England Journal of Medicine. 2006; 355: 1572-82.
Artículo clásico que describe la morbimortalidad en una gran cohorte de supervivientes al cáncer pediátrico. Fundamental para comprender la implicación de este diagnóstico en la vida adulta.
- Minard-Colin V, Brugières L, Reiter A, Cairo MS, Gross TG, Woessmann W, et al. Non-Hodgkin lymphoma in children and adolescents: Progress through effective collaboration, current knowledge, and challenges ahead. Journal of Clinical Oncology. 2015; 33: 2963-74.
Artículo excelente, que resume el tratamiento de los LNH en Pediatría y el progreso que ha habido a lo largo del tiempo.
| Caso clínico |
|
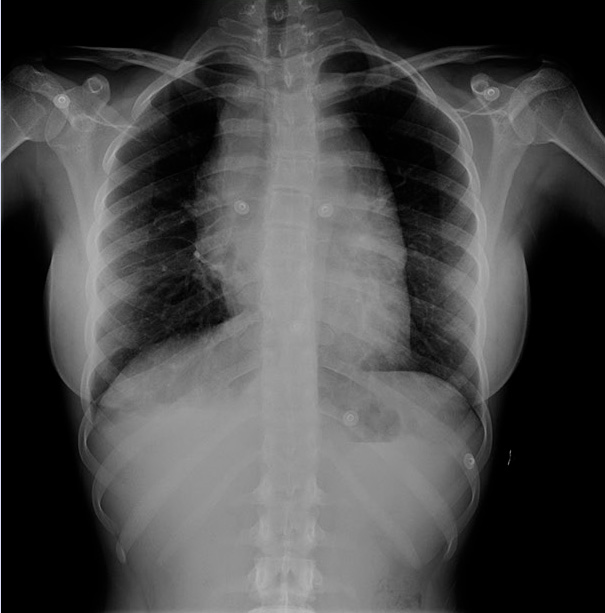
Adolescente mujer de 15 años, que acude a urgencias por dolor centro-torácico de 4 días de evolución. Asocia disfagia, con sensación de ocupación en región esternal. Sin fiebre, sin pérdida de peso, ni sudoración nocturna. A la exploración física, no se palpan adenopatías periféricas, se palpa bultoma en región supra-esternal y polo de bazo. En Urgencias, se solicita analítica completa con marcadores de lisis tumoral, radiografía de tórax y ecografías abdominal y cervical: • Analítica: hemograma sin alteraciones significativas. Bioquímica con parámetros de lisis tumoral sin alteraciones. LDH: 400 UI/l. VSE: 25 mm/h. • Radiografía de tórax (Fig. 6): ensanchamiento mediastínico.
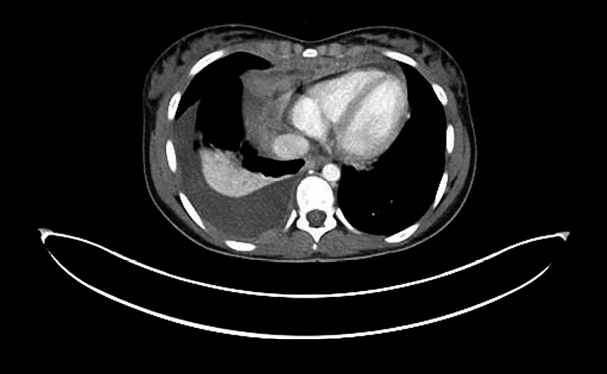
Figura 6. Radiografía de tórax al diagnóstico donde se observa una masa mediastínica. • Ecografía abdominal: nódulos corticales bilaterales compatibles con infiltración renal. • Ecografía cervical: masa mediastínica heterogénea multinodular, probablemente tímica, que se extiende desde horquilla esternal, desplazando lateralmente ambas confluencias yúgulo-subclavias, así como a la vena innominada. Todas ellas están disminuidas de calibre, aunque permeables. Ante los hallazgos, se realiza una ecocardiografía, donde se objetiva una masa mediastínica anterior con compresión moderada de la vena cava superior y derrame pericárdico ligero. Ingresa a cargo de Hemato-Oncología por sospecha de linfoma. Como estudio de extensión, se realiza PET/TC, donde se objetiva masa de gran tamaño en mediastino anterior, compatible con tejido tumoral viable, con extensión por contigüidad a la superficie pleural derecha (Fig. 7).
Figura 7. TC torácico de la paciente, donde se objetiva masa mediastínica con extensión por contigüidad a la superficie pleural derecha y derrame pleural asociado. Ganglios laterocervicales derechos, en ambas fosas claviculares y en axila derecha, compatibles asimismo con tejido tumoral viable. Imágenes dudosas en ambos riñones. La paciente presenta leve taquipnea, sin otra sintomatología. Precisa oxigenoterapia con gafas nasales a 2 lpm. Previo a la realización de biopsia, se inicia tratamiento con prednisona a 60 mg/m2 ante el riesgo anestésico. Tras inicio de corticoide, la paciente presenta una LDH máxima de 542 UI/l y un ácido úrico de 8,2 mg/dl que precisa una dosis de rasburicasa. Se recibe el diagnóstico anatomo-patológico compatible con linfoma linfoblástico T y se inicia tratamiento específico.
|

 How to suspect cancer in Primary Care
How to suspect cancer in Primary Care