 |
| Temas de FC |
A.J. Cartón Sánchez*, F. Gutiérrez-Larraya**
*Facultativo especialista. **Jefe de Servicio. Servicio de Cardiología Pediátrica. Hospital Universitario La Paz. Madrid
| Resumen
Con el término miocardiopatía (MC) se designan enfermedades muy heterogéneas en origen, manifestaciones y curso clínico que afectan de manera principal al músculo cardiaco. En esta revisión, aplicamos la clasificación morfológica más general (MC dilatada, hipertrófica, restrictiva y no compactada) a la edad pediátrica y exponemos, de manera diferenciada, enfermedades de presentación específica a estas edades, que se manifiestan con clínica cardiaca y extracardiaca, de sospecha para el pediatra de Atención Primaria e indicación de derivación. La sintomatología más expresiva de las MC pediátricas es la insuficiencia cardiaca, pero existen subtipos donde se llega al diagnóstico en situación asintomática, por cribaje familiar, o tras el hallazgo de pruebas complementarias anormales (radiografía de tórax, ECG). El diagnóstico de MC tiene implicaciones para la familia del niño afecto, y las limitaciones del estudio genético para aclarar su situación deben conocerse. Si bien el manejo último recae en especialistas, el pediatra de Atención Primaria tiene un papel fundamental en el diagnóstico inicial, en el conocimiento de situaciones de riesgo o de descompensación del niño con MC, y en la participación en las medidas de inmunoprofilaxis y estilo de vida específicas. |
| Abstract
The term cardiomyopathy (CM) defines a set of diseases of the cardiac muscle that show a wide variability in their causes, clinical manifestations and course. Here, we use the general classification of CM (dilated, hypertrophic, restrictive and non-compaction CM) to review the different subtypes that present in children, some in a specific manner, raising the index of suspicion of cardiac and extracardiac signs detected by the primary care paediatrician that warrant further assessment. Heart failure may signal the presence of a paediatric CM, but other cases are detected in asymptomatic children, through family screening, or after abnormal findings in ancillary tests such as the chest X-ray, or the ECG. Diagnosis of a CM may imply the need to study other members of the child’s family, and limitations of genetic testing should be acknowledged. Ultimately, specialised management is required in paediatric CM, but primary care paediatricians play a central role, referring a possible diagnosis at its onset, knowing risks and decompensation occurrences, and collaborating in immunoprophylaxis and lifestyle advices. |
Palabras clave: Miocardiopatías; Miocardiopatía familiar; Miocardiopatía dilatada; Miocardiopatía hipertrófica; Pediatría.
Key words: Inherited Cardiovascular Disease; Cardiomyopathies; Cardiomyopathy Dilated; Cardiomyopathy Hypertrophic; Pediatrics.
Pediatr Integral 2021; XXV (8): 427 – 436
Miocardiopatías
Panorámica general
Las miocardiopatías afectan de manera principal al músculo cardiaco, y se clasifican según sus características morfológicas. Su frecuente origen genético implica un abordaje integral del niño y de su familia.
Las miocardiopatías (MC) son enfermedades heterogéneas del músculo cardiaco. Ocasionan cambios estructurales en el corazón que producen un fallo en su función de bomba y la aparición de arritmias, sobre todo. Suelen tener una base genética y un componente hereditario, y el diagnóstico requiere excluir otras causas de afectación miocárdica (enfermedad coronaria, hipertensión arterial, valvulopatía o cardiopatía congénita), pudiendo implicar el estudio tanto del niño como de sus familiares más relacionados.
Las MC tienen una morbimortalidad alta y suponen una causa principal de insuficiencia cardiaca (IC) en la infancia e indicación del trasplante cardiaco (TXC; hasta el 40% del global de las formas sintomáticas en los 2 primeros años tras el diagnóstico). Los registros pediátricos (Pediatric Cardiomyopathy Registry [PCMR](1,2), National Australian Childhood Cardiomyopathy Study [NACCS])(3) informan de incidencias de al menos 1:100 000 en menores de 20 años, siendo el grupo de niños menores de 1 año el de mayor incidencia (3-4 veces más), del orden de magnitud similar a otras enfermedades pediátricas, como los linfomas, el tumor de Wilms o el neuroblastoma.
La clasificación de las MC es compleja y evoluciona con los conocimientos de sus mecanismos. El elemento clave es qué tipo de manifestaciones morfofuncionales (fenotipo) presenta el músculo cardiaco. La clasificación más comúnmente empleada (Tabla I), aunque con ligeras diferencias, por las sociedades científicas (European Society of Cardiology [ESC], American Heart Association/American College of Cardiology [AHA/ACC]), habla de los siguientes tipos principales(4):
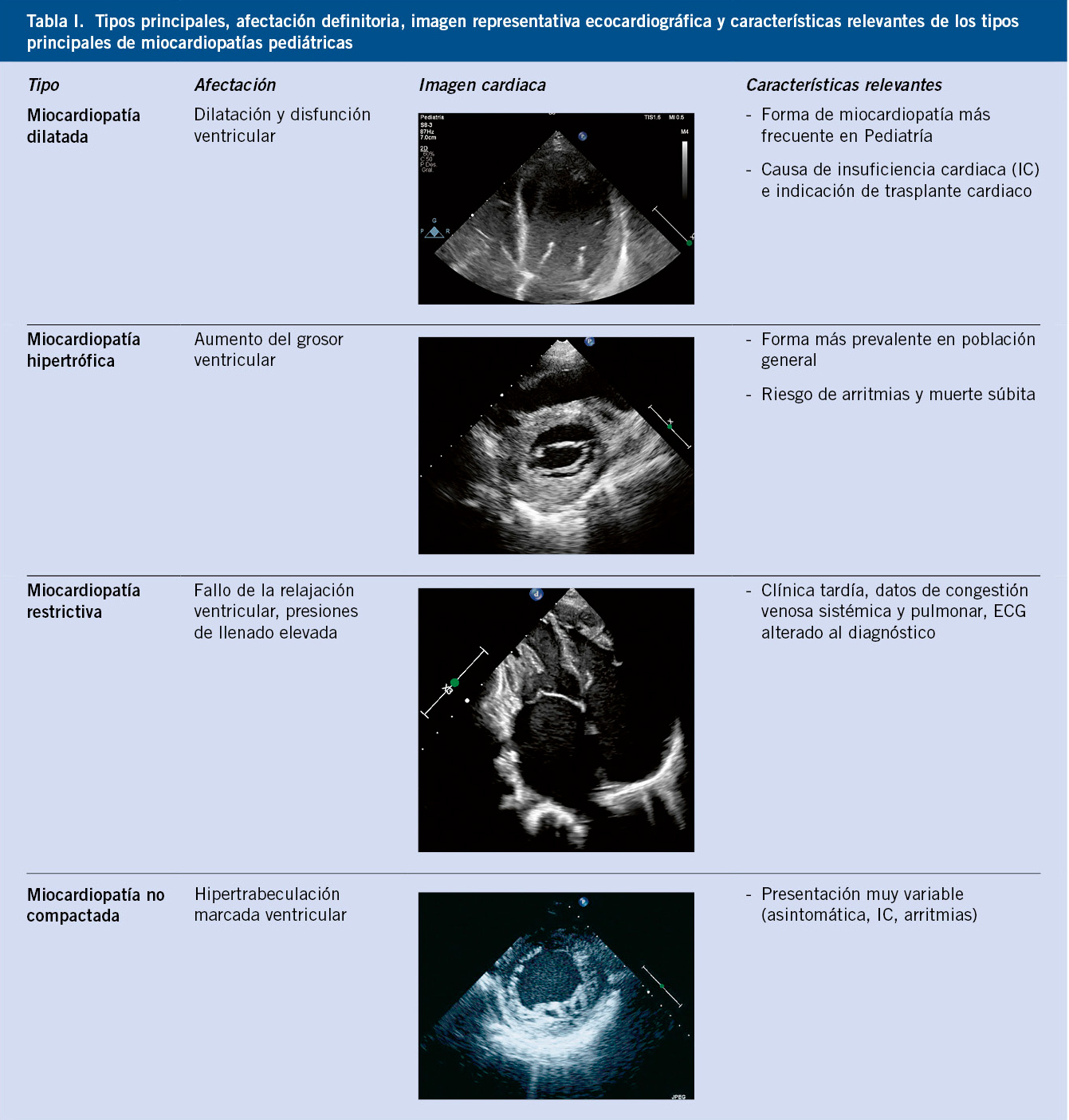
• Miocardiopatía dilatada (MCD; predomina el aumento del volumen cardiaco y la disfunción sistólica).
• Miocardiopatía hipertrófica (MCH; existe un aumento del grosor del músculo cardiaco que puede conducir a disfunción diastólica y sistólica, y aparición de arritmias).
• Miocardiopatía restrictiva (MCR; hay una rigidez de miocardio que obliga a presiones elevadas de llenado).
• Miocardiopatía no compactada (MCNC; alteraciones en el desarrollo del músculo ventricular con aumento de las trabeculaciones).
Debido a la heterogeneidad de las MC, se tiende a describir e integrar su complejidad con clasificaciones más recientes, como la MOGE(S)(5), que emplea 5 características: el fenotipo morfofuncional (M), la participación de otros órganos (O), el patrón genético o de herencia familiar (G), la descripción etiológica del defecto genético subyacente, si se identifica (E), y S como estado funcional (que no se suele emplear en niños). Esta clasificación flexibiliza dar cuenta de la presentación de algunos pacientes y sus familias (fenotipos solapados, mixtos y de idéntico origen genético, grado de afectación extracardiaca, causa y eventual patrón de transmisión).
El origen genético de numerosas MC conlleva el estudio familiar (comenzando por anamnesis, al menos, de 3 generaciones) y diversos escenarios(6). Así, el niño asintomático, hijo de un progenitor afecto, puede necesitar que se derive para evaluación específica y determinar la presencia de alteraciones compatibles, y de su potencial causa genética, en ciertas ocasiones, con persistencia de la incertidumbre en la situación: los resultados genéticos negativos no excluyen el diagnóstico evolutivo ni la posibilidad de afectación familiar, que puede no ser idéntica en expresión del tipo de MC a la del caso original (expresividad variable). Por otro lado, en ausencia de datos de imagen de MC, a pesar de encontrar mutaciones genéticas que implican enfermedad (variantes patogénicas), no suele indicarse, salvo excepciones que se mencionarán, el tratamiento médico ni la restricción de la actividad física, y algunos casos ni siquiera llegan a desarrollar la enfermedad (penetrancia incompleta). Finalmente, en el otro extremo, no se suele considerar en riesgo ni efectuar seguimiento al niño que no presenta la mutación familiar patogénica ni tiene alteraciones cardiológicas tras ser evaluado.
Miocardiopatía dilatada
La miocardiopatía dilatada es la forma de MC más frecuente en la infancia.
En la MCD, existe una dilatación y disfunción ventricular, predominantemente izquierda (VI), que puede conducir a IC, arritmias y muerte súbita. Es la MC más frecuente en la infancia (0,58 casos por 100.000 niños), con una edad mediana al diagnóstico entre 1 y 2 años (en menores de 1 año, puede ser hasta 8 veces más frecuente que en el global de los niños)(7,8).
Etiopatogenia
Las causas más frecuentes de MCD tienen una base genética, con posible afectación extracardiaca orientativa, pero también existen causas infecciosas, sobre todo, víricas, con mejor pronóstico.
Las MCD se deben más frecuentemente a anomalías de genes que codifican proteínas del citoesqueleto, proteínas de anclaje o del sarcómero cardiaco; en otras ocasiones, son el resultado de daño miocárdico directo por un agente externo. Las clasificaciones actuales (AHA/ACC) encuadran las causas genéticas como MCD primarias, y las causas inflamatorias (infecciosas o no infecciosas), tóxicas y endocrino-metabólicas, como MCD secundarias. La MCD idiopática (diagnóstico de exclusión) tiene una base presumida, no identificada, genética, y es de peor pronóstico que otras formas; los niños clasificados bajo este tipo pueden reasignarse a una categoría de MCD familiar en la ampliación del estudio(4).
Dentro de las causas genéticas encontramos:
• MCD familiar (30-50%): donde hay otros miembros relacionados en primer grado, con o sin una mutación genética encontrada (genes que codifican proteínas del citoesqueleto o del disco Z, p. ej.: sarcoglicanos, desmina, lámina A/C, titina, actinina…); el patrón de herencia más común es autosómico dominante (AD).
• MCD asociada con mutaciones de genes sarcoméricos (10-20%): actina, troponina, cadena pesada de la miosina β, proteína C de unión a la miosina, α-tropomiosina, fosfolambano, canal de sodio SCN5A…
• MCD asociada con enfermedad neuromuscular: sobre todo, las distrofias de Duchenne y de Becker, el síndrome de Barth (también como MCR o MCNC), la ataxia de Friedreich (como MCH) y las distrofias musculares de cintura y extremidades. Tienen patrón de herencia variable (autosómico dominante o recesivo, o ligado a X). Estos pacientes deben evaluarse cardiológicamente al diagnóstico de la enfermedad y durante su seguimiento de despistaje (en la enfermedad de Duchenne, p. ej., la afectación cardiaca es más evidente a partir de la segunda década de la vida), y tienen la mayor mortalidad y la menor tasa de trasplante a los 5 años, tras el diagnóstico de MCD.
• MCD por laminopatías: suele haber manifestaciones extracardiacas, como neuropatía periférica, enfermedades del músculo esquelético y progerias. De manera anticipada a las alteraciones estructurales, estos pacientes pueden presentar, en edades pediátricas, arritmias diversas (desde trastornos de conducción AV o disfunción sinusal hasta taquicardias ventriculares).
• MCD por enfermedad mitocondrial: en genes del DNA mitocondrial o nuclear que son relevantes en el metabolismo energético. Ejemplos: déficit de la transferasa II carnitina palmitoil, síndrome de Kearns Sayre, síndrome MELAS (con encefalopatía y acidosis láctica), síndrome MERFF (con epilepsia mioclónica), déficit en la reductasa NADH-coenzima Q (complejo I) y déficit de la oxidasa del citocromo C.
Las causas infecciosas de MCD incluyen, sobre todo, virus (adenovirus, enterovirus, herpes simple, varicela zóster, citomegalovirus, Ebstein-Barr, herpes humano 6, influenza A y B, inmunodeficiencia humana, parvovirus). También se mencionan, más raramente, causas bacterianas (Chlamydia, C. diphteriae, Legionella…), fúngicas (Actinomyces, Aspergillus, Candida…), ricketsiosis y Borrelia. Las MCD atribuidas a miocarditis vírica (miocarditis linfocitaria) tienen un 70% de supervivencia global y libre de TXC a los 5 años, tras el diagnóstico. Las causas inflamatorias no infecciosas son las enfermedades autoinmunes y reacciones de hipersensibilidad (dermatomiositis, síndrome hiperesoinofílico, miocarditis, lupus y artritis reumatoide). Se han involucrado tóxicos y fármacos como penicilina, ampicilina, cefalosporinas, tetraciclinas y sulfonamidas; los niños expuestos a quimioterapia para su cáncer (sobre todo, antraciclinas para leucemias, linfomas y sarcomas) pueden desarrollar, a largo plazo, MCD e IC. Las causas metabólicas, finalmente, son raras en niños, y suelen existir otras manifestaciones que orientan el diagnóstico: alteraciones tiroideas (hiper e hipotiroidismo), adrenérgicas (tumor secretor de catecolaminas), hipocalcemia crónica (hipoparatiroidismo), etc.
Clínica
La MCD puede presentarse como insuficiencia cardiaca grave de repercusión multisistémica.
La presentación sintomática típica, y lo más frecuente al diagnóstico, es la de IC, con manifestaciones variables que dependen de la edad: en niños pequeños, fallo de medro, problemas de alimentación, infecciones respiratorias que descompensan la situación basal; en niños mayores, la clínica más característica es de intolerancia al esfuerzo o disnea; en todos los casos, el shock cardiogénico o la muerte súbita son potenciales consecuencias finales. El antecedente de cuadro vírico o catarral puede faltar o no reconocerse en las secundarias a miocarditis, haciéndolas indistinguibles de las idiopáticas o genéticas. La asociación con hipoglucemia, acidosis metabólica, hipotonía o afectación hepática, orienta a un error congénito del metabolismo. La sintomatología neurológica o muscular esquelética se asocia con enfermedades mitocondriales o una distrofia muscular. Casi la mitad de casos, al diagnóstico, puede precisar cuidados intensivos y soporte inotrópico, así como asociarse con comorbilidad de otros órganos o sistemas (insuficiencia renal, hepatopatía, trombosis vascular…). Otros casos pueden detectarse en pacientes asintomáticos, como resultado de cribaje familiar.
Diagnóstico
El diagnóstico de MCD es por imagen cardiaca, pero pruebas como la radiografía de tórax o el ECG, permiten sospecharla con clínica sugerente.
Las alteraciones diagnósticas de MCD se basan en la imagen cardiaca, sobre todo, la ecocardiografía, y no son específicas de su causa. El criterio fundamental es la dilatación, ajustada por tamaño corporal del niño, del VI, que se acompaña habitualmente de disfunción sistólica (disminución de la función contráctil). La resonancia magnética (RM) cardiaca puede cuantificar de manera más precisa los volúmenes y la función biventricular, y caracteriza mejor el tejido afecto (edema, inflamación o fibrosis que pueden ser etiológica o pronósticamente informativas).
Antes de estas pruebas, la radiografía de tórax puede constituir un alto índice de sospecha, al mostrar cardiomegalia como signo que orienta la presencia de MCD, si hay clínica de presentación similar a una infección respiratoria; menos frecuentemente, el hallazgo es casual. En el ECG, pueden observarse alteraciones no específicas de MCD, pero merecedoras de estudio con clínica dudosa (en concreto, sospechar de taquicardias sinusales no apropiadas a la situación fisiológica del niño como índice de IC) como hipertrofia ventricular o auricular, anomalías no específicas de la onda T, arritmias supraventriculares y ventriculares. Las pruebas de laboratorio se orientan, por un lado, a evaluar la repercusión en otros órganos o sistemas (elevación de transaminasas, afectación de la función renal) al diagnóstico, y por otro lado, a tratar de establecer la etiología, sobre todo, infecciosa (serologías y PCR específicas), aunque otras alteraciones bioquímicas pueden ser también sugerentes; así, la elevación de la CPK podría sugerir una enfermedad neuromuscular relacionada con la distrofina, una laminopatía o una miopatía miofibrilar, o la elevación de TSH orienta a causa endocrina. Las pruebas microbiológicas específicas (serología de virus, Chagas, Borrelia) establecen su diagnóstico.
El estudio genético establece la etiología en el caso pediátrico afecto y el potencial estado de portador en los familiares (hasta un 50% podría presentar una mutación patogénica); el conocimiento de este estado podría contribuir a estrategias de prevención o detección precoz en otros familiares.
Tratamiento
El tratamiento es sintomático de insuficiencia cardiaca, y algunos niños pueden precisar trasplante cardiaco.
Comprende el tratamiento de la causa, cuando se identifica, y el tratamiento de apoyo y sintomático de la IC y las arritmias. En fase aguda, si predomina la retención de líquidos, con buena perfusión (forma caliente y húmeda), la base son los diuréticos y los vasodilatadores sistémicos; si hay mala perfusión, se emplean inótropos. En fase crónica, el tratamiento especializado consiste en inhibidores de la enzima convertidora de angiotensina (IECA), bloqueantes beta-adrenérgicos, antagonistas de los mineralcorticoides y furosemida, con papel seleccionado de digoxina e ivabradina; en estudio se encuentran otros fármacos de uso ya estándar en adultos, como los inhibidores de angiotensina y del receptor de nesiprilina (ANRI). En fases finales de IC, pueden ser candidatos a un TXC, ayudados o no de manera previa con una asistencia ventricular de corta (ECMO) o larga duración (p. ej., Berlin Heart, Heartware).
Miocardiopatía hipertrófica
La MCH es el segundo tipo de MC en edad pediátrica, con relativo mejor pronóstico, pero riesgo de muerte súbita.
En la MCH, se objetiva un aumento del grosor ventricular (hipertrofia del VI, HVI) sin explicación hemodinámica aparente (HTA, obstrucción a la salida del VI [OTSVI]). Ciertas adaptaciones fisiológicas en el niño y adolescente (corazón del deportista), pueden plantear dudas al inicio; en estos casos, podría ayudar el desacondicionamiento deportivo durante unos meses, así como la ausencia de alteraciones en otras pruebas, para identificar si hay MCH realmente.
La incidencia pediátrica estimada es de 0,3 a 0,5 casos por 100.000, y supone la segunda MC por frecuencia (25-40% de casos de MC). Sin embargo, la MCH es la enfermedad cardiaca genética más prevalente en adultos (1:500 personas, en algunas estimaciones hasta 1:250, siendo sintomáticos menos de 1:3.000). La edad de aparición es muy variable, desde el periodo neonatal hasta la adolescencia (en el grupo pediátrico). En general, hay mejor pronóstico que en otras MC (supervivencia libre de TXC de 90% a los 5 años), pero se debe considerar el riesgo de muerte súbita y que algunos subgrupos (lactantes con IC a la presentación, niños mayores con fisiología restrictiva, HVI grave al diagnóstico, el síndrome de Noonan…) evolucionan peor(9-11).
Etiología y clasificación
Las causas más frecuentes de MCH son genéticas; en Pediatría, hay síndromes específicos que cursan con MCH.
Algunas sociedades (AHA/ACC)(10) no consideran como MCH las formas donde hay una causa sistémica de la HVI, en concreto síndromes multisistémicos de origen genético, no sarcoméricos que, en general, son enfermedades de presentación infantil específica (hasta un 25% de los casos a esta edad; p. ej., las RASopatías, como el síndrome de Noonan); esas formas se denominarían de manera alternativa fenocopias, y no propiamente MCH. La ESC(11) sí las incluye en la denominación de MCH (causas secundarias), nomenclatura más empleada y que adoptamos aquí.
Las MCH primarias son las de origen sarcomérico (8 genes explican el 30-60% de los casos globalmente; MYH7 y MYBCP3 explican el 70% de las causas sarcoméricas), y son las de mayor prevalencia en la población general, pero no en el período de la infancia, donde es frecuente que se llegue al diagnóstico en un niño asintomático con un familiar afecto. Las causas no sarcoméricas (secundarias, sistémicas, fenocopias) son las más frecuentes en menores de 1 año y abarcan:
• Enfermedades por almacenamiento anormal de glucógeno: tipos más frecuentes asociados con MCH:
- Tipo II o enfermedad de Pompe; déficit de α-1, 4 glucosidasa. Debe sospecharse en un lactante que presenta hipotonía muscular, cardiomegalia radiológica y alteraciones ECG características (intervalo PR corto, voltajes muy elevados del complejo QRS).
- Tipo IIb o enfermedad de Danon (ligado a X, mutaciones en gen LAMP2). Hay MCH, miopatía de músculo esquelético y retraso intelectual leve. Los varones suelen tener afectaciones más precoces y graves.
- MC por PRKAG2. Presenta: MCH, preexcitación ventricular y enfermedad progresiva del sistema de conducción.
• Enfermedades por depósito lisosomal anormal. Incluye: síndrome de Hurler (talla baja, rasgos faciales toscos, anomalías esqueléticas), enfermedad de célula I (mucolipidosis, enfermedad de Leroy) y enfermedad de Anderson-Fabry (presentación en la adolescencia con angiokeratomas y dolor abdominal).
• MCH sindrómica. Incluye las RASopatías (síndrome de Noonan, síndrome de Noonan con lentiginosis [antiguo síndrome de LEOPARD], síndrome de Costello, síndrome cardiofaciocutaneo); frecuencia estimada de 1:1.000 a 2.500 niños. Pueden asociar cardiopatía congénita (sobre todo, estenosis pulmonar) y, típicamente, participación extracardiaca: dismorfias craneofaciales, hipotonia, desarrollo neurológico alterado, talla baja, predisposición a cáncer, anomalías cutáneas, musculares y oculares.
• Déficits de oxidación de ácidos grasos (déficit de la deshidrogenasa de la acil-CoA de cadena larga). Pueden tener arritmias ventriculares y muerte súbita.
• Hiperinsulinismo. La presentación es neonatal. En las formas primarias, hay una hiperproducción de insulina por hiperfunción de las células beta pancreáticas (focal como adenoma, o difusamente); clínicamente, pueden debutar como hipoglucemia grave, crisis epilépticas, hipotermia, hipotonía y letargia; son niños grandes, pero no necesariamente macrosómicos. El tratamiento es médico o quirúrgico. En las formas secundarias, el hiperinsulinismo es secundario a hiperglucemia materna (DM materna o gestacional), el RN suele ser macrosómico y la hipoglucemia inicial suele responder a suplementos; la HVI suele resolverse espontáneamente en el primer año de vida.
Patogenia
La MCH implica alteraciones cardiacas mecánicas y riesgo de arritmias.
Aunque no está claro cómo conduce al fenotipo la alteración del sarcómero, se observan cambios miocárdicos con aumento del grosor y fibrosis, que dan lugar a un ventrículo pequeño y rígido, disfunción diastólica y, posteriormente, sistólica. La HVI localizada en el tracto de salida del ventrículo izquierdo puede condicionar obstrucción a la salida de sangre del VI (OTSVI) y anomalías de la función valvular mitral (movimiento sistólico anterior e insuficiencia valvular). Otros elementos alterados son: arterias coronarias intramurales (isquemia), elongamiento del aparato subvalvular mitral o anomalías congénitas… Histológicamente, se produce una desorganización de miocitos y miofibrillas, fibrosis miocárdica, y enfermedad de pequeño vaso, que pueden ser la base del riesgo arrítmico.
Clínica
Es frecuente que el niño con MCH esté asintomático y esté siendo estudiado por otro motivo.
Lo más habitual es la situación asintomática de los niños. En otras ocasiones, la presencia de soplo (OTSVI, insuficiencia mitral por movimiento sistólico anterior) puede motivar la derivación al especialista. En un 10-15% de los casos, la presentación es de IC (fases finales) o arritmias, estas últimas como síncope de características cardiogénicas (durante esfuerzo, sin pródromos claros, aunque pueden describirse palpitaciones o malestar general durante el ejercicio) o atípico (no claro perfil vasovagal, presentación fuera de esfuerzo). Las formas de MCH asociadas a errores congénitos del metabolismo o malformaciones sindrómicas suelen tener debut más precoz y más clínica de IC al diagnóstico, siendo la muerte súbita más rara.
Las formas sindrómicas pueden presentar síntomas o signos clínicos orientativos de su causa genética:
• Problemas de aprendizaje o retraso mental (enfermedades mitocondriales, síndrome de Noonan, LEOPARD, síndrome de Costello, enfermedad de Danon).
• Sordera neurosensorial (enfermedad mitocondrial, sobre todo, con diabetes; enfermedad de Fabry-Anderson, síndrome de LEOPARD).
• Alteraciones visuales: retinopatía (enfermedad mitocondrial), retinitis pigmentosa (Danon), cataratas, opacidades corneales (Anderson-Fabry).
• Alteraciones de la marcha: ataxia de Friedreich.
• Parestesias, anomalías sensoriales: Anderson Fabry.
• Hipotonía muscular: enfermedad mitocondrial, glucogenosis, ataxia de Friedreich.
• Ptosis palpebral: enfermedad mitocondrial, Noonan/LEOPARD, distrofia miotónica.
• Lentiginosis, manchas café con leche: LEOPARD, Noonan.
• Angiokeratoma, hiposudoración: Anderson-Fabry.
• Dismorfias/anomalías cutáneas: RASopatía.
Diagnóstico
En niños, la MCH se diagnostica con imagen durante cribado familiar y a partir de alteraciones cardiacas y extracardiacas detectables clínicamente o en pruebas complementarias.
Se basa en la confirmación por imagen de la hipertrofia miocárdica, prueba a la que llega el paciente tras una clínica como la descrita, incluyendo eventos sincopales, por despistaje familiar o por hallazgo de elementos alterados en un ECG casual o indicado por síntomas.
El ECG es una prueba accesible desde Atención Primaria, y en más del 90% pacientes se presentan anomalías, por lo demás, elementos índice suficientes de sospecha: HVI por voltaje asociados, o no, con strain (inversión de la onda T, más específico; a veces, la inversión, en derivaciones precordiales izquierdas V4-V6, puede darse sin el crecimiento eléctrico), ondas P anchas (crecimiento auricular izquierdo), ondas Q patológicas (duración mayor de 40 ms o amplitud superior al 25% del QRS asociado… Un intervalo PR corto o preexcitación es frecuente en la enfermedad de Pompe, la MCH por PRKAG2 o la enfermedad de Danon, que pueden también presentar crecimiento ventricular izquierdo extremo.
El ecocardiograma suele bastar para confirmar el diagnóstico (grado y distribución de la HVI) y determinar la presencia de alteraciones asociadas (la OTSVI, la IM; la disfunción diastólica, alteraciones preclínicas de la contractilidad regional). Cuando la ecocardiografía no es concluyente (más que mala ventana en niños, para las localizaciones atípicas de HVI), la RM cardiaca es útil y también permite evaluar la presencia de fibrosis, que podría relacionarse con riesgo arrítmico.
El Holter ECG tiene utilidad durante el seguimiento, si el niño refiere palpitaciones o presíncopes, y para detectar la aparición de arritmias que confieran un riesgo mayor de eventos adversos. En la ergometría, se puede evaluar la situación funcional del niño y observar si sucede hipotensión en esfuerzo, dato clásico de peor pronóstico.
En las formas metabólicas de niños más pequeños, determinados hallazgos analíticos contribuyen a buscar la etiología: hipoglucemia con acidosis metabólica aparece en enfermedades del glucógeno y mitocondriales. Si no hay acidosis, la HVI puede encontrarse en hiperinsulinismo congénito, síndromes de resistencia a insulina y defectos de la oxidación de ácidos grasos.
El estudio genético, que se beneficia de la orientación dada por hallazgos previos anormales, confirma el origen de la MCH, pero no es necesario encontrar una mutación genética patogénica para que el niño con el fenotipo compatible sea diagnosticado.
Tratamiento
El manejo de la MCH puede implicar el implante de un desfibrilador y muchos niños pueden no necesitar medicación inicialmente.
El objetivo general es mejorar la calidad de vida y la supervivencia. Solo el implante de desfibrilador (DAI) mejora el pronóstico en los grupos de alto riesgo (más del 6% de eventos adversos a los 5 años) de muerte súbita de formas sarcoméricas; en las formas no sarcoméricas, la estimación de riesgo es más incierta. Los pacientes con MCH supervivientes de una muerte súbita o que han experimentado una taquiarritmia ventricular con síncope tienen indicación de implante (prevención secundaria). En el resto de casos, incluso en ausencia de cualquier síntoma, se estratifica el riesgo (prevención primaria) mediante factores clínicos reconocidos (síncope inexplicado, grosor septal, aparición de taquicardias ventriculares [TV] no sostenidas…), con calculadoras de riesgo o sumación de factores presentes (para niños: HCM-Kids Risk Calculator, puntuación Primacy; en mayores de 16 años, calculadora de la ESC)(11-13). El DAI reconoce arritmias ventriculares y es capaz de proporcionar terapias eléctricas para abortarlas. Su implante es similar al de un marcapasos endovenoso clásico; también hay dispositivos de implante subcutáneo (para pacientes mayores seleccionados). El niño debe evitar actividades de contacto que supongan un traumatismo sobre el dispositivo o desplazamiento del cable, aparte de las restricciones que le aplique su cardiólogo. La familia recibe información sobre eventuales interacciones electromagnéticas, incluyendo, por ejemplo, pruebas médicas como la RM (muchos DAI son actualmente compatibles) o el uso de bisturí eléctrico en intervenciones quirúrgicas (precisa reprogramación transitoria).
La medicación consiste en betabloqueantes (propranolol, atenolol, bisoprolol) para tratar de reducir la OTSVI, si está presente (hay formas no obstructivas que no los precisan); en caso de mala tolerancia o contraindicación, se propone el uso de antagonistas de los canales de calcio. Antiarrítmicos como la amiodarona pueden añadirse ante la aparición de arritmias ventriculares. Los diuréticos no se suelen emplear (las situaciones de hipovolemia relativa, como deshidratación en gastroenteritis, pueden tolerarse mal). La digoxina está contraindicada en presencia de OTSVI. Las opciones quirúrgicas se plantean en formas graves de OTSVI (miectomías septales; la ablación septal con alcohol en centros de experiencia y en pacientes mayores). En las fases finales de la enfermedad, con fallo de bomba e IC terminal, se indica el TXC, en general con buenos resultados.
Miocardiopatía restrictiva
La MCR es poco frecuente en niños, pero tiene mal pronóstico.
En las MCR, se produce rigidez y un fallo de relajación ventricular (disfunción diastólica); morfológicamente, la dilatación no se produce en los ventrículos, que suelen ser más bien de menor tamaño, sino en las aurículas. Es la MC de peor pronóstico (con mortalidad a los 5 años cercana al 30%) y de las menos frecuentes (0,03-0,04 por 100.000), aunque su incidencia aumenta con la edad, siendo raras las presentaciones en menores de 1 año. Las características de ciertos casos de MCR se solapan con la MCH: los niños con formas puras de MCR suelen trasplantarse más frecuentemente(4).
Etiopatogenia
Es característica de la MCR la congestión venosa, transmitida por fallo de relajación ventricular.
Las causas primarias son genéticas (proteínas del sarcómero, desmina, filamina C); las causas secundarias son infiltrativas (amiloidosis, muy rara en niños), por depósito lisososomal (Anderson-Fabry, estados de sobrecarga de hierro) y la fibrosis endomiocárdica (en parasitosis de niños procedentes de zonas de África, Asia y Sudamérica, enfermedades autoinmunes, síndromes con hipereosinofilia, déficits metabólicos o tóxicos).
Histológicamente, pueden existir anomalías en los miocitos o en la matriz intercelular (infiltración o fibrosis), pero el dato de restricción es funcional y requiere evidencia de presiones de llenado elevadas, confirmadas invasivamente (cateterismo) o de forma indirecta por la presencia de signos de congestión venosa sistémica.
Clínica y diagnóstico
La clínica de insuficiencia cardíaca sucede tarde en la evolución, y suele presentar alteraciones detectables en el ECG.
Al inicio, en las formas con afectación exclusivamente cardiaca, los síntomas son inespecíficos: fatiga general, intolerancia gastrointestinal o al esfuerzo. En la evolución, al aumentar las presiones venosas pulmonares y sistémicas elevadas, puede darse edema periférico, hepatomegalia, edema e hipertensión pulmonar (disnea o equivalente). La disfunción sistólica es tardía, así como los síntomas por bajo gasto cardiaco. La presentación sincopal es de mal pronóstico (arritmias, isquemia coronaria y evento tromboembólico).
El ECG puede ser la prueba complementaria inicial que oriente hacia un origen cardiaco de los síntomas, por estar alterado en la presentación, prácticamente en casi todos los niños: datos de crecimiento biauricular, con afectación variable de la amplitud de los complejos QRS y alteraciones de repolarización asociadas. En la ecocardiografía, el hallazgo casi patognomónico es la dilatación biauricular con ventrículos de tamaño normal o reducido. La analítica de sangre puede mostrar elevación del propéptido natriurético cerebral N-terminal (NT-proBNP) y orientar inicialmente la presencia de MC si la clínica es dudosa. Con el cateterismo cardiaco, se confirman las presiones de llenado ventricular elevadas y se obtiene información discriminante de otras situaciones fisiopatológicas similares, como la pericarditis constrictiva, muy rara en niños. La biopsia endomiocárdica tiene un rendimiento limitado y no se suele hacer de rutina. El estudio genético permite establecer el origen primario de la MCR.
Tratamiento
El tratamiento de la MCR es sintomático de insuficiencia cardiaca.
El tratamiento es sintomático de la IC en fases ya avanzadas, donde habitualmente se considera la inclusión en lista de trasplante. Es dudoso el papel de iniciar tratamiento anticongestivo en fases iniciales oligosintomáticas.
Miocardiopatía no compactada
La MCNC es poco frecuente, pero puede encontrarse en lactantes sin síntomas.
En la MCNC, morfológicamente, se observa una trabeculación prominente del VI, que expresa un fallo de maduración o del desarrollo del miocardio (miocardio más trabeculado = más primitivo) y que puede darse en combinación con características propias de otras MC. Las incidencias pediátricas referidas son de 0,11 por 100.000 (0-10 años), más frecuentemente en lactantes(4).
Se identifica una causa genética en un 30-45% de los casos (lo más frecuente: mutaciones sarcoméricas); también se ha relacionado la MCNC con otros genes no sarcoméricos y con el síndrome de Barth, una rara metabolopatía infantil que cursa con fallo de medro y neutropenia cíclica (también como MCR). Las formas aisladas parecen tener mejor pronóstico, y la asociación con arritmias (bloqueo AV) o fenotipos mixtos lo tienen peor(4,14).
Las presentaciones asintomáticas son frecuentes (hallazgo durante cribaje), pero también puede darse, de manera variable: insuficiencia cardiaca congestiva (ICC), arritmias o un evento tromboembólico. La imagen diagnóstica característica se suele obtener por ecocardiografía.
El tratamiento es sintomático y puede requerir, en fases finales de IC, inclusión en lista de TXC.
Miocardiopatía asociada con sustrato arrítmico
La miocardiopatía arritmogénica es una miocardiopatía con muy alto riesgo de arritmias graves para la vida, y detectarla en fase asintomática o por estudio genético tiene implicaciones muy relevantes.
En la miocardiopatía arritmogénica (MCA; históricamente, displasia arritmogénica del ventrículo derecho, nombre en desuso: puede haber afectación del VI, y se clasifica como MC) aparece, de forma característica, una sustitución fibrograsa del tejido miocárdico, aunque en fases iniciales, como las propias de niños y adolescentes, las alteraciones morfológicas pueden ser menos aparentes y el debut clínico consistir en arritmias graves para la vida. Es una MC muy poco frecuente (no se suele pensar en ella en menores de 10 años), pero muy relevante, por causar aproximadamente un 10% de casos de muerte súbita en jóvenes y de hasta un 22% en atletas. Una combinación de alteraciones de imagen, del ECG o la histología cardiaca, la presencia de arritmias ventriculares o una historia familiar confirmada, además o junto con la presencia de una mutación patogénica de un gen relacionado (proteínas relacionadas con el desmosoma cardiaco, zona de unión intercelular) son la base de los criterios diagnósticos(15). Al estudio de la enfermedad en niños, se llega por despistaje de un progenitor afecto, la presencia de arritmias ventriculares (a veces, solo extrasistolia ventricular aislada, pero frecuente), la evaluación de episodios sincopales o tras una muerte súbita resucitada. Los portadores de mutaciones genéticas asociadas con enfermedad, aun sin expresar sus alteraciones, deben restringir su actividad física, por ser esta un desencadenante de arritmias y elemento favorecedor para desarrollar el fenotipo. El tratamiento se dirige a los síntomas de IC o por arritmias, y una proporción importante de pacientes es candidato a DAI y se le implanta.
A los niños con canalopatías (síndrome de QT largo congénito, síndrome de Brugada, taquicardia ventricular polimorfa catecolaminérgica), clásicamente, se les ha considerado sin alteraciones propias de MC, aunque pueden tenerlas por el solapamiento de sus causas genéticas con las de MC. Por último, cabe mencionar dos situaciones de MCD asociada con arritmias:
1. El niño portador de marcapasos, por la estimulación crónica, puede experimentar dilatación y disfunción ventricular y referir clínica de IC.
2. En las taquicardias supraventriculares incesantes o muy prolongadas de lactantes, tras un comienzo, a veces, inadvertido, la clínica de IC (expresada de manera inespecífica como: mal estado general, rechazo de tomas, vómitos, aspecto incluso séptico) indica la evolución hacia disfunción ventricular (taquimiocardiopatía, generalmente reversible), y es un primer contacto con su pediatra lo que puede orientar el diagnóstico, al detectar la taquicardia, sobre todo, en ausencia de datos de infección.
Función del pediatra de Atención Primaria
Las MC, como grupo global, son entidades raras, pero desde Atención Primaria se puede contribuir a los cuidados de los niños afectos en varias cuestiones (Fig. 1):

Figura 1. Papel del pediatra de Atención Primaria: resumen gráfico de intervenciones en el niño con MC.
• Al diagnóstico. Se debe conocer el componente familiar de muchas MC y facilitar el acceso al estudio en cascada (de primer grado –padres a hijos, e hijos a padres– a más lejano): muchos niños están en situación asintomática y es solo el diagnóstico en un progenitor lo que inicia el proceso. En cuanto a las presentaciones sintomáticas, las MC no son las causas más frecuentes de clínica cardiovascular atribuible en la infancia, pero debe prestarse atención a elementos muy sugerentes (insuficiencia cardiaca, a veces, solo como taquicardia mantenida en rangos fisiológicos altos, en ausencia de infección, anemia o proceso intercurrente claro, que puede asociarse con síntomas de intolerancia digestiva, vómitos…; síncopes de presentación atípica o durante el esfuerzo…). Asimismo, las alteraciones en pruebas complementarias realizadas con otro motivo (cardiomegalia radiológica, ECG con anomalías del tipo crecimiento de cavidades o alteraciones de repolarización) pueden justificar la derivación al especialista. En el niño con signos de afectación extracardiaca diversa (dismorfias, retraso mental, hipotonía, fallo de medro, alteraciones metabólicas), esta derivación puede formar parte de la estrategia de estudio.
• Durante el seguimiento. Debe conocer la particular vulnerabilidad del niño con MC a procesos intercurrentes, sobre todo, procesos respiratorios (también gastroenteritis con deshidratación o trastornos iónicos), que ocasionen descompensaciones (IC). El niño con MC puede considerarse en el grupo de enfermedades crónicas cardiacas (por IC, alteraciones hemodinámicas asociadas o hipertensión pulmonar) al que hay que aplicar, junto a los convivientes, lo expuesto por el Comité de Vacunas de la Asociación Española de Pediatría(16):
- Vacunación antigripal (tetravalentes inactivadas) anual. Son pacientes de alto riesgo de complicaciones(17).
- Vacunación antineumocócica secuencial (incluyendo VNC13 y la VNP23).
- Vacunación antivaricela, con 2 dosis según pauta de calendario. Niños susceptibles no vacunados deben ser vacunados lo antes posible con dos dosis (intervalo 1-3 meses). Bajo tratamiento con aspirina, si es posible, al menos, dejando 6 semanas sin tratamiento para vacunar; no obstante, el síndrome de Reye se ha evidenciado con la variante salvaje del virus y no con la vacunal, por lo que en riesgo de exposición, también se recomienda vacunar.
- Además, algunos niños menores de 2 años pueden beneficiarse de la profilaxis estacional de VRS (Palivizumab), de aplicación hospitalaria(18). En lo referente a la covid-19, por ser un campo en evolución, que este trabajo no representaría de manera actualizada, remitimos al lector a fuentes en constante revisión.
• Sobre recomendaciones generales sobre estilo de vida. Aparte de la aplicación general de consejos saludables sobre la dieta y la evitación de tóxicos (tabaco), es oportuno estar familiarizado con lo referido al ejercicio físico y deporte del niño con MC. Existe una responsabilidad directa del especialista en proporcionar esta información, pero el pediatra de Atención Primaria es un acceso más directo al que pueden recurrir las familias en situaciones de duda, y algunas situaciones deben ser de conocimiento general. Se tiende a la discusión individualizada y decisión final compartida con el paciente y su familia sobre el nivel de ejercicio, entendiendo que la ausencia de factores de riesgo conocidos no confiere inmunidad frente a arritmias de riesgo vital y que, en una importante proporción de casos, estas pueden ocurrir durante el sueño, en reposo o actividades de baja exigencia. No obstante, es posible que niveles bajos a moderados de actividad física se acuerden y permitan. Un niño portador de una mutación familiar sin expresión fenotípica no tiene descalificación automática del deporte intenso, salvo si es una MCA o una MCD por genotipos donde está involucrada la lámina A/C o la filamina C, donde de entrada sí se aplica. Los deportes de alta intensidad (p. ej., muchos deportes de equipo practicados competitivamente como fútbol, baloncesto, balonmano, rugby, hockey…; o actividades de resistencia como el ciclismo de grandes distancias, el remo, la natación competitiva, y algunas variedades de atletismo) se suelen desaconsejar en niños con MCH, MCD o MCNC que presentan marcadores de riesgo (síntomas durante esfuerzo o historia de síncope, aparte de otros hallazgos determinados en las pruebas complementarias, p. ej.: disfunción ventricular, presencia de arritmias ventriculares en esfuerzo…)(19).
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, et al. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin. 2010; 6: 401-13.
2. Wilkinson JD, Westphal JA, Bansal N, Czachor JD, Razoky H, Lipshultz SE. Lessons learned from the Pediatric Cardiomyopathy Registry (PCMR) Study Group. Cardiol Young. 2015; 25: 140-53.
3. Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG; National Australian Childhood Cardiomyopathy Study. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003; 348: 1639-46.
4.*** Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation. 2019; 140: e9-e68.
5.** Arbustini E, Narula N, Tavazzi L, Serio A, Grasso M, Favalli V, et al. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol. 2014;64:304-18. Erratum in: J Am Coll Cardiol. 2014; 64: 1186. Bonow, Robert D [Corrected to Bonow, Robert O].
6.** Landstrom AP, Kim JJ, Gelb BD, Helm BM, Kannankeril PJ, Semsarian C, et al. Genetic Testing for Heritable Cardiovascular Diseases in Pediatric Patients: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2021:HCG0000000000000086.
7. Alexander PM, Daubeney PE, Nugent AW, Lee KJ, Turner C, Colan SD, et al; National Australian Childhood Cardiomyopathy Study. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population-based study of childhood cardiomyopathy. Circulation. 2013; 128: 2039-46.
8. Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006; 114: 2671-8.
9. Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, et al. Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed During Childhood: Results from a National Population-Based Study. Circulation. 2018; 138: 29-36.
10.** Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020; 76: e159-e240.
11.** Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014; 35: 2733-79.
12. Norrish G, Ding T, Field E, Ziólkowska L, Olivotto I, Limongelli G, et al. Development of a Novel Risk Prediction Model for Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy (HCM Risk-Kids). JAMA Cardiol. 2019; 4: 918-27.
13. Miron A, Lafreniere-Roula M, Steve Fan CP, Armstrong KR, Dragulescu A, Papaz T, et al. A Validated Model for Sudden Cardiac Death Risk Prediction in Pediatric Hypertrophic Cardiomyopathy. Circulation. 2020; 142: 217-29.
14. Sabaté Rotés A, Huertas-Quiñones VM, Betrián P, Carretero J, Jiménez L, Girona J, et al. Miocardiopatía no compactada: características clínicas, evolutivas y pronósticas en edad pediátrica. Resultados de un estudio multicéntrico An Pediatr (Barc). 2012; 77: 360-5.
15. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010; 31: 806-14.
16.** Comité asesor de Vacunas. Capítulo 17. Vacunación en niños con enfermedades crónicas. En: Manual de vacunas en línea de la AEP. Acceso el 1 de octubre de 2021. Disponible en: https://vacunasaep.org/documentos/manual/cap-17.
17.** AAP Committee on Infectious Diseases Recommendations for Prevention and Control of Influenza in Children, 2021-2022. Pediatrics. 2021; 148: e2021053745.
18.** American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics. 2014; 134: 415-20. Erratum in: Pediatrics. 2014; 134: 1221.
19.*** Pelliccia A, Sharma S, Gati S, Bäck M, Börjesson M, Caselli S, et al; ESC Scientific Document Group. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J. 2021; 42: 17-96. Erratum in: Eur Heart J. 2021; 42: 548-9.
20. Centeno Malfaz F, Alcalde Martín C. Miocardiopatías. Pediatr Integral. 2016; XX(8): 548-58.
Bibliografía recomendada
– Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation. 2019; 140: e9-e68.
No es una guía clínica, sino un consenso de expertos que expone de manera muy exhaustiva: clasificación, alteraciones, causas, evaluación diagnóstica, estudio genético y pronóstico de los diferentes tipos de miocardiopatía en edad pediátrica.
– Pelliccia A, Sharma S, Gati S, Bäck M, Börjesson M, Caselli S, et al; ESC Scientific Document Group. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J. 2021; 42: 17-96. Erratum in: Eur Heart J. 2021; 42: 548-49.
Guías de práctica clínica de la Sociedad Europea de Cardiología, que define al individuo en riesgo durante el ejercicio y los tipos de ejercicio según el riesgo, con recomendaciones sobre el nivel permisible, en decisión conjunta con el paciente y su familia, para diferentes enfermedades cardiovasculares, incluyendo miocardiopatías y cardiopatías congénitas.
| Caso clínico |
|
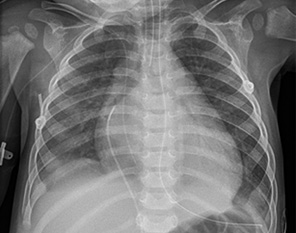
Recién nacido de 9 días de vida, que ingresa para observación por decaimiento general y rechazo de tomas, sin vómitos, diarrea, febrícula o contacto infeccioso conocido. Antecedentes personales Embarazo controlado, sin incidencias. Nacido a término con peso adecuado. Periodo neonatal inmediato normal, con alta a domicilio a las 48 h de vida. Antecedentes familiares Primer hijo de dos progenitores no consaguíneos, sanos. Sin historia de cardiopatías en menores de 50 años, arritmias o muertes súbitas. Evolución Al ingreso, llama la atención la presencia de una perfusión capilar enlentecida y de una taquicardia mantenida en 170 lpm, sin variabilidad apreciable (sinusal). El resto de constantes no está alterado, existe soplo sistólico a la exploración y el tono muscular es normal. Durante las primeras horas del ingreso, experimenta deterioro hemodinámico con hipotensión arterial y, secuencialmente, se inicia soporte ventilatorio mecánico e inotrópico. La radiografía de tórax muestra cardiomegalia y congestión pulmonar (Fig. 2), y en el ECG se observa taquicardia sinusal con alteraciones extensas de repolarización (T negativas en precordiales izquierdas) y presencia de ondas Q patológicas en cara lateral izquierda. La ecocardiografía confirma la presencia de una miocardiopatía dilatada con disfunción ventricular severa (fracción de eyección [FE] menor del 20%; normal mayor de 55%), insuficiencia mitral masiva y derrame pericárdico.
Figura 2. Radiografía de tórax al ingreso. El paciente termina precisando soporte circulatorio mecánico (ECMO) durante 7 días, tras los cuales, es posible su destete progresivo. Se evidencia recuperación parcial de la función ventricular, que permanece en niveles de disfunción leve a moderada al alta, con insuficiencia cardiaca controlada con diuréticos orales y captopril. El estudio etiológico incluye: hemocultivos, serologías y PCR de virus en sangre y respiratorios, estudio metabólico (ácidos orgánicos, carnitinas y acilcarnitina) y extracción de muestra genética. Se detecta finalmente en sangre, una PCR positiva para enterovirus; el resto de pruebas es negativo, incluyendo la detección de mutaciones patogénicas de un panel de genes relacionados con miocardiopatía dilatada. El paciente tiene actualmente 5 años y se encuentra sin signos de insuficiencia cardiaca aparentes, con muy buena evolución de medro y sin descompensaciones intercurrentes respiratorias. El ventrículo izquierdo solo está ligeramente dilatado, y persisten una disfunción moderada (FE 35%) y alteraciones en el ECG (presencia de ondas Q en cara inferior y lateral izquierda) (Fig. 3). Mantiene tratamiento con captopril y recibe vacunación antigripal anual. No está incluido en lista de trasplante cardiaco.
Figura 3. Alteraciones ECG persistentes.
|


 Syncope
Syncope