|
| Temas de FC |
I. Sánchez Pérez
Servicio de Cardiología Pediátrica y Cardiopatías Congénitas y Unidad de Arritmias, Hospital Universitario Ramón y Cajal. Madrid
| Resumen
Con el aumento de la monitorización prenatal y postnatal, se han hecho más evidentes una serie de arritmias en niños sanos previamente infradiagnosticadas(1). La mayoría son benignas. |
| Abstract
The increase in pre- and postnatal monitoring has determined that several previously underdiagnosed arrhythmias (most of them benign) have become evident in “apparently” healthy children(1). |
Palabras clave: Arritmias; Bradicardia; Bloqueo; Taquicardia supraventricular; Taquicardia ventricular
Key words: Arrhythmias; Bradycardia; Blockage; Supraventricular tachycardia; Ventricular tachycardia
Pediatr Integral 2016; XX (8): 527-538
Arritmias más frecuentes en la población infantojuvenil
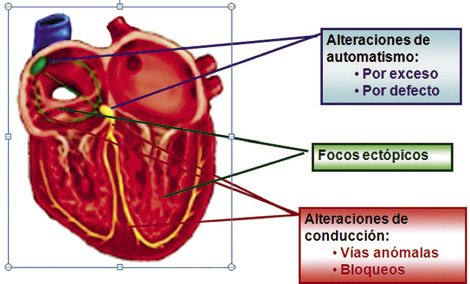
Figura 1. Mecanismos arritmias.
Ritmos lentos
Se denomina bradiarritmia a la alteración del ritmo cardiaco regular o irregular con frecuencias cardiacas inferiores a las correspondientes a la edad (Tabla I)(2,3), por alteración del automatismo sinusal o bloqueo de la conducción sinoauricular o auriculoventricular de mayor o menor grado.

Generalmente, son secundarias a otra patología subyacente o postquirúrgicas, aunque pueden ser de etiología congénita, siendo la más frecuente el bloqueo auriculoventricular completo congénito. Se diagnostican generalmente mediante electrocardiografía y requieren tratamiento cuando son sintomáticas o repercuten hemodinámicamente.
Depresión de la función del nodo sinusal
• Bradicardia sinusal: en la mayoría de las ocasiones es fisiológica sin repercusión clínica y no requiere tratamiento. Puede deberse a: hipertono vagal, hipertensión intracraneal, apnea obstructiva del sueño, fármacos, hipotermia, hipoxia o infecciones cardiacas o postoperatorio de determinadas cardiopatías (comunicación interauricular tipo seno venoso, defecto de septo auriculoventricular, cirugía de Mustard o Senning o Fontán). Cuando se sospecha una causa congénita y está presente al nacimiento, puede ser debida a hipotiroidismo o disfunción sinusal, generalmente ligada a determinadas cardiopatías congénitas con dilatación auricular derecha (Ebstein, heterotaxia [poliesplenia], ventrículo único…). La mayoría de las veces es asintomática, aunque puede manifestarse con: astenia, cansancio en relación con el ejercicio, mareo o síncope. El diagnóstico es electrocardiográfico, puede requerirse un Holter para evaluar las frecuencias cardiacas inferiores, ergometría para valorar la respuesta cronotrópica o test farmacológicos bradicardizantes o taquicardizantes (atropina) o bloqueo autonómico con propanolol y atropina para evaluar la frecuencia cardiaca intrínseca. Mucho menos frecuente es que se requiera un estudio electrofisiológico para su diagnóstico. Generalmente, tienen buen pronóstico y solo en casos sintomáticos con repercusión hemodinámica, es necesario el implante de un marcapasos para estimulación permanente, siempre y cuando no exista una causa externa subyacente tratable. Según las últimas guías publicadas, la recomendación de estimulación cardiaca permanente se establece en aquellos niños con frecuencia ventricular menor de la que le corresponde por edad y sintomáticos (clase I, nivel de evidencia B). También está recomendado en pacientes con cardiopatía congénita y bradicardia, como prevención de taquicardia por reentrada auricular y hemodinámicamente deteriorados por la bradicardia, con frecuencias inferiores a 40 lpm o pausas ventriculares mayores de 3 segundos (clase IIa, nivel de evidencia C)(4).
• Arritmia sinusal: al igual que la bradicardia puede ser fisiológica, en los niños la más frecuente es la respiratoria. Cuando no es respiratoria, puede asociarse a determinadas cardiopatías congénitas o correcciones quirúrgicas que cursan con dilatación auricular derecha. En el electrocardiograma se objetiva una variabilidad PP mayor de 0,12 s con PR normal. Generalmente, su pronóstico es bueno y no necesitan tratamiento.
• Paro sinusal: es clínicamente significativa y se considera patológica cuando produce pausas superiores a 3 segundos; en la mayoría de las ocasiones obedece a isquemia aguda, accidente cerebrovascular o intoxicación digitálica. Es raro que sea de causa congénita, a veces, se asocia a la corrección tipo Mustard de la transposición de grandes arterias. El tratamiento, cuando es sintomática, es el implante de marcapasos.
Bloqueo sinoauricular
En la mayoría de las ocasiones es de causa degenerativa, en el niño puede observarse en el contexto de una infección miocárdica connatal. Esta entidad es difícil de valorar electrocardiográficamente, ya que la actividad eléctrica del nodo sinusal no tiene expresión en el electro de superficie y, a menudo, se confunde con alguna de las anteriores entidades. Se distingue del paro sinusal en que la pausa siempre es un múltiplo del intervalo PP. Puede ser de 3 tipos:
1. Primer grado: enlentecimiento de la conducción del impulso del nodo sinusal a la aurícula, pero siempre se conduce. No expresión electrocardiográfica.
2. Segundo grado: algunos impulsos se bloquean y no llegan a la aurícula.
- Tipo I (Wenckebach): acortamiento del intervalo PP progresivo hasta que uno se alarga bruscamente.
- Tipo II: no acortamiento PP, pausa súbita que es múltiplo de PP.
3. Tercer grado: ningún impulso se transmite. Ritmo de escape auricular, nodal…
Generalmente, no requiere tratamiento y solo en bradicardias sintomáticas, se requerirá implante de marcapasos.
Bloqueo auriculoventricular
El intervalo PR es la expresión de la activación auricular, nodo AV, ramas del haz de His y fibras de Purkinge. Cualquier alteración a estos niveles se traducirá en un trastorno de la conducción auriculoventricular de mayor o menor grado.
Al igual que los bloqueos sinoauriculares, pueden ser:
1. De primer grado: todos los impulsos son conducidos, aunque más lentamente. Esta entidad puede encontrarse en determinadas cardiopatías congénitas, como: Ebstein, drenaje venoso anómalo, comunicación interauricular o transposición de grandes arterias. También en el hipotiroidismo y determinadas alteraciones iónicas, como la hipopotasemia. Puede aparecer en el postoperatorio de cirugías de aurícula o peri nodo auriculoventricular. Generalmente, no tienen repercusión clínica y no necesitan tratamiento, salvo corregir la causa subyacente.
2. De 2º grado tipo Mobitz I (Wenckebach): el intervalo PR se alarga hasta que una P no conduce. Puede encontrarse en sujetos sanos con hipertono vagal y en enfermedades inflamatorias miocárdicas o degenerativas. No suele tener repercusión clínica ni progresar a bloqueo de tercer grado y rara vez requieren tratamiento.
3. De 2º grado tipo Mobitz II: no existe alargamiento PR, sino que súbitamente una onda P no conduce. Suele encontrarse en enfermedades degenerativas, infecciosas (chagas), isquemia y postoperatorio de cirugía cardiaca. Pueden dar clínica de mareo o síncope y evolucionar a bloqueo de tercer grado. En ocasiones, requieren el implante de un marcapasos: bloqueos de segundo grado avanzado con bradicardia sintomática o disfunción ventricular y en postoperatorio no resueltos tras 7 días (clase I, nivel de evidencia B) (4).
4. Bloqueo auriculoventricular de tercer grado: ausencia de conducción auriculoventricular. En el electrocardiograma de superficie, se observa la presencia de un ritmo auricular, en general más rápido que el ventricular, por lo que hay más ondas P que QRS, con intervalos R-R constantes y PR variables. Existen ritmos de escape nodal o ventricular. Puede encontrarse en enfermedades degenerativas, isquémicas o postoperatorias de cirugía cardiaca. También existe una forma familiar asociada al cromosoma 3p21 (enfermedad de Lenegre) y también en el cromosoma 19q13. La causa más frecuente es la primaria. En la población infantil, la causa más frecuente es la congénita, que puede aparecer de forma aislada o familiar, presentando una incidencia variable entre 1/10.000 y 1/20.000 nacidos vivos. Puede presentarse de forma aislada o asociarse a malformación cardiaca (entre 25-50%). Las más frecuentes son: transposición de las grandes arterias, síndromes poliesplénicos, defecto del septo auriculoventricular completo y ventrículo único. Puede deberse a dos causas: malformación del tejido de conducción cardiaca a ese nivel o destrucción del mismo por distintos agentes como infecciones (miocarditis, endocarditis), hemorragias, presencia de enfermedades autoinmunes maternas y/o colagenosis, como: lupus eritematoso sistémico (LES), síndrome de Sjögren, artritis reumatoide o dermatomiositis. Estas son las más frecuentes, se cree que causan el 60-70% de todos los bloqueos cardiacos congénitos. Con la ecocardiografía puede observarse bradicardia fetal extrema y con el modo M puede verificarse la disociación auriculoventricular(15). En algunos casos, se tolera bien y no da síntomas hasta el nacimiento. En otros casos, puede producir disfunción ventricular severa con insuficiencia cardiaca, cardiomegalia, hidrops, ascitis, derrame pericárdico y sufrimiento fetal, que puede llegar a ocasionar la muerte intraútero. En lactantes mayores, pueden presentar terrores nocturnos, cansancio e irritabilidad. También se observan niños de mayor edad, que están asintomáticos. Algunos de ellos pueden presentar mareos con o sin síncope llamados episodios de Stokes-Adams. El tratamiento de urgencia, cuando existe disfunción ventricular o bradicardia severa, es la perfusión intravenosa de isoproterenol y el implante de marcapasos temporal. El tratamiento definitivo es el implante de un marcapasos, indicado en: bloqueos auriculoventriculares de tercer grado con bradicardia sintomática o disfunción ventricular, ritmo de escape QRS ancho o ectopia ventricular; en postoperatorio no resueltos tras 7 días (clase I, nivel de evidencia B); en lactantes con bloqueo AV de 3 grado congénito con frecuencia ventricular media inferior a 55 lpm y 70 lpm cuando se asocia a cardiopatía congénita (clase I, nivel de evidencia C); y en niños mayores de 1 año, cuando la frecuencia ventricular media es inferior a 50 lpm, pausas ventriculares abruptas (mayores de 2 a 3 ciclos) o síntomas o síncopes de causa no aclarada postcirugía con bloqueo auriculoventricular de 3 grado transitorio y bloqueo fascicular residual (clase IIa, nivel de evidencia B)(4).
El manejo agudo de las bradiarritmias será común para todos ellos y quedaría resumido en el siguiente algoritmo (Algoritmo 2)(5). En cuanto al tratamiento crónico, sería el seguimiento y observación en los casos en los que no esté indicado el implante de un marcapasos permanente descrito en cada una de las bradiarritmias previamente.
Ritmos rápidos
Supraventriculares (TSV)
Se define como ritmo cardíaco anormalmente rápido, el que se origina por encima del haz de His, a menudo (pero no siempre), con un complejo QRS estrecho. Las dos formas más comunes de TSV en los niños, son reentrada auriculoventricular (TRAV)(73%), incluyendo el síndrome de Wolff-Parkinson-White (WPW), y la reentrada nodal (TRNAV)(13%). En el adolescente, aumenta sensiblemente el porcentaje de TRNAV y disminuye TRAV (Tabla II).

La prevalencia de la TSV no está bien definida, pero se estima que entre uno de 250 y 25.000 niños, y es la alteración del ritmo más común en niños. La prevalencia es mucho mayor en los niños gravemente enfermos con cardiopatía congénita (Ebstein) o adquirida, tratados en una unidad de cuidados intensivos pediátrica(6).
Del nodo sinusal
Los ritmos originados en el nodo sinusal tienen dos características: siempre hay una onda P delante de cada complejo QRS, con un intervalo PR regular. El eje de la onda P está entre 0º y +90º.
1. Taquicardia sinusal: frecuencia mayor de 166 lpm en la primera semana de vida y de 176 lpm en el resto del 1º mes de vida, 125 lpm a los 2 años, 115 lpm a los 4 años y 100 lpm en mayores de 6 años en reposo. Casi siempre, debida a alteración extracardiaca y raramente sintomáticas, se tratará la causa de la taquicardia (anemia, fiebre, etc.).
2. Arritmia sinusal o respiratoria: benigna. La frecuencia cardiaca aumenta durante la inspiración y disminuye con la espiración. Esta arritmia no tiene ningún significado hemodinámico, ya que se trata de un fenómeno normal debido a las variaciones de la frecuencia con las fases de la respiración.
Ritmos originados en las aurículas
Tienen dos características: las ondas P son anormales en número (número de ondas P ? al de QRS) o morfología (eje de P anormal). Complejos QRS normales, ocasionalmente anchos por aberrancias.
1. Extrasístoles auriculares: el QRS ocurre prematuramente precedido de una onda P anormal. No hay pausa compensadora (es incompleta). Son las arritmias más frecuentes en recién nacidos sanos. No tienen significado hemodinámico y no precisan tratamiento.
2. Marcapasos auricular “migratorio”: consiste en cambios graduales en las morfologías de las ondas P y los intervalos R-R. El complejo QRS es normal. Se ve en niños sanos, no tiene significación clínica y no precisa tratamiento.
3. Taquicardia auricular: las taquicardias ectópicas son raras. Su mecanismo no es del todo conocido, un foco único o múltiple a nivel auricular o microrreentradas auriculares podrían ser sus causantes. La frecuencia ventricular (QRS) varía, pues hay ondas P bloqueadas. En neonatos y lactantes, existe un tipo denominado incesante que característicamente, mantiene en ritmo auricular ectópico el 90% del tiempo, rebelde al tratamiento antiarrítmico habitual con digital, b-bloqueantes o amiodarona, y tampoco respondedora a la cardioversión y que pueden desaparecer espontáneamente hasta un 90% antes de los 6 meses. Cuando no es así y existe repercusión hemodinámica (taquimiocardiopatía) el tratamiento será la ablación(7).
4. Flutter auricular: se caracteriza por una frecuencia auricular rápida (>300/min) y ondas P características en forma de “dientes de sierra” (ondas F). El ventrículo responde con bloqueo de diferentes grados 2:1,3:1, 4:1, y QRS normal. Es una de las arritmias más comunes en el periodo neonatal, se debe a una reentrada auricular, generalmente bien tolerada por el neonato. Su tratamiento será la cardioversión síncrona.
5. Fibrilación auricular: se caracteriza por una frecuencia auricular extremadamente rápida (350-600/min) con “ondas F” y una respuesta ventricular irregularmente irregular con QRS normal. La fibrilación sugiere una patología subyacente significativa. La no sincronía entre la aurícula y el ventrículo dará lugar a una disminución del gasto cardíaco. En casos de inestabilidad, el tratamiento será la cardioversión síncrona y, si está estable, digoxina asociada o no a procainamida.
Ritmos que implican al nodo AV
La onda P puede estar ausente u ondas P (-) pueden seguir al QRS. El QRS es normal en morfología y duración. Pueden ser:
1. Extrasístoles de la unión: un QRS ocurre prematuramente con onda P ausente o (-) retrógrada (detrás del QRS). La “pausa compensadora” puede ser completa o incompleta. Se ve en niños sanos. No tiene significación clínica.
2. Ritmo nodal acelerado: en presencia de una frecuencia sinusal normal y conducción AV normal, si el nodo AV (región nodo AV –Haz de His) presenta un “automatismo” aumentado, toma la función de marcapasos. Las ondas P están ausentes o son (-) retrógradas. En el neonato y lactante sano, la causa más frecuente es la miocarditis. No suele tener significación clínica ni precisar tratamiento.
3. Taquicardia con implicación nodal: la frecuencia ventricular varía de 220-280 lpm en neonatos a 180-240 lpm en niños más mayores y adolescentes. El QRS es generalmente normal, pero pueden ocurrir “aberrancias”. Podemos clasificarlas en:
a. Ectópicas (por automatismo), se caracterizan por tener una frecuencia ventricular mayor o igual que la auricular, fenómeno de “calentamiento” en su inicio y “enfriamiento” en su cese. Al igual que la automática auricular, también puede ser incesante y ser rebelde al tratamiento farmacológico requiriendo ablación con catéter.
b. Reentrada (“reciprocantes”), “paroxísticas”, el nodo AV participa en el circuito de reentrada y son las taquicardias más frecuentes en la edad pediátrica, menos frecuentes en el neonato. Se producen por la falta de regresión de conexiones anómalas a nivel auriculoventricular (más frecuente posteroseptal y lateral izquierda) o intranodales (vía lenta en septo interatrial posterior, junto a la desembocadura del seno coronario; más propias del adolescente o adulto) presentes durante la vida fetal. Característicamente, se inician bruscamente precedidas de un extrasístole y su cese también es brusco. Durante la taquicardia, podremos observar una onda p retrógrada tras QRS estrecho (conducción ortodrómica) o ancho en el menor de los casos <5% (antidrómica) en las TRAV, y ausente o inmediatamente posterior al QRS en las TRNAV. Clínicamente, se suele manifestar como: palpitaciones, dolor torácico, fatiga y mareo (que es debido a la hipotensión asociada con la frecuencia cardíaca rápida). La mayoría de los niños toleran bien los episodios de taquicardia. Sin embargo, los episodios prolongados por la difícil identificación en niños más pequeños, pueden precipitar la insuficiencia cardiaca: tos, palidez, irritabilidad, mala alimentación, cianosis e inquietud (19-50% de los neonatos con SVT de 24 a 36 horas y más de 48 h, respectivamente)(8).
El síncope puede ser una presentación poco común de TSV. Síncope asociado con el síndrome de WPW y RR preexcitado <220 ms en niños es una señal de advertencia grave, y puede ser señal de la fibrilación auricular (FA) con conducción AV rápida a lo largo de la vía accesoria. Se encuentran en mayor riesgo de desarrollar fibrilación auricular y muerte súbita cardíaca, por lo que serían candidatos a estudio electrofisiológico y posible ablación(9,10).
Un tipo de TRAV es la permanente reciprocante de la unión (Coumel), en la que a diferencia del resto de TRAV, la conducción retrógrada por la vía es lenta al igual que la anterógrada por el nodo AV, creando un ciclo estable de reentrada, comportándose de manera incesante y pudiendo desencadenar taquimiocardiopatía e insuficiencia cardiaca(11).
En cuanto a la historia natural de TAV, está en parte relacionada con la edad en la presentación inicial. Los bebés que se presentan con TSV en el período neonatal tienen una probabilidad de 30 a 70% de ser asintomáticos y no requieren tratamiento al año de edad, aunque algunos pueden tener recurrencia posterior en una media de edad de ocho años. En contraste, TSV persiste en el 78% de los pacientes que se presentaron después de los cinco años de edad. Se especula qué cambios en el tono autonómico durante la infancia puede dar lugar a este patrón de desaparición y reaparición, alterando la vía accesoria y la conducción y refractariedad del nodo AV(12).
El tratamiento inicial es común para todas las TSV y queda reflejado en el siguiente algoritmo (Algoritmo 3)(5).
Con lo que respecta al tratamiento crónico, no existe unanimidad; por lo general, se tomará una actitud expectante en niños mayores de 1 año y <5 años con un único episodio, sin miocardiopatía ni inestabilidad hemodinámica tras observación durante 24 horas. En lactantes, por falta de reconocimiento de la arritmia, se preferirá el tratamiento farmacológico durante el primer año de vida, aunque los estudios realizados hablan de similar efectividad del tratamiento crónico con digital y la actitud expectante con maniobras vagales durante la crisis. Se tratarán farmacológicamente a largo plazo los niños con episodios frecuentes, recurrentes muy sintomáticos y, generalmente, los fármacos elegidos serán flecainida y b-bloqueantes(4,13-15).
En cuanto a la ablación con radiofrecuencia, es una técnica con alta tasa de éxito (90%), pero no exenta de complicaciones, tales como: bloqueo auriculoventricular, perforación, lesión del plexo braquial, embolia o neumotórax, mayores cuanto menor es la edad del paciente (<15 kg), tal como queda descrito en la tabla III, por ello es recomendable ceñirse a las indicaciones actualmente establecidas que son las siguientes(4):

• Clase I:
- Síndrome de WPW tras un episodio abortado de muerte súbita.
- Síndrome de WPW asociado con síncope e intervalo RR corto durante la fibrilación auricular (RR preexcitado < 250 ms) o con período refractario efectivo de la vía accesoria corto < 250 ms durante estimulación auricular.
- TSV crónica o recurrente asociada con disfunción ventricular.
• Clase IIa:
- TSV recurrente y/o sintomática asociada con disfunción ventricular.
- Pacientes con cardiopatía congénita, en los que la cirugía cardíaca puede imposibilitar o dificultar el acceso vascular o cardíaco al sustrato arrítmico.
- TSV incesante crónica (>6-12 meses tras un evento inicial) con función ventricular normal.
- Taquicardia por reentrada auricular crónica o con frecuentes recurrencias.
- Palpitaciones con TSV sostenida inducida durante el estudio electrofisiológico.
• Clase IIb:
- Preexcitación asintomática (patrón de WPW en el ECG), edad > 5 años, sin taquicardia reconocida, y cuando el riesgo/beneficio del procedimiento y de la arritmia ha sido claramente explicitado.
- TSV en niños > 5 años, cuando el tratamiento antiarrítmico ha controlado eficazmente la taquicardia.
- TSV en niños < 5 años de edad, refractaria al tratamiento antiarrítmico, incluido el sotalol y la amiodarona.
- Taquicardia por reentrada auricular, con 1-3 episodios por año, que requieren intervención médica.
- Ablación del nodo AV e implantación de marcapasos como alternativa al tratamiento médico en pacientes con taquicardia por reentrada auricular refractaria.
• Clase III
- Síndrome de WPW asintomático en < 5 años.
- TSV controlada médicamente en niños < 5 años.
- Episodios de TSV no sostenida que no requieren otro tipo de terapia y/o son mínimamente sintomáticos.
Actualmente, se disponen de nuevas técnicas como mapeo de activación y crioablación, que ya se han demostrado más seguras minimizando las complicaciones secundarias al procedimiento, útiles en niños menores de 20 kg y arritmias originadas en regiones próximas al nodo auriculoventricular disminuyendo el riesgo de bloqueo, aunque con una mayor tasa de recidivas(17,18).
Ventriculares
Se trata de ritmos originados en el miocardio ventricular o células de Purkinje por debajo de la bifurcación del haz de His. Electrocardiográficamente, se caracterizan por QRS anchos (>80 ms) y “abigarrados”, con morfología de bloqueo de rama con ondas T en dirección opuesta. Los QRS están disociados con respecto a las ondas P.
Ritmos originados en los ventrículos
1. Extrasístoles ventriculares: consisten en complejos QRS que se adelantan. Tienen la onda T en sentido opuesto al QRS. Tienen, por lo general, “pausa compensadora” completa. Se pueden producir “latidos de fusión” (complejos QRS intermedios entre el QRS sinusal y el extrasístole, generalmente precedidos de onda P y con un PR corto). Son frecuentes en niños sanos. No precisan estudios adicionales.
Pueden clasificarse de diversas formas:
• Por la interrelación de los extrasístoles:
- “Bigeminismo”: cada complejo QRS anormal alterna con un QRS normal regularmente.
- “Trigeminismo”: cada QRS anormal (extrasístole) es seguido de 2 QRS normales.
- “Parejas”: 2 QRS anormales (extrasístoles) seguidos.
- “Tripletes extrasístoles seguidos”: 3 o más extrasístoles se denomina de forma arbitraria como taquicardia ventricular.
• Según la “similitud” de los QRS, los extrasístoles se dividen en:
- “Monomórficos” o unifocales: los QRS tienen la misma morfología en la misma derivación.
- “Polimórficos” o multifocales: los QRS son de morfología diferente.
Los extrasístoles ventriculares son frecuentes en niños sanos, también pueden verse en: miocarditis, falsos tendones ventriculares, lesiones miocárdicas, tumores, displasias, etc.
Los extrasístoles aislados son benignos, sobre todo si son monomórficos y disminuyen con el ejercicio. Son signos de “mal pronóstico”: si se asocian a cardiopatía, si hay antecedentes familiares de muerte súbita, si aumentan con el ejercicio, si son “multifocales”, si hay rachas con síntomas, si hay episodios frecuentes de taquicardia ventricular paroxística o si son incesantes. Si son aislados y con características de benignidad, no precisan de más estudios. Extrasístoles monomórficos frecuentes, incluyendo bigeminismo y trigeminismo, no necesitarán tratamiento. Sí les realizaremos ECG para valorar el QTc y posibles alteraciones de ST-T. Ecocardiografía para descartar anomalías estructurales, miocarditis, etc. Prueba de esfuerzo, pues puede inducir o exacerbar la arritmia, lo cual orientaría sobre una posible cardiopatía subyacente, y puede revelar un QTc largo durante el período de recuperación. Los niños con extrasístoles polimórficos o multifocales, además de las pruebas antes mencionadas, precisan de un registro electrocardiográfico de 24 horas (Holter), para detectar la severidad y la extensión de la arritmia ventricular. Tienen que ser evaluados periódicamente.
2. Taquicardia ventricular (TV): se define como 3 o más extrasístoles ventriculares a una frecuencia de 120-200/min. Los QRS son anchos con ondas T opuestas al QRS. Los QRS pueden ser monomórficos o pueden variar de forma fortuita (polimórficos). La torsada de punta es una forma de TV polimórfica, caracterizada por una TV paroxística, durante la cual hay cambios progresivos en la amplitud y polaridad del QRS, separados por un complejo de transición estrecho. Pueden ocurrir en el síndrome QT largo. Las TV, a veces, son difíciles de distinguir de una TSV con aberrancia. Los siguientes hallazgos son de ayuda en diferenciar la conducción ventricular aberrante de los extrasístoles ventriculares: a) un patrón rsR´en V1 recuerda al bloqueo de rama derecha y sugiere aberrancia. En un extrasístole, la morfología es rara y no recuerda a la forma clásica de bloqueo de rama derecha ni izquierda. b) QRS anchos aislados siguiendo a ondas P con intervalos R-R regulares, sugieren aberrancia. c) la presencia de “latidos de fusión” es un signo de extrasistolia. Una taquicardia con un QRS ancho en un niño deberá ser considerada y tratada como una taquicardia ventricular, mientras no se demuestre lo contrario.
Las causas de las TV generalmente indican una patología miocárdica seria. En niños, se ven con mayor frecuencia:
1. Asociadas a cardiopatía congénita reparada o no y dentro de ellas en tetralogía de Fallot, sobre todo si existen lesiones residuales (hasta un 12%)(19), obstrucciones del tracto de salida del ventrículo izquierdo y disfunción ventricular, cuando estas cardiopatías tienen un QRS mayor de 180 ms y en edades más avanzadas. Se ha hablado de la utilidad de estudios con estimulación ventricular en el postoperatorio de estos pacientes para identificar los de mayor riesgo de desarrollar arritmias ventriculares y muerte súbita. En este tipo de pacientes, se tratará la arritmia, cuando la TV es sostenida y/o sintomática. En una primera instancia, se corregirán los defectos hemodinámicos residuales si los hubiera. No existe un consenso claro en cuanto al mejor tratamiento, pero sí parece haberse visto la falta de respuesta al tratamiento farmacológico (amiodarona, procainamida, sotalol)(20) y la alta tasa de recidivas y ausencia de reducción de muerte súbita postablación(21) (indicada en TV sostenida monomorfa estable); por ello, cada vez se amplían más las indicaciones de implante de desfibrilador (DAI) en este tipo de pacientes que, actualmente, según las guías son: pacientes resucitados de muerte súbita por TV de causa no reversible (clase I evidencia B), TV sostenida tras ablación no exitosa (clase I evidencia C), síncope no explicado asociado a disfunción ventricular (clase IIa evidencia C)(4).
2. Trastornos genéticos. Se pueden clasificar como canalopatías (trastornos que afectan el movimiento de iones) o trastornos de miocardiopatía.
• Síndrome de QT largo (SQTL): es un trastorno de la repolarización del miocardio que se caracteriza por una prolongación del intervalo QT y anomalías de la onda T en el ECG, síncope y muerte súbita cardíaca. La mayoría de los pacientes tienen una pérdida de función de un canal de potasio (p. ej., KCNQ1 y KCNH2) o una ganancia de función de un sodio canal (p. ej., SCN5A) (Tabla IV).

Se han descrito 2 fenotipos de SQTL. El más común de forma autosómica dominante, el síndrome de Romano-Ward, tiene un fenotipo puramente cardiaco. La forma autosómica recesiva, el síndrome de Jervell-Lange-Nielsen, se asocia con SQTL y sordera neurosensorial, y tiene un curso clínico más maligno. SQTL se asocia con un mayor riesgo de arritmias potencialmente mortales, conocidas como torsades de pointes. También existen numerosas causas de SQTL adquirido (Tabla IV). Los pacientes con SQTL se presentan con síncope, convulsiones o paro cardiaco. Una historia familiar de muerte súbita o convulsiones debe hacer sospechar esta entidad. La tasa de mortalidad se ha reducido con la identificación más temprana de los pacientes afectados y el tratamiento, incluida la terapia con ß-bloqueantes, la restricción de los deportes, la evitación de los medicamentos que prolongan el intervalo QTc, y/o implante de DAI en sujetos de alto riesgo: en muerte súbita resucitada concomitante con ß-bloqueantes (clase I evidencia A), síncope o TV a pesar de tratamiento con ß-bloqueantes (clase IIa evidencia B)(4,22).
• El síndrome de Brugada: canalopatía poco común, se asocia con los hallazgos ECG característicos de elevación del segmento ST en las derivaciones V1 a V3 y bloqueo de rama derecha. Se hereda en un patrón autosómico dominante con expresividad variable. Las mutaciones genéticas de SCN5A, que codifica para la subunidad de un canal de sodio cardíaco, dando lugar a una pérdida de la función del canal de sodio, se ha encontrado en 10 a 30%(23).
Los pacientes afectados presentan arritmias ventriculares potencialmente mortales. La muerte súbita cardíaca puede ser el primer acontecimiento clínico, ocurre en hasta un tercio de los pacientes. La muerte generalmente ocurre durante el sueño y en horas de la madrugada. Una historia familiar de muerte súbita temprana debe hacer sospecharla. El tratamiento con DAI estará indicado en muerte súbita resucitada (clase I evidencia C) y es razonable con ECG espontáneo de Brugada y síncope o TV asociada (clase IIa evidencia C)(4,22).
• Taquicardia ventricular polimórfica catecolaminérgica (CPVT): están en riesgo de TV, fibrilación ventricular y muerte súbita, especialmente en relación con el estrés o el ejercicio. En reposo, el ECG suele demostrar frecuentes extrasístoles ventriculares y TV polimórfica no sostenida, se hereda en un patrón autosómico dominante o autosómico recesivo y también ocurre en casos esporádicos como una mutación de novo. Diversas mutaciones de los loci de genes que codifican proteínas implicadas en la regulación del calcio en el retículo sarcoplásmico se han identificado en pacientes con CPVT. Estos incluyen mutaciones del gen RyR2 (que codifica el receptor de rianodina) y del gen de la calsecuestrina 2 (que codifica una proteína principal del reservorio de calcio dentro del retículo sarcoplásmico). Mayor prevalencia durante la infancia por mayor intensidad de ejercicio físico. Aproximadamente, el 30% de los pacientes tienen antecedentes familiares de muerte súbita, a menudo en la adolescencia. El pronóstico es malo, incluso en pacientes tratados con ß-bloqueantes o DAI, cuyas indicaciones son similares al SQTL(4,22).
• Miocardiopatía hipertrófica: la miocardiopatía hipertrófica es la causa más frecuente de muerte súbita en personas jóvenes en los Estados Unidos. Es una enfermedad genética del sarcómero cardíaco que es causada por mutaciones en uno de los más de 50 genes que codifican la mayoría de las proteínas contráctiles del miocardio, que condicionan inestabilidad eléctrica y TV. Estará indicado el tratamiento con DAI en TV sostenida o FV (clase I evidencia B)(4,22).
• Miocardiopatía arritmogénica del ventrículo derecho (MAVD): es un trastorno del ventrículo derecho se caracteriza por el reemplazo fibroadiposo del miocardio, con formación de aneurismas y anormalidades del movimiento de la pared (evidenciado mediante resonancia magnética); en algunos casos, también del tabique ventricular. Representa el 3 a 5% de las muertes súbitas sin explicación en adolescentes y adultos jóvenes. Ambas formas, autosómica dominante y recesiva, de la MAVD se han reportado con mutaciones identificadas en varios genes de la proteína desmosómica. La MAVD autosómica recesiva es menos común y puede ser parte de un síndrome llamado enfermedad de Naxos, que también incluye hiperqueratosis de las palmas y plantas, y alteraciones del pelo. Los pacientes afectados presentan mareos, palpitaciones y síncope debido a TV. La TV tiene un patrón de bloqueo completo de rama izquierda. Estará indicado el implante de DAI en muerte súbita resucitada asociada a TV o FV (clase I evidencia B) o en enfermedad extensa con disfunción ventricular, antecedentes familiares o síncope sin exclusión de TV o FV, como causa (clase IIa evidencia C)(4,22).
3. Enfermedad cardíaca adquirida: menos frecuentes, algunas de ellas son: enfermedad cardiaca coronaria (Kawasaki), miocarditis y enfermedad de Chagas (causada por el parásito Trypanosoma cruzi, que se encuentra principalmente en América Central y del Sur; aunque es raro en los niños, pueden desarrollar una miocardiopatía dilatada y se asocia con bloqueo cardíaco y TV durante la fase crónica de la infección) alteraciones metabólicas (hipopotasemia, hipomagnesemia) la anorexia y enfermedad intracraneal (accidente cerebrovascular).
4. Causa idiopática:
• Ritmo idioventricular acelerado (RIVA): TV monomórfica relativamente lenta, 120-200 lpm, que se observa con más frecuencia en los recién nacidos. Se distingue de otras formas más malignas de la TV, debido a su ritmo más lento y a la ausencia de síntomas. Desaparece con el ejercicio. Generalmente, no necesita tratamiento(24).
• Taquicardia idiopática del tracto de salida VD (TVTSVD): condición benigna que se resuelve espontáneamente. Esta alteración del ritmo se cree que es una actividad mediada por el AMPc activa que surge del tracto de salida del ventrículo derecho. Frecuencia lenta de 140 a 190 latidos por minuto, con un patrón de QRS de bloqueo de rama izquierdo y eje inferior. La mayoría de los niños con TVTSVD son asintomáticos y se presentan como un hallazgo incidental. El tratamiento de la TVTSVD está indicado en pacientes sintomáticos y en los pacientes que desarrollan cardiomiopatía inducida por taquicardia. La terapia incluye fármacos antiarrítmicos y la ablación con catéter que ha demostrado ser muy exitosa. El pronóstico es excelente(25).
• TV pueden originarse en el tracto de salida del ventrículo izquierdo (TSVI), y es similar a la taquicardia del TVTSVD en sus manifestaciones clínicas, pronóstico y manejo. El ECG muestra TV monomórfica con un bloqueo de rama derecha y eje inferior.
• Taquicardia ventricular izquierda o taquicardia fascicular Belhassen: es una TV benigna, el ECG muestra un bloqueo de rama derecha con eje superior o izquierdo. Se origina en el fascículo posterior izquierdo y se produce en ausencia de cardiopatía estructural, aunque puede estar relacionada con falsos tendones de ventrículo izquierdo.
El mecanismo de la TV se cree que es la reentrada, ya que se inicia con la estimulación auricular y ventricular, es sensible al verapamilo.
5. Fibrilación ventricular: son QRS irregulares de diverso tamaño y configuración. La frecuencia es rápida e irregular. La situación hemodinámica es grave, fatal. Precisa de maniobras de resucitación inmediatas, incluyendo la “desfibrilación” (4 jul/kg).
El tratamiento agudo de todas las taquicardias ventriculares y fibrilación ventricular queda reflejado en el algoritmo 5; en cuanto al tratamiento crónico, ya ha quedado reflejado en cada apartado(5).
Bibliografía
1. Geggel RL and Flyer DC. History, growth, nutrition, physical examination, and routine laboratory studies. In Nadas’ Pediatric Cardiology, 2nd ed, Keane JF, Lock JE, Flyer DC (Eds), Saunders Elsevier, Philadelphia; 2006: p. 129.
2. Michaelson M, Engle MA. Congenital complete heart block: An international study of the natural history. In: Cardiovascular Clinics, Brest AN, Engle MA (Eds), FA Davis, Philadelphia; 1972: p. 85.
3. Kugler JD. Sinus node dysfunction. In: Pediatric Arrhythmias: Electrophysiology and Pacing, Gillette PC, Garson AG Jr (Eds), WB Saunders, Philadelphia; 1990: p. 250.
4. Brugada J, Blom N, Sarquella-Brugada G, et al. Pharmacological and non-pharmacological therapy for arrhythmias in the pediatric population: EHRA and AEPC-Arrhythmia Working Group joint consensus statement. Europace. 2013; 15: 1337-82.
5. Pediatric Advanced Life Support Provider Manual. Ralston, M, et al (Eds), American Heart Association, Subcommittee on Pediatric Resuscitation, Dallas; 2006: p. 116.
6. Hoffman TM, Wernovsky G, Wieand TS, et al. The incidence of arrhythmias in a pediatric cardiac intensive care unit. Pediatr Cardiol. 2002; 23: 598.
7. Blomstrom-Lundqvist C, Scheinman MM, Aliot EM, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias–executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Supraventricular Arrhythmias). Circulation. 2003; 108: 1.
8. Gilljam T, Jaeggi E, Gow RM. Neonatal supraventricular tachycardia: outcomes over a 27-year period at a single institution. Acta Paediatrica. 2008; 97: 1035-9.
9. Paul T, Guccione P, Garson A Jr. Relation of syncope in young patients with Wolff-Parkinson-White syndrome to rapid ventricular response during atrial fibrillation. Am J Cardiol. 1990; 65: 318.
10. Harahsheh A, Du W, Singh H, et al. Risk factors for atrioventricular tachycardia degenerating to atrial flutter/fibrillation in the young with Wolff-Parkinson-White Pacing Clin Electrophysiol. 2008; 31: 1307-12.
11. Cruz FE, Cheriex EC, Smeets JL, et al. Reversibility of tachycardia-induced cardiomyopathy after cure of incessant supraventricular tachycardia. J Am Coll Cardiol. 1990; 16: 739.
12. Ko JK, Deal BJ, Strasburger JF, et al. Supraventricular tachycardia mechanisms and their age distribution in pediatric patients. Am J Cardiol. 1992; 69: 1028.
13. Kugler JD, Danford DA. Management of infants, children, and adolescents with paroxysmal supraventricular tachycardia. J Pediatr. 1996; 129: 324.
14. Paul T, Bertram H, Bokenkamp R, et al. Supraventricular tachycardia in infants, children and adolescents: diagnosis, and pharmacological and interventional therapy. Paediatr Drugs. 2000; 2: 171.
15. Blomstrom-Lundqvist C, Scheinman MM, Aliot EM, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias–executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Supraventricular Arrhythmias). Circulation. 2003; 108: 1871.
16. Friedman RA, Walsh EP, Silka MJ, et al. NASPE Expert Consensus Conference: Radiofrequency catheter ablation in children with and without congenital heart disease. Report of the writing committee. North American Society of Pacing and Electrophysiology. Pacing Clin Electrophysiol. 2002; 25: 1000.
17. Miyazaki A, Blaufox AD, Fairbrother DL, et al. Cryo-ablation for septal tachycardia substrates in pediatric patients: mid-term results. J Am Coll Cardiol. 2005; 45: 581.
18. Collins KK, Dubin AM, Chiesa NA, et al. Cryoablation versus radiofrequency ablation for treatment of pediatric atrioventricular nodal reentrant tachycardia: initial experience with 4-mm cryocatheter. Heart Rhythm. 2006; 3: 564.
19. Ghai A, Silversides C, Harris L, et al. Left ventricular dysfunction is a risk factor for sudden cardiac death in adults late after repair of Tetralogy of Fallot. J Am Coll Cardiol. 2002; 40: 1675.
20. Walsh EP. Arrhythmias in patients with congenital heart disease. Card Electrophysiol Rev. 2002; 6: 422.
21. Morwood, JG, Triedman JK, Berul CI, et al. Radiofrequency catheter ablation of ventricular tachycardia in children and young adults with congenital heart disease. Heart Rhythm. 2004; 1: 301.
22. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006; 114: e385.
23. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992; 20: 1391.
24. Reynolds JL, Pickoff AS. Accelerated ventricular rhythm in children: a review and report of a case with congenital heart disease. Pediatr Cardiol. 2001; 22: 23.
25. Harris KC, Potts JE, Fournier A, et al. Right ventricular outflow tract tachycardia in children. J Pediatr. 2006; 149: 822.
26. Sarquella-Brugada G, Campuzano O, Brugada R. Trastornos del ritmo cardiaco más frecuentes en pediatría: síndrome de QT largo. Pediatr Integral. 2012; XVI(8): 617-21.
| Caso clínico |
|
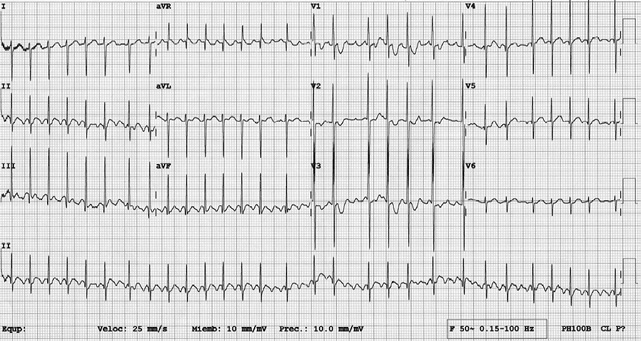
Recién nacido de 4 días de vida con letargia y disminución de ingesta y diuresis. A la exploración, mala perfusión y taquicardia (Fig. 2).
La mayoría de los niños toleran bien los episodios de taquicardia, es fundamental mantener la calma, realizar un EKG y valorar la estabilidad hemodinámica, las maniobras vagales y ATP tienen una efectividad del 75% (Fig. 3).
|