 |
| Temas de FC |
A. Escudero López
Instituto de Genética Médica y Molecular (INGEMM). Hospital Universitario La Paz
| Resumen
El cáncer infantil abarca un conjunto de enfermedades muy heterogéneas que aparecen tras la acumulación de una serie de cambios genéticos en una o varias células, conocidas como mutaciones somáticas. La aparición de estas alteraciones genéticas, en la mayoría de los casos, es desconocida. Sin embargo, existe hasta un 10% de pacientes pediátricos que presentan adicionalmente a las mutaciones somáticas, otro tipo de alteraciones genéticas que afectan a todas las células del organismo conocidas como mutaciones germinales. Dichas mutaciones en línea germinal, van a aumentar el riesgo a desarrollar un tumor y su presencia confirma el diagnóstico de síndrome de predisposición a cáncer (SPC). La identificación de las mutaciones germinales es importante para el diagnóstico precoz de segundas neoplasias y tienen una gran relevancia, tanto en el seguimiento del paciente como en sus familiares, ya que pueden ser heredadas. Por otra parte, la detección de mutaciones somáticas resulta de gran utilidad en situaciones de refractariedad al tratamiento o en recaída, ya que permite utilizar terapias dirigidas en función de las características genéticas del tumor. |
| Abstract
Cancer comprises a group of heterogeneous diseases characterized by the accrual of genetic alterations in one or several cells, known as somatic mutations. Its etiology is unknown in most cases. However, 10% of pediatric patients have been reported to harbor additional genetic abnormalities affecting all of their cells, known as germline mutations, which increase the risk of tumor development and confirm the diagnosis of a cancer predisposing syndrome (CPS). Identifying germline mutations is relevant for the early diagnosis of secondary neoplasias as well as for the patient and relatives’ follow-up since they could be inherited. On the other hand, detecting somatic mutations can be helpful, especially in patients that relapse or that are refractory to treatment, given that their identification could provide pertinent information regarding targeted therapies based on the tumor’s genetic profile. |
Palabras clave: Cáncer infantil; Genética; Predisposición; Medicina personalizada.
Key words: Child cancer; Genetics; Predisposition; Personalized medicine.
Pediatr Integral 2021; XXV (6): 276 – 282
Bases genéticas y moleculares del cáncer infantil
Introducción
El cáncer infantil es una enfermedad rara de base genética, cuyo origen multifactorial es en gran parte desconocido.
El cáncer infantil, a diferencia del cáncer de adultos, es una enfermedad rara de la que se diagnostican unos 155 casos por millón de niños al año. Comprende principalmente tumores hematológicos, del sistema nervioso central y linfomas, y supone la primera causa de muerte por enfermedad en menores de 18 años.
El cáncer, independientemente de la edad de aparición, es una enfermedad de base genética causada por la acumulación de cambios en el ADN que promueven la transformación de una célula sana en una célula tumoral, en un proceso complejo que requiere varias etapas(1). Dichos cambios o mutaciones genéticas aparecen a nivel somático, es decir, en un pequeño grupo de células que, posteriormente, dará lugar al tumor y afectan a genes que participan en diversos procesos moleculares del ciclo celular, como los protooncogenes, genes supresores de tumores o genes relacionados con la reparación del ADN. Existen factores intrínsecos y extrínsecos que contribuyen a la acumulación de mutaciones genéticas(2). En relación a los primeros, las mutaciones pueden aparecer de forma esporádica como consecuencia de la división celular, en la que tiene lugar la replicación del ADN. Durante la replicación, la polimerasa amplifica por completo todo genoma contenido en el núcleo de una célula y, aunque la tasa de error es muy baja, dado que el genoma tiene un tamaño de más de 3 billones de pares de bases, la aparición de errores de forma aleatoria es inevitable. Además, los factores extrínsecos también desempeñan un papel importante en la aparición de mutaciones genéticas. Entre estos factores, se incluye la exposición a radiación, a carcinógenos y a otros factores ambientales que pueden aumentar el riesgo a desarrollar cáncer. Sin embargo, la temprana aparición de los tumores infantiles sugiere que los factores extrínsecos, aunque pueden favorecer el desarrollo de cáncer en edad pediátrica, podrían desempeñar un papel menos relevante comparado con la aparición de tumores en adultos.
Síndromes de predisposición genética a cáncer infantil (Tabla I)
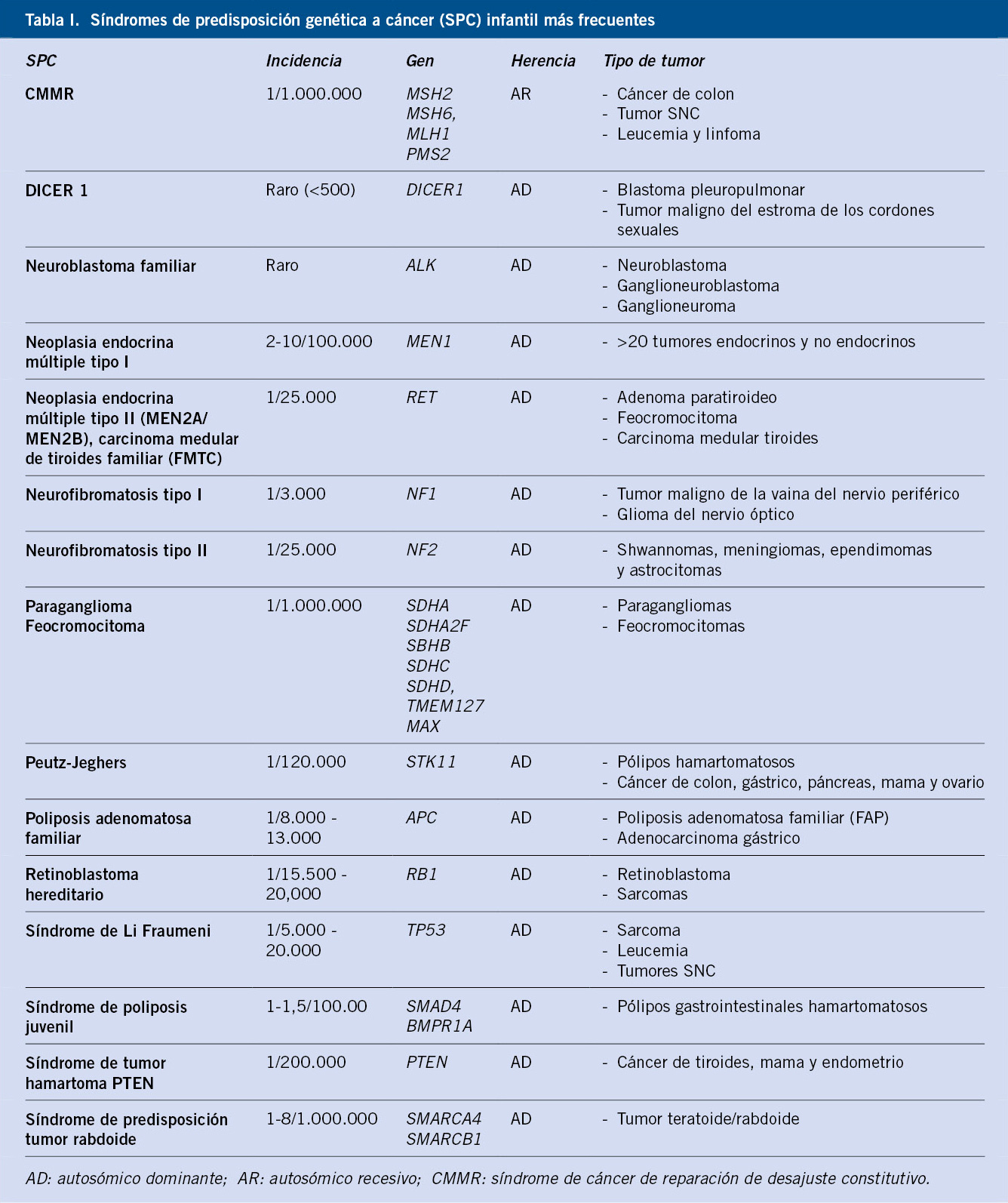
Un 10% de pacientes oncológicos pediátricos presentan algún tipo de síndrome de predisposición a cáncer, causado por la presencia de mutaciones en línea germinal.
La gran mayoría de tumores pediátricos se originan por la aparición de mutaciones somáticas de forma esporádica o desconocida, sin embargo, un pequeño número de pacientes presentan mutaciones germinales; es decir, en todas las células del organismo, que van a aumentar el riesgo de desarrollo de tumores en edad pediátrica. En 2015, el Hospital de St. Jude (Memphis, EE.UU.) demostró la presencia de factores genéticos predisponentes al cáncer en un 8,5% de pacientes oncológicos pediátricos, un porcentaje significativamente mayor al 1% detectado en individuos sanos(3). Estos resultados fueron posteriormente confirmados por el ICGC (International Cancer Genome Consortium), el NIH (National Cancer Institute) o el Baylor College of Medicine, tras la publicación de varios artículos en los que detectaban la presencia de mutaciones genéticas predisponentes en un 7-12% de pacientes diagnosticados de cáncer infantil(4-6).
Los Síndromes de Predisposición a Cáncer (SPCs), causados por la aparición de mutaciones germinales predisponentes, engloban un heterogéneo grupo de enfermedades, las más frecuentes se resumen en la tabla II.

En general, suelen aparecer como consecuencia de mutaciones germinales en una de las dos copias de un gen supresor tumoral y obedecen a un patrón de herencia autosómico dominante(7). Esto significa que cada hijo tiene un 50% de probabilidad de heredar la copia mutada y, por lo tanto, podrá desarrollar la enfermedad. Sin embargo, debido al fenómeno de penetrancia incompleta, no todos los portadores que presenten la mutación desarrollarán un tumor(2).
La identificación de un SPC pediátrico es compleja y puede pasar desapercibida por varios motivos. Uno de ellos es el mencionado fenómeno de penetrancia incompleta; es decir, mutaciones que pueden presentarse en individuos que no desarrollan la enfermedad o la aparición de mutaciones de novo. Ambos, van a contribuir a la ausencia de historia familiar de cáncer. Además, los SPCs que asocian anomalías fenotípicas o malformaciones congénitas, como puede ser el síndrome de Noonan, asociado a un mayor riesgo de padecer Leucemia Mielomonocítica Juvenil (LMMJ), en algunos casos debutan al nacer con rasgos clínicos sutiles difícilmente reconocibles, si no es evaluado por un especialista. Para facilitar el correcto diagnóstico de los SPCs, Jogmans y cols., publicaron en 2016 un cuestionario, en el que se indican los criterios más importantes a tener en cuenta a la hora de hacer la evaluación de cualquier niño diagnosticado de cáncer (Fig. 1)(8).
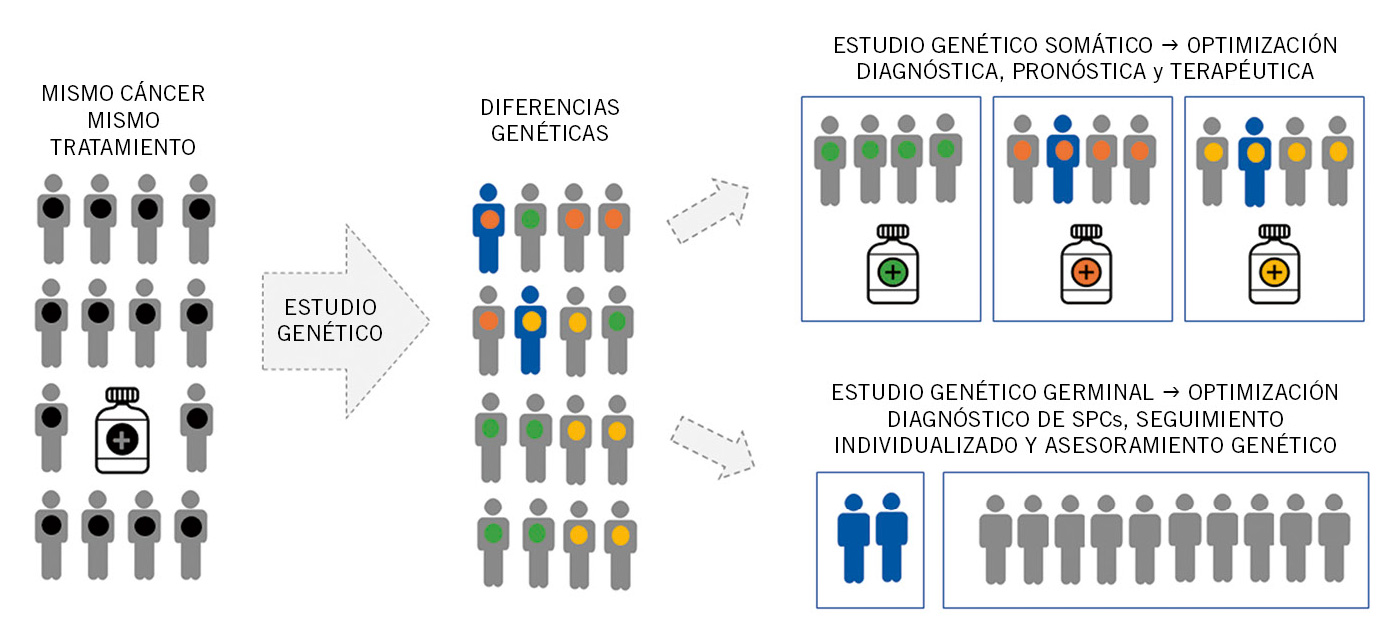
Figura 1. Papel de los estudios genéticos en la optimización diagnóstica y terapéutica del cáncer infantil. SPCs: Síndromes de Predisposición al Cáncer.
La optimización diagnóstica de los SPCs es clave para mejorar el manejo clínico y seguimiento de los pacientes, dado que este grupo de pacientes puede beneficiarse de estrategias de seguimiento y tratamiento individualizado (Fig. 1).
Un claro ejemplo son los pacientes con retinoblastoma hereditario, causado por mutaciones que inactivan una de las dos copias del gen RB1. Los pacientes con mutaciones en dicho gen, tienen un 90% de probabilidad de desarrollar retinoblastoma, el tumor intraocular más frecuente durante la edad pediátrica, y en algunos casos, también pueden desarrollar otros tipos de tumores, principalmente tumores de partes blandas y especialmente a partir de la adolescencia. Esta enfermedad, además, se caracteriza por presentar una penetrancia del 90%, por lo que existe un 10% de portadores de mutaciones en el gen RB1 que no van a desarrollar la enfermedad, pero sí pueden transmitirla a su descendencia. Un estudio completo que incluya a todos los familiares de primer grado y un estrecho seguimiento clínico de todos aquellos familiares que puedan estar en riesgo, permite realizar el diagnóstico precoz de futuros tumores y, además, puede resultar de gran utilidad de cara a la planificación de futuros embarazos(9).
En algunos casos, la identificación de un SPC ayuda a optimizar el manejo clínico. Es el caso de los pacientes con síndrome de deficiencia de reparación de desajuste constitutivo o (constitutional mismatch repair, CMMR), una enfermedad autosómica recesiva causada por mutaciones que inactivan las dos copias de alguno de los siguientes genes, cuyo papel es esencial en la reparación del ADN: MLH1, MSH2, MSH6, PMS2 o EPCAM. Los pacientes que sufren este tipo de síndromes pueden experimentar multitud de complicaciones clínicas severas tras la dosis de quimioterapia o radioterapia habitual. En cambio, un tratamiento adaptado e individualizado evita el desarrollo de toxicidades o resistencia a los tratamientos convencionales, mejorando así el manejo clínico y su supervivencia(10).
Medicina de precisión aplicada a la Oncología pediátrica
La medicina de precisión supone una novedosa alternativa terapéutica para los tumores pediátricos en recaída o refractarios a las terapias convencionales.
Las mutaciones somáticas son aquellas alteraciones genéticas específicas del tumor. Actualmente, la identificación de algunas de ellas, conocidas como mutaciones drivers, se recomienda en las diferentes guías desarrolladas para el tratamiento de tumores pediátricos sólidos y hematológicos; ya que, junto con los datos clínicos, resultan de gran utilidad a la hora de establecer el diagnóstico, el pronóstico y la aproximación terapéutica de los pacientes. Un ejemplo sería la detección de alteraciones estructurales o cromosómicas en tumores hematológicos. Por ejemplo, las leucemias que presentan hiperdiploidía (aumento en el número de cromosomas) o el gen de fusión MLL-AF4 se van a asociar a un mejor y peor pronóstico, respectivamente, y su monitorización mediante técnicas genéticas clásicas, va a permitir determinar la respuesta de los pacientes a la terapia administrada(11). Sin embargo, la información obtenida de los análisis rutinarios, a veces, es insuficiente, ya que existen pacientes considerados de buen pronóstico que, posteriormente, recaen o son refractarios a las terapias convencionales.
En los últimos años, los grandes avances relacionados con el desarrollo de las tecnologías de alta resolución, como la secuenciación masiva o next-generation secuencing (NGS)(12), han favorecido la detección de alteraciones secundarias que cooperan con los eventos primarios de la tumorogénesis, modulando su pronóstico y abriendo un nuevo abanico de posibilidades en cuanto al tratamiento(13). En este contexto, la caracterización de los eventos secundarios, además de los primarios o drivers, podría optimizar la estratificación y el manejo clínico de los pacientes, sobre todo, en aquellos casos que no se curan y no disponen de tratamientos alternativos(14) (Fig. 1).
En relación a la estratificación, a medida que conocemos mejor la genética del tumor, vamos a poder diferenciar nuevos subtipos moleculares en tumores aparentemente idénticos, tras ser analizados con técnicas genéticas clásicas. Un ejemplo es la leucemia aguda linfoblástica de linaje B (LLA-B). Si se analizan pacientes clasificados como LLA-B con tecnologías de alta resolución, se puede diferenciar un subgrupo genético conocido como leucemia Ph like o B-other y los pacientes que la presenten van a tener un peor pronóstico comparado con el resto(15).
Además, se ha descrito recientemente la presencia de mutaciones sensibles o resistentes a fármacos dirigidos en más de un 30% de pacientes oncológicos pediátricos, lo que supone un gran avance en el conocimiento y tratamiento del cáncer infantil(4,13,16). Un ejemplo sería la presencia de fusiones génicas que afecten a la familia de genes NTRK, por ejemplo, que pueden conferir sensibilidad tumoral a inhibidores tirosín kinasa específicos, como larotrectinib, en el caso de pacientes diagnosticados de fibrosarcoma congénito(17).
A pesar de que estas técnicas aún no están del todo implementadas en la práctica clínica diaria, los resultados obtenidos hasta ahora han sido alentadores, lo que plantea un cambio de paradigma y una revolución en cuanto al abordaje del cáncer infantil(18).
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de la autora.
1. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. doi:10.1038/nature07943. Nature. 2009; 458: 719-24.
2. Taeubner J, Wieczorek D, Yasin L, Brozou T, Borkhardt A, Kuhlen M. Penetrance and Expressivity in Inherited Cancer Predisposing Syndromes. doi:10.1016/j.trecan.2018.09.002. Trends in cancer. 2018; 4: 718-28.
3.** Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. doi:10.1056/NEJMoa1508054. N Engl J Med. 2015; 373: 2336-46.
4. Chang W, Brohl AS, Patidar R, Sindiri S, Shern JF, Wei JS, et al. MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. doi:10.1158/1078-0432.CCR-15-2717. Clin Cancer Res. 2016; 22: 3810-20.
5.** Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. doi:10.1038/nature25480. Nature. 2018; 555: 321-7.
6.Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. doi:10.1001/jamaoncol.2015.5699. JAMA Oncol. 2016; 2: 616-24.
7.*** Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. American journal of medical genetics. doi:10.1002/ajmg.a.38142. Part A. 2017; 173: 1017-37.
8.*** Jongmans MC, Loeffen JL, Waanders E, Hoogerbrugge PM, Ligtenberg MJ, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. doi:10.1016/j.ejmg.2016.01.008. Eur J Med Genet. 2016; 59: 116-25.
9. Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, et al. Retinoblastoma. Nature reviews. doi:10.1038/nrdp.2015.21. Disease primers. 2015; 1: 15021.
10. Abedalthagafi M. Constitutional mismatch repair-deficiency: current problems and emerging therapeutic strategies. doi:10.18632/oncotarget.26249. Oncotarget. 2018; 9: 35458-69.
11. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. doi:10.3324/haematol.2015.141101. Haematologica. 2016; 101; 407-16.
12. Santos Simarro F, Vallespín García E, Palomares Bralo M. Nuevas metodologías en el estudio de enfermedades genéticas y sus indicaciones. Pediatr Integral. 2019; XXIII(5), 241-8.
13. Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. doi:10.1001/jamaoncol.2015.5689. JAMA Oncol. 2016; 2: 608-15.
14. Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients – The INFORM pilot study. doi:10.1016/j.ejca.2016.06.009. Eur J Cancer. 2016; 65: 91-101.
15. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. doi:10.1056/NEJMoa1403088. N Engl J Med. 2014; 371: 1005-15.
16.Harttrampf AC, Lacroix L, Deloger M, Deschamps F, Puget S, Auger N, et al. Molecular Screening for Cancer Treatment Optimization (MOSCATO-01) in Pediatric Patients: A Single-Institutional Prospective Molecular Stratification Trial. doi:10.1158/1078-0432.CCR-17-0381. Clin Cancer Res. 2017; 23: 6101-12.
17. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. doi:10.1038/s41571-018-0113-0. Nat Rev Clin Oncol. 2018; 15: 731-47.
18.*** Mody RJ, Prensner JR, Everett J, Parsons DW, Chinnaiyan AM. Precision medicine in pediatric oncology: Lessons learned and next steps. doi:10.1002/pbc.26288. Pediatr Blood Cancer; 2017. p. 64.
Bibliografía recomendada
– Jongmans MC, Loeffen JL, Waanders E, Hoogerbrugge PM, Ligtenberg MJ, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. doi:10.1016/j.ejmg.2016.01.008. Eur J Med Genet. 2016; 59: 116-25.
Recomendaciones para la realización de pruebas genéticas, con el fin de optimizar el diagnóstico de síndromes de predisposición a cáncer en población pediátrica.
– Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. American journal of medical genetics. doi:10.1002/ajmg.a.38142. Part A. 2017; 173: 1017-37.
Revisión sobre los síndromes de predisposición a cáncer más frecuentes que aparecen en edad pediátrica.
– Mody RJ, Prensner JR, Everett J, Parsons DW, Chinnaiyan AM. Precision medicine in pediatric oncology: Lessons learned and next steps. doi:10.1002/pbc.26288. Pediatr Blood Cancer; 2017. p. 64.
Revisión que explica las ventajas y limitaciones de la medicina de precisión aplicada al cáncer infantil.
| Caso clínico |
|
Antecedentes personales Varón de 14 años de edad trasladado de otro hospital con sospecha de leucemia. Refiere astenia y pérdida de peso (3-4 kg) desde hace aproximadamente un mes. Presenta, además, dolores óseos, y en las últimas 48 h refiere dolor abdominal y vómitos. Tras realizar ecografía en su centro de salud, detectan esplenomegalia. Se realiza un hemograma donde se evidencia una trombopenia grave (13 x 106/mm3) con el resto de series normales. La bioquímica básica muestra elevación de LDH (485 UI/L), sin otras alteraciones electrolíticas importantes. Se realiza estudio de coagulación que es normal y un frotis de sangre periférica, en el que se observa un 39% de células inmaduras de hábito blástico. A su llegada, a la exploración física, impresionaba de buen estado general. Se realiza un aspirado de médula ósea con el que se llevan a cabo las siguientes pruebas: • Citología: se observan células de características blásticas, que constituyen entre el 45-55% de la celularidad global, compatible con leucemia aguda linfoblástica tipo L2 según la clasificación FAB (clasificación según el grupo cooperativo Franco-Americano-Británico). • Citometría: se observa una población que supone aproximadamente el 38% de los eventos de tamaño y complejidad similar a los linfocitos residuales, que expresan marcadores compatibles con el diagnóstico de leucemia aguda linfoide B-II común, según la clasificación EGIL (Grupo Europeo de Clasificación Inmunológica de Leucemia). • Citología de líquido cefalorraquídeo: ausencia de infiltración en SNC. • Cariotipo y array-CGH: presencia de un clon con cariotipo hipodiploide de 35 cromosomas, con pérdida de los cromosomas 2, 3, 4, 7, 12, 13, 15, 16, 17,18 y 20. Compatible leucemia linfoblástica aguda con cariotipo hipodiploide. • FISH: el estudio de hibridación fluorescente in situ (FISH) detectó deleción TEL en el 45% de las células. • Se realiza placa de tórax que descarta la presencia de masa mediastínica. • Se realiza ecografía abdominal que confirma la presencia de hepatoesplenomegalia. Evolución clínica Una vez confirmado el diagnóstico, previa información a la familia y obtención del consentimiento informado, se inicia tratamiento de quimioterapia según protocolo LAL-SEHOP-PETHEMA 2013. El paciente recibe tratamiento citostático hasta cumplir 2 años desde la fecha del diagnóstico, encontrándose en el momento actual en remisión de su enfermedad de base. Como complicaciones relevantes durante el tratamiento, destacan: una neuropatía grave por vincristina al inicio de la inducción, así como complicaciones derivadas del tratamiento esteroideo, como una diabetes mellitus y una osteonecrosis del cóndilo femoral que ha requerido el implante de una prótesis. Estudio genético El paciente fue diagnosticado de leucemia linfoblástica con cariotipo hipodiploide. Por este motivo, incluido en los criterios de Jongmans (Tabla II), se deriva a la consulta de predisposición a cáncer infantil, en la que se decide realizar estudio genético en línea germinal mediante secuenciación masiva, utilizando un panel de diseño propio que incluye 150 genes previamente asociados a cáncer infantil. Los resultados del estudio revelan la presencia de una variante en heterocigosis en el gen TP53 (NM_000546): c.559+1G>T, p.? que es clasificada como probablemente patogénica. Se descarta la presencia de dicha variante en los padres y hermanos del paciente, indicando que se trata de una mutación de novo, es decir, no heredada. Asesoramiento genético y plan de seguimiento La mutación probablemente patogénica identificada en el paciente en el gen TP53, confirma el diagnóstico de síndrome de Li-Fraumeni. Los pacientes de Li-Fraumeni presentan un mayor riesgo a desarrollar otros tipos de cáncer a lo largo de la vida. Además, tienen una probabilidad del 50% de transmitir la mutación y la predisposición a cáncer a su descendencia. Se recomienda que siga las revisiones y el tratamiento relacionado con la leucemia aguda linfoblástica. Se valora la realización de un examen físico cada 3-4 meses, que incluye examen dermatológico y neurológico. Se propone la realización de un body-TAC/TAC tóraco-abdominal anual. Además, se realizará una colonoscopia cada 2-5 años a partir de los 25 años. Se le explican las opciones reproductivas futuras, que incluyen el diagnóstico prenatal y preimplantacional, en el caso en que desee descendencia. Se entregan recomendaciones de hábitos de vida saludables.
|


 How to suspect cancer in Primary Care
How to suspect cancer in Primary Care