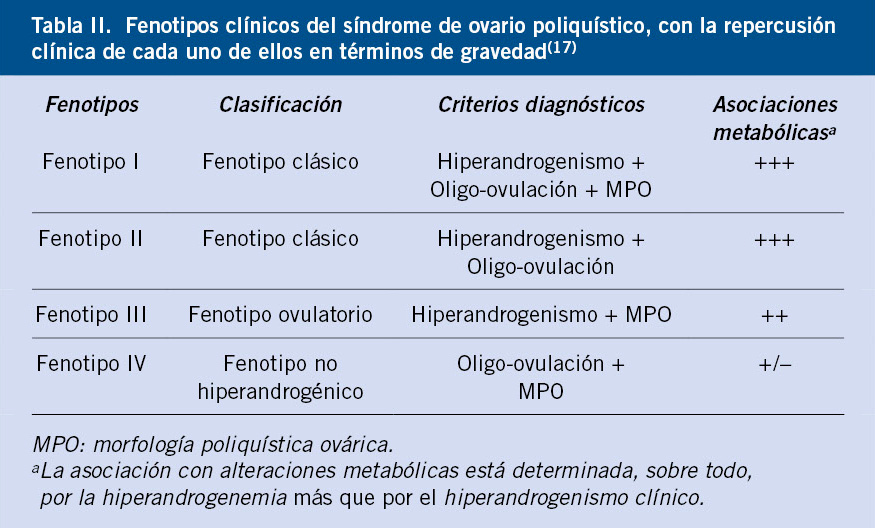
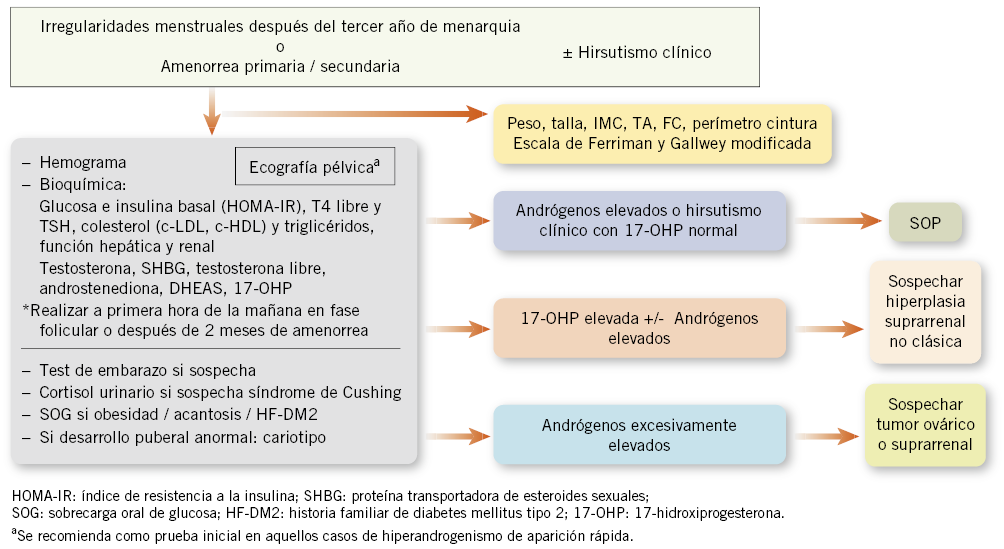
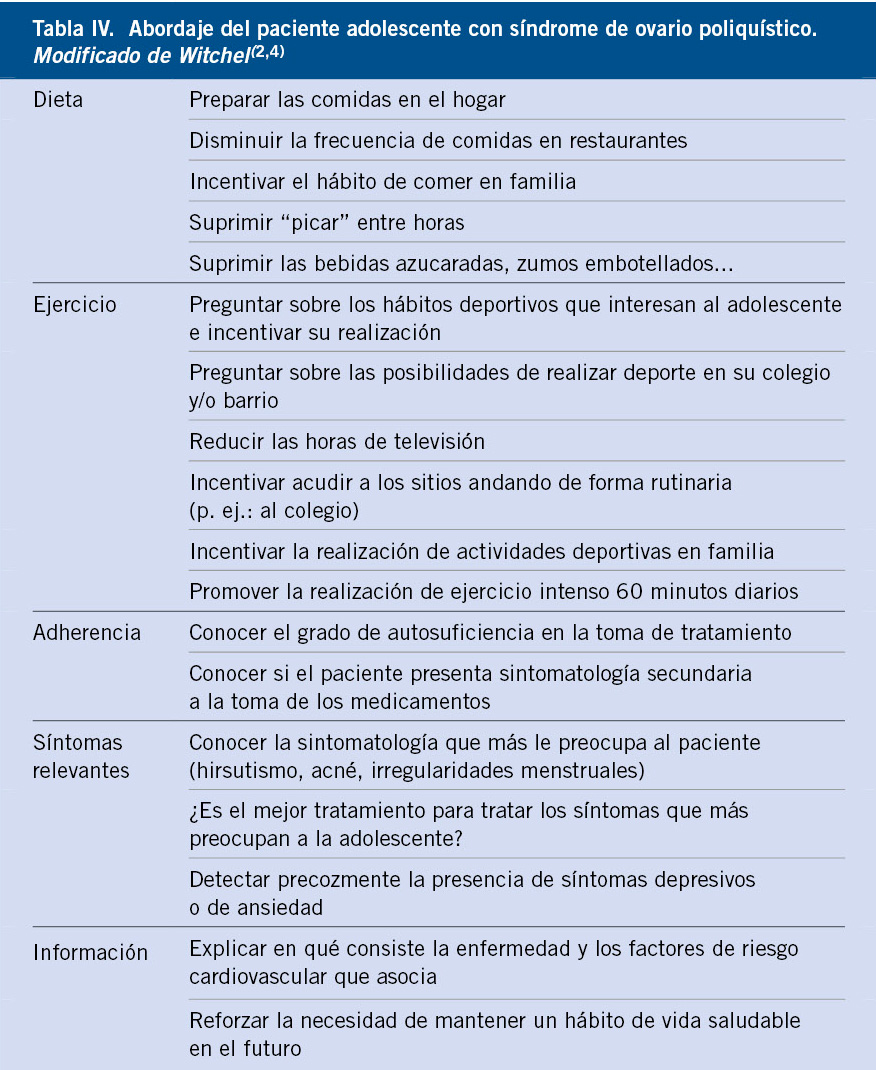
Gráficas de crecimiento en España. Cuál y cómo utilizarlas
https://doi.org/10.63149/j.pedint.60
Introducción
El crecimiento humano es un proceso dinámico de regulación multifactorial. El crecimiento es un indicador del estado de salud infantil, siendo la talla y el índice de masa corporal (IMC) indicadores del estado de bienestar de una población. La mejora en las condiciones de vida nutricionales y socioeconómicas ha determinado una aceleración secular del crecimiento en los países desarrollados con incremento de la talla y del IMC.
El crecimiento humano es un proceso dinámico, continuo pero no lineal, que está sometido a una regulación multifactorial y supone la expresión fenotípica de un potencial genético modulado por la interacción de factores exógenos, dependientes del entorno del individuo (nutrición, influencia psicosocial, afectividad), y endógenos, propios de cada sujeto (hormonales, metabólicos), que abarcan tanto el crecimiento prenatal como postnatal hasta llegar a la talla adulta. Los múltiples factores implicados en este proceso hacen que su valoración constituya un indicador sensible, aunque no específico, del estado de salud y bienestar de cada sujeto y de la comunidad a la que pertenece. Entre los indicadores del estado de bienestar de una población se encuentran la talla, el índice de masa corporal (IMC), la esperanza de vida y la prevalencia de enfermedades crónicas. La evolución de los mismos refleja el progreso humano en los últimos siglos y, para algunos autores, este proceso de mejora no habría concluido. El crecimiento es, pues, un indicador del estado de salud de un niño/a(1,2).
La valoración del crecimiento consiste en el estudio de los cambios que se producen a lo largo del tiempo en el tamaño, forma y composición del organismo. Es un proceso complejo en su conjunto y, por ello, se seleccionan una serie de parámetros clínicos, accesibles a la exploración, que son informativos de los cambios somáticos que se producen. La exploración del mismo en un sujeto se debe comparar con unos estándares de referencia. Mientras que las gráficas de crecimiento describen cómo es el crecimiento, los estándares de crecimiento describen cómo deberían crecer los niños/as(3,4).
El crecimiento infantil y la talla adulta han variado a lo largo de la historia en función de las características genéticas y ambientales en las diferentes poblaciones, en lo que se ha denominado aceleración o tendencia secular del crecimiento. La mejora en las condiciones de vida, la desaparición de muchas enfermedades infecciosas, gracias en gran parte a los programas de vacunación, y la mayor disponibilidad de nutrientes han comportado una aceleración secular del crecimiento en los países desarrollados, junto con un aumento del sobrepeso y la obesidad. Las condiciones nutricionales, psicosociales o socioeconómicas desfavorables condicionan una peor talla adulta y un desarrollo puberal más tardío; por el contrario, unas condiciones favorables asocian tallas más altas y desarrollo puberal más temprano(3). En España, la talla media de los reclutas pasó de 163,4 cm en el año 1910 a 166,3 cm en 1960, a 174,6 cm en 1990 y a 177,3 cm en el 2008(5,6). Estos datos muestran que la talla adulta de los varones ha aumentado alrededor de 14 cm en el último siglo. Si estos datos son extrapolables o no a la población femenina, no se puede afirmar, pero es muy probable que así haya ocurrido. Igualmente, en España, a principios del siglo XX, había diferencias en la talla adulta en función de la región, que podían llegar a ser de hasta 5 cm y, en la actualidad, no hay diferencias(2).
Auxología
La auxología es la disciplina que estudia el crecimiento y desarrollo humanos. La antropometría conlleva la medición de unos parámetros clínicos y su comparación con estándares de referencia adecuados. La talla es un dato estático y la velocidad de crecimiento, reflejo de la evolución de la talla a lo largo del tiempo, se considera el mejor parámetro de control de salud de un niño/a.
La interpretación de la velocidad de crecimiento como normal o patológica puede ser difícil dadas las variaciones normales de la misma, especialmente en la primera infancia y antes de iniciar la pubertad. El perímetro cefálico es un parámetro de crecimiento importante en la primera infancia. En ocasiones, puede ser necesaria la medición de los segmentos corporales, que varían en función de la edad, estadio puberal y etnia. La valoración del crecimiento se debe completar con la evaluación del estado nutricional, siendo el índice de masa corporal (IMC) el parámetro clínico más utilizado, que exige una correcta interpretación y la estimación de la maduración biológica a través de la edad ósea y el desarrollo puberal.
La valoración del crecimiento y desarrollo del niño es fundamental en la práctica diaria del pediatra. La auxología es la ciencia que estudia el crecimiento y desarrollo humanos y la antropometría hace referencia a las diferentes técnicas biométricas aplicadas al estudio y valoración del crecimiento e incluye el estudio comparativo de las medidas del cuerpo humano(3,7).
Valoración del crecimiento
Los indicadores de crecimiento son aquellos que valoran preferentemente el aumento de tamaño del organismo en su conjunto o de determinados segmentos u órganos. Los más importantes en la práctica clínica son la medición de la talla, el peso, el perímetro cefálico (PC) y aquellos parámetros que valoran los segmentos corporales, como son talla sentada (TS), braza, segmento inferior, segmento superior o la relación entre ellos. La utilización de los parámetros antropométricos conlleva la realización de una medición rigurosa de los mismos con una metodología adecuada y la comparación de los datos recogidos con unos estándares de referencia adecuados para la población estudiada, considerando edad, sexo, etnia y país de origen. Con ello se podrá saber si el niño/a se encuentra o no dentro de los límites de la normalidad, y ello se expresa en forma de percentiles o desviación estándar (DE) de la media (Z-score). Según las propiedades de la distribución normal, entre ±1 DE de distancia de la media se encuentra aproximadamente el 68 % de la población, entre ±2 DE el 95 % y entre ±3 DE el 99,7 % de la población. Como idea general, cuanto más alejada esté una medición de la media poblacional, mayor probabilidad hay de encontrar una causa patológica.
La talla y sus incrementos en el tiempo, la velocidad de crecimiento (VC), son los parámetros antropométricos más importantes en la valoración clínica del crecimiento. La talla es un dato estático puntual y la VC, reflejo de la evolución de la talla a lo largo del tiempo, es considerado como el mejor parámetro de control de salud del niño. Los niños/as, desde el nacimiento hasta la edad de 2 años, deben medirse en decúbito supino (longitud) y, a partir de entonces, en bipedestación (talla). La diferencia media entre ambas mediciones en un niño de más de 20 meses puede ser de 1-2 cm, a favor de la talla en decúbito. Es importante conocer la evolución de la talla a lo largo del tiempo, es decir, la reconstrucción de la gráfica de la talla para la edad cronológica, y establecer el patrón de crecimiento. La velocidad de crecimiento es el parámetro antropométrico más difícil de interpretar y el intervalo de observación ideal es de un año y nunca inferior a 6 meses, ya que el error se incrementaría como resultado de las grandes fluctuaciones que, en periodos más cortos de tiempo, puede experimentar el ritmo de crecimiento. La interpretación de la VC como normal o patológica puede ser difícil, dadas las variaciones normales de la VC, que dependen de la naturaleza cíclica del crecimiento, del potencial genético de crecimiento y del tempo madurativo. Los niños/as con talla baja constitucional o tempo madurativo lento tienden a crecer durante la prepubertad por debajo de la media, al contrario de lo que ocurre en niños/as con talla alta constitucional o tempo madurativo rápido. Igualmente, hay que considerar que, en los primeros años de la vida, se produce el fenómeno de canalización del crecimiento, proceso por el cual el niño/a adquiere su carril genético hacia los 2-3 años. Este fenómeno explicaría la existencia de variaciones normales de la VC en la primera infancia, mediante aceleraciones o deceleraciones del crecimiento a partir del segundo semestre, que dependen del potencial de crecimiento y/o ritmo de maduración heredado (talla familiar alta/media/baja y/o maduración rápida/media/lenta). Igualmente, antes de realizar el brote puberal, se produce un enlentecimiento fisiológico de la VC, en lo que se ha denominado deceleración prepuberal de la VC, que puede ser muy intenso y manifiesto en niños/as con tempo madurativo lento o retrasado, pudiendo llegar a ser de 2-3 cm/año. Todos estos factores limitan la interpretación de la VC, que, si bien es un parámetro muy sensible y precoz en la valoración de los trastornos del crecimiento, tiene aisladamente una baja especificidad, y por ello se requieren periodos largos de tiempo de observación (2-3 años) para poder establecer con seguridad su anormalidad. A su vez, determinados hipocrecimientos patológicos, sobre todo aquellos ligados a causas genéticas o sindrómicas, pueden mantener durante periodos largos de tiempo una VC dentro de la normalidad, antes de experimentar una pérdida de talla. El PC es un parámetro de crecimiento importante en los primeros años de la vida, ya que refleja de manera indirecta el crecimiento cerebral; en condiciones normales, su ritmo de crecimiento es máximo durante la vida fetal, disminuye rápidamente después del nacimiento y la mayor parte de su crecimiento ocurre durante los primeros 4 años de vida postnatal, especialmente durante el primer año(3,7).
En ocasiones, puede ser necesario realizar la medición de los segmentos corporales para poder establecer si el sujeto tiene una talla baja proporcionada o desproporcionada. Entre los parámetros utilizados se incluye la TS, braza, ratio TS/talla, ratio braza/talla y ratio segmento superior/segmento inferior, siendo el segmento inferior la medida entre la sínfisis del pubis y el suelo, y el segmento superior la diferencia entre talla y segmento inferior. Los segmentos corporales varían en función de la edad, estadio puberal y etnia, y estos factores deben ser considerados. Las proporciones corporales difieren entre poblaciones como consecuencia de factores genéticos y ambientales ligados a la etnia. Existen diferencias raciales, de modo que los asiáticos tienen un segmento inferior (altura de las extremidades inferiores) más corto que los caucásicos, y estos, a su vez, más corto que los de raza negra. Para una misma talla, en EE.UU., las personas de raza negra tienen menor TS y mayor altura de las extremidades inferiores en comparación con la población caucásica(8). La ratio TS/talla es menor en población negra no-hispana que en la población blanca no-hispana y que en la población hispanoamericana. La población afroamericana tiene una relativa mayor longitud de las extremidades inferiores y de la braza que los europeos, y estos, a su vez, mayor que la de los japoneses. Además de los factores raciales, existen también factores ambientales que influyen en los segmentos corporales; históricamente, los niños que han sufrido abusos y negligencias tienen una mayor cortedad de las extremidades inferiores, que se modifica con la intervención social. Igualmente, la aceleración secular de la talla se debe a un mayor incremento de la longitud de las extremidades inferiores en comparación con la altura del tronco(9,10).
Existe una asociación positiva entre talla, TS y longitud de extremidades inferiores y negativa entre la ratio TS/talla y talla; de manera que los niños con talla baja tienen un cociente TS/talla superior, indicando una cortedad relativa de las extremidades inferiores. En la infancia, el tronco representa el 68 % de la longitud, desciende al 57 % a los 3 años de edad y, durante la pubertad, la altura del tronco supone el 52 % de la talla. Entre los 10 y los 15 años se produce un incremento de la longitud de las extremidades inferiores y la ratio TS/segmento inferior desciende desde 2,1 en el primer año a 1,05 y 1,1 a los 20 años de edad en varones y mujeres, respectivamente. En ambos sexos, para una misma edad, el cociente TS/talla es mayor en estadio prepuberal (Tanner I) que en pubertad (Tanner II-III), lo que indica que durante la pubertad hay más crecimiento de los huesos largos que de la columna. Ello está indicando que el tempo madurativo también influye en la relación de los segmentos corporales y este factor debe ser considerado en la evaluación del crecimiento. Niños/as con retraso puberal presentan un cociente TS/talla inferior a lo esperado por su edad, y a la inversa, niños/as con pubertad precoz tendrán un cociente mayor a lo esperado por su edad(9,10).
La ratio de TS/talla es un parámetro útil en la práctica clínica para el despistaje de la talla baja desproporcionada y la clasificación de las displasias óseas en acortamiento de tronco o de extremidades. La talla en bipedestación es la suma de dos componentes, el tronco, que equivale a la TS, y las extremidades. En la población normal, la ratio entre TS/talla es de aproximadamente 0,7 al nacimiento y se sitúa en 0,5 al alcanzar la madurez esquelética. El estudio longitudinal de Zaragoza ha establecido los valores de normalidad de la ratio TS/talla desde el nacimiento hasta la edad adulta, elaborando unas tablas y gráficas percentiladas(11). El valor TS/talla desciende, tanto en varones como en mujeres, desde el nacimiento (0,656 y 0,647) hasta el inicio de la pubertad (0,514 y 0,519) y experimenta un ligero aumento al llegar a talla adulta (0,52 y 0,53, respectivamente). Estos datos son muy similares a los encontrados en la población alemana, donde se establecen valores de referencia en función de la edad y sexo de 0 a 21 años para TS, ratio TS/talla y longitud de las extremidades inferiores (diferencia entre talla y TS)(12). En este estudio, la ratio TS/talla es de 0,68 en la infancia y disminuye a 0,52 en la adolescencia, indicando que en la prepubertad el crecimiento se produce más en las extremidades inferiores que en el tronco. Los valores de normalidad del estudio centroeuropeo establecen, como puntos de corte de la ratio TS/talla para diferenciar una talla baja patológica, con acortamiento del segmento inferior, o una talla alta patológica, con incremento del segmento inferior, de variantes de la normalidad en +2,5 DE y -2,2 DE, respectivamente. Se han desarrollado curvas de referencia del cociente TS/talla para población americana en función de su origen étnico, no-hispánico (raza negra y raza blanca) o hispánico(9,10).
Valoración del estado nutricional
Los parámetros antropométricos más importantes en la valoración del estado nutricional son, además de la talla y el PC, el peso, el perímetro braquial y los pliegues cutáneos(3,5). El peso para la edad, de manera aislada, tiene poco valor y, por ello, se usa la relación peso/talla y existen curvas percentiladas de la relación peso/talla, si bien estas curvas solamente pueden ser utilizadas durante el tiempo en que la distribución del peso para la talla es independiente de la edad, lo que ocurre entre los 2 años y el inicio de la pubertad. El índice más empleado que relaciona ambos parámetros es el índice de masa corporal (IMC) [peso (kg) / talla2 (m)]. El IMC varía en función de la edad, sexo y estadio puberal. Pese a que sus valores no siguen una distribución normal, por lo que la percentilación o aplicación del Z-score no sería matemáticamente correcta, con frecuencia se utiliza la DE para edad y sexo con objeto de hacer una valoración del estado nutricional, así como del grado de sobrepeso/obesidad o delgadez/malnutrición, por la relación existente entre severidad y aparición de comorbilidades. Se acepta, como criterio de sobrepeso y obesidad, los valores del IMC para edad y sexo superiores al percentil 90 y 97, respectivamente. El peso y la talla o los índices derivados no aportan información sobre la composición corporal y ello impide afirmar si el exceso de peso o del IMC de un paciente está motivado por un incremento de tejido graso o de masa magra. Existen diferentes metodologías para valorar la composición corporal (DEXA, impedanciometría, ecografía, RMN, etc.), pero en la práctica clínica los pliegues cutáneos y los perímetros corporales son los parámetros más utilizados. El pliegue cutáneo es un indicador de la grasa subcutánea que es aproximadamente el 50 % de la grasa total; se mide en diferentes puntos, pero lo más utilizado es el pliegue subcutáneo tricipital y subescapular, y así, de manera indirecta, estimar si la distribución de la grasa es generalizada o de predominio troncular, respectivamente. El perímetro abdominal, circunferencia de la cintura, es un excelente indicador de la cantidad de grasa abdominal. Existen curvas de normalidad del perímetro abdominal en la infancia y adolescencia y se ha demostrado que también es buen indicador de obesidad central y es útil como predictor de riesgo metabólico y cardiovascular, cuando sus valores se sitúan por encima del percentil 90. El perímetro braquial mide simultáneamente el componente muscular y graso(13,14).
Valoración del nivel de maduración
En el estudio antropométrico, además de la valoración del crecimiento y del estado nutricional, se deben estudiar otros parámetros que sirven para estimar la maduración o edad biológica, y entre ellos los más usados son la edad ósea y el nivel de desarrollo puberal. El ritmo de maduración es algo individual que no siempre va paralelo a la edad cronológica y a la maduración biológica, estando regulado por una compleja interacción hormonal. El único indicador aceptado de maduración es la edad ósea, que refleja la edad biológica. Una valoración de la edad ósea puntual tiene menos valor que el ritmo de cambio de la misma; un desfase entre edad ósea y edad cronológica no siempre es constante a lo largo del tiempo, pudiendo cambiar tanto por motivos fisiológicos (adrenarquia, inicio puberal) como patológicos (obesidad, retraso de crecimiento intrauterino, hipotiroidismo, hipertiroidismo, hipercortisolismo, etc.). La estimación de la edad ósea se realiza mediante métodos basados en el estudio de la radiografía de la mano izquierda. El método de Greulich-Pyle es un método comparativo de la radiografía del caso con las radiografías de un atlas de niños/as normales y es el método más utilizado en la práctica clínica. El método de Tanner-Whitehouse se basa en el uso de puntuaciones numéricas para los diferentes huesos del carpo y de la mano. Más recientemente, se han implementado métodos automatizados, como el BoneXpert. El cálculo del pronóstico de talla adulta basado en la maduración esquelética (edad ósea) es un proceso muy común en la práctica diaria. Existen diversos métodos, pero los más extendidos son el método de Bayley-Pinneau y el de Tanner-Whitehouse, basados en la lectura de la edad ósea por Greulich-Pyle y por Tanner-Whitehouse, respectivamente. El más utilizado por su sencillez es el primero, que se basa en el porcentaje de talla adulta alcanzada a una edad ósea determinada. Si el pronóstico de talla adulta se encuentra entre ±5 cm (equivalente a ±1 DE) de la talla diana o genética, se asume que existe una concordancia entre la talla del niño/a con la de sus padres, mientras que si se aleja ±10 cm (equivalente a ±2 DE), orienta a una probable alteración del crecimiento. El nivel de desarrollo puberal se valora mediante la exploración de los caracteres sexuales secundarios, de acuerdo a los estadios de Tanner. Durante la pubertad, se produce una aceleración de la velocidad de crecimiento y la ganancia neta de talla durante la pubertad es de 26 cm en los varones y 20 cm en las mujeres(13), siendo la magnitud del estirón similar tanto en sujetos de talla alta familiar como de talla baja familiar.
Monitorización del crecimiento
La monitorización del crecimiento exige dos tipos de gráficas: gráficas de distancia, para valorar parámetros como talla, peso, IMC y PC según edad y sexo, y las gráficas de la VC, que indican el incremento de la talla según edad y sexo. Cuanto más alejado esté un parámetro clínico de la media poblacional, mayor probabilidad hay de encontrar una causa patológica. La interpretación de los percentiles tiene un diferente significado en función de si se trata de gráficas de distancia o gráficas de velocidad de crecimiento. Una VC mantenida en el tiempo ≤ al percentil 25 debe considerarse como patológica, no así en el caso de las gráficas de distancia. La distancia de la talla de un sujeto con respecto a la talla de sus padres es un parámetro clínico muy útil y de alta sensibilidad en la detección de una talla baja patológica y para ello es necesario un correcto cálculo e interpretación de la talla genética. Es difícil diferenciar un crecimiento normal de un crecimiento patológico, existiendo mucha variabilidad en la práctica clínica. Los parámetros más útiles en el diagnóstico de un retraso de crecimiento patológico son la severidad de la talla baja y de la distancia de la talla en relación a la talla genética, y la combinación de talla baja y VC disminuida. Las curvas para niños/as con síndromes genéticos o enfermedades específicas permiten valorar el crecimiento con más precisión y detectar en ellos trastornos de crecimiento adquiridos.
Para que un estudio antropométrico sea objetivo y válido debe seleccionar parámetros o indicadores clínicos suficientemente sensibles y reproductibles; ser preciso en la recogida de las medidas mediante una metodología adecuada; emplear estándares o curvas de referencia adecuadas; y, finalmente, realizar una interpretación correcta de los resultados obtenidos. Una de las características del crecimiento es que es dinámico; de manera que es necesario valorar no solamente el crecimiento en un momento determinado (talla), sino que es necesario efectuar su seguimiento (velocidad de crecimiento)(1-3,14). Para monitorizar el crecimiento, el pediatra necesita dos tipos de gráficas:
1. Gráficas de talla, peso, IMC y PC según edad y sexo, obtenidas preferentemente mediante estudios transversales, son las denominadas gráficas de distancia. No puede olvidarse que son curvas acumulativas y reflejan la evolución desde el nacimiento, no lo que está ocurriendo en el momento de la observación. Por ello, nunca puede descartarse una alteración del crecimiento con un único examen, cualquiera que sea el percentil en que se encuentre el sujeto en la curva de distancia para la talla, si no se conoce la velocidad de crecimiento. Carecen, por lo tanto, de sensibilidad para detectar cambios o anomalías recientes, a no ser que estos trastornos tengan una intensidad importante y una duración prolongada. Aunque existen diferencias de criterios en la interpretación de los datos, deben considerarse patológicos los que están alejados más de 3 DE por debajo de la media o se sitúan por debajo del percentil 1; los casos que están por debajo de 2 DE de la media o por debajo del percentil 3 no siempre indicarán un estado de patología.
2. Gráficas de velocidad de crecimiento según edad y sexo, obtenidas a través de estudios longitudinales. Los incrementos o diferencias entre las distintas edades que se pueden extraer de los estudios transversales expresan la diferencia de las medias y no permiten valorar los percentiles ni la morfología real de las inflexiones que se producen en momentos de aceleración o enlentecimiento del ritmo de crecimiento, como sucede en la primera infancia o en la pubertad. La mayoría de los niños/as con problemas de crecimiento tienen una VC baja y el control de esta es un método sensible para detectar trastornos de crecimiento, pero poco específico, ya que el proceso de crecimiento no es lineal y sí dinámico, pudiendo un niño normal tener fases de crecimiento lento, especialmente en la primera infancia y en la edad prepuberal. En el estudio longitudinal suizo se describe que hasta dos tercios de los sujetos normales experimentaron un cruce de percentiles superior a 1 DE, especialmente durante la pubertad, que se explica por los diferentes patrones de maduración. La curva de VC hay que utilizarla con rigor, ya que de lo contrario se pueden cometer errores de interpretación. En primer lugar, los controles deben ser anuales, entre un periodo de 9 y 15 meses, nunca inferior a 6 meses, ya que en periodos más cortos, el error de la doble medida sumada al efecto estacional sobre el crecimiento, introduce una gran imprecisión. El segundo aspecto a tener en cuenta es el diferente significado que tienen los percentiles de VC con los de las curvas de distancia. En las curvas de distancia, lo normal es que, a partir de los 2 años, el niño/a se mantenga en ese carril o posición, independientemente del percentil en que se sitúe la talla. Por el contrario, en las curvas de VC, si un niño crece en un percentil 25 de velocidad, ello no es suficiente para que se mantenga en el mismo carril en la curva de talla y, progresivamente, se va alejando y perdiendo talla. Por eso, en la práctica clínica, una VC inferior al percentil 10 se debe considerar patológica y, si está en el percentil 25 (~1 DE), se puede considerar inicialmente normal, pero si se mantiene en él durante un periodo prolongado, superior a 2-3 años, debe ser considerada patológica, ya que se desvía del canal inicial y perderá talla y su repercusión dependerá de su posición inicial. Finalmente, la valoración de la VC debe tener en consideración el tempo o ritmo madurativo del niño/a, sobre todo en las edades peripuberales, ya que no todos los niños/as realizan el desarrollo y brote puberal a la misma edad, existiendo una variabilidad importante.
En las gráficas de crecimiento, además de la talla y la VC, existen otros parámetros que deben ser valorados para aumentar su sensibilidad y especificidad. La relación de la talla con la talla de sus padres es un parámetro muy útil en la práctica clínica, y para algunos autores sería el de mayor sensibilidad, basándose en el hecho de que existe una correlación elevada (r = 0,63) entre la talla de los hijos y la talla de los padres. Para aumentar la sensibilidad de la distancia de la talla de la media poblacional, es útil recurrir a las curvas de distancia en función de la talla de los padres, en las que una desviación de la talla del niño/a superior a 2 DE de la talla genética es muy significativa de patología. Pero para ello, es necesario hacer un cálculo previo de la talla genética o talla diana. La talla genética se calcula a partir de la talla media de los padres y permite conocer el potencial genético del niño/a. Existen diferentes fórmulas para el cálculo de la talla genética, pero la más utilizada es la fórmula de Tanner, que ajusta la talla media parental en función del sexo, añadiendo o restando 13 cm si es varón o mujer, respectivamente, que es la diferencia de la talla adulta entre ambos sexos. En el caso del varón, la talla genética sería [(talla padre + talla madre + 13 cm)/2] y en el caso de la mujer [(talla padre + talla madre – 13 cm)/2](15). Otros autores, además, ajustan la talla media parental, en función de la aceleración secular del crecimiento, y consideran un factor de corrección de +4,5 cm o de +3,8 cm(16). El cálculo de la talla genética por la fórmula de Tanner es el más utilizado en la clínica diaria, pero esta fórmula infraestima la talla genética en casos de talla baja familiar, ya que el niño/a adquiere una talla final superior a la estimada, y a la inversa, sobreestima la talla genética en casos de talla alta familiar, porque el niño/a no alcanza dicha talla. Por ello, algunos autores han realizado fórmulas de corrección que no dependen del sexo ni de la aceleración secular y que se podrían utilizar en aquellos casos donde las tallas de los padres se encuentran en posiciones muy extremas. Estas fórmulas tienen en consideración el emparejamiento selectivo entre personas de la misma talla y el fenómeno de regresión a la media de la talla. Con estas consideraciones, la talla genética corregida expresada en DE equivaldría a [(talla padre en DE + talla madre en DE)/2 x 0,72](17).
Es difícil diferenciar un crecimiento patológico de un crecimiento normal y diversos estudios han mostrado una variabilidad en la práctica clínica y en el uso de gráficas de crecimiento diferentes. Una encuesta realizada entre pediatras de Atención Primaria en Europa informa que un 69 % utilizan gráficas nacionales, mientras que el 29 % utilizan las gráficas de la Organización Mundial de la Salud (OMS)(18) y solamente un 21 % de los mismos dicen tener un algoritmo establecido para definir un retraso de crecimiento. Por el contrario, a nivel de pediatras endocrinos de sociedades científicas, el 50 % utilizan gráficas nacionales o locales y un 39 % dicen tener un protocolo de estudio de retraso de crecimiento(19). La monitorización del crecimiento en la población infantil en la edad escolar ha mostrado ser coste-efectivo para aumentar el rendimiento diagnóstico y mejorar la salud de patologías, como déficit de GH o síndrome de Turner(20). No hay consenso en relación a cómo definir un crecimiento anormal indicativo de una patología secundaria en contraposición con las tallas bajas variantes de la normalidad. La experiencia indica que existen muchas derivaciones para el estudio de talla baja que obedecen a variantes de la normalidad y que no requieren estudios complementarios, por lo que sería altamente beneficioso disponer de algoritmos clínicos de derivación que permitiesen identificar una talla baja patológica y evitar así pruebas innecesarias. Se conocen 4 guías de consenso y 3 protocolos de decisión clínica que intentan establecer criterios clínicos para definir un crecimiento patológico(21). El nivel de validación de las diferentes guías y protocolos es muy bajo y tienen un nivel de sensibilidad y especificidad variables(22). Los indicadores o parámetros auxológicos utilizados varían entre ellos y se han utilizado hasta 8 indicadores auxológicos con diferentes criterios y puntos de corte. Los criterios auxológicos más utilizados son: severidad de la talla baja (≤ -2 DE; < -3 DE), distancia de la talla genética (< a -1,3 DE; < a -1,6 DE; < a -2 DE), pérdida de talla (>0,25 DE/año; >0,5/año; ≥1 DE), VC disminuida (< a -1 DE; < a -1,5 DE; < a -2 DE) y otros, como pérdida del IMC, presencia de síntomas asociados, antecedentes patológicos y fenotipo especial, ya que hasta un 25 % de los trastornos de crecimiento se presentan en ausencia de talla baja. Los parámetros más útiles para el diagnóstico de un retraso de crecimiento patológico son la severidad de la talla baja, distancia de la talla en relación a la talla genética y la combinación de talla baja y VC disminuida(21,23).
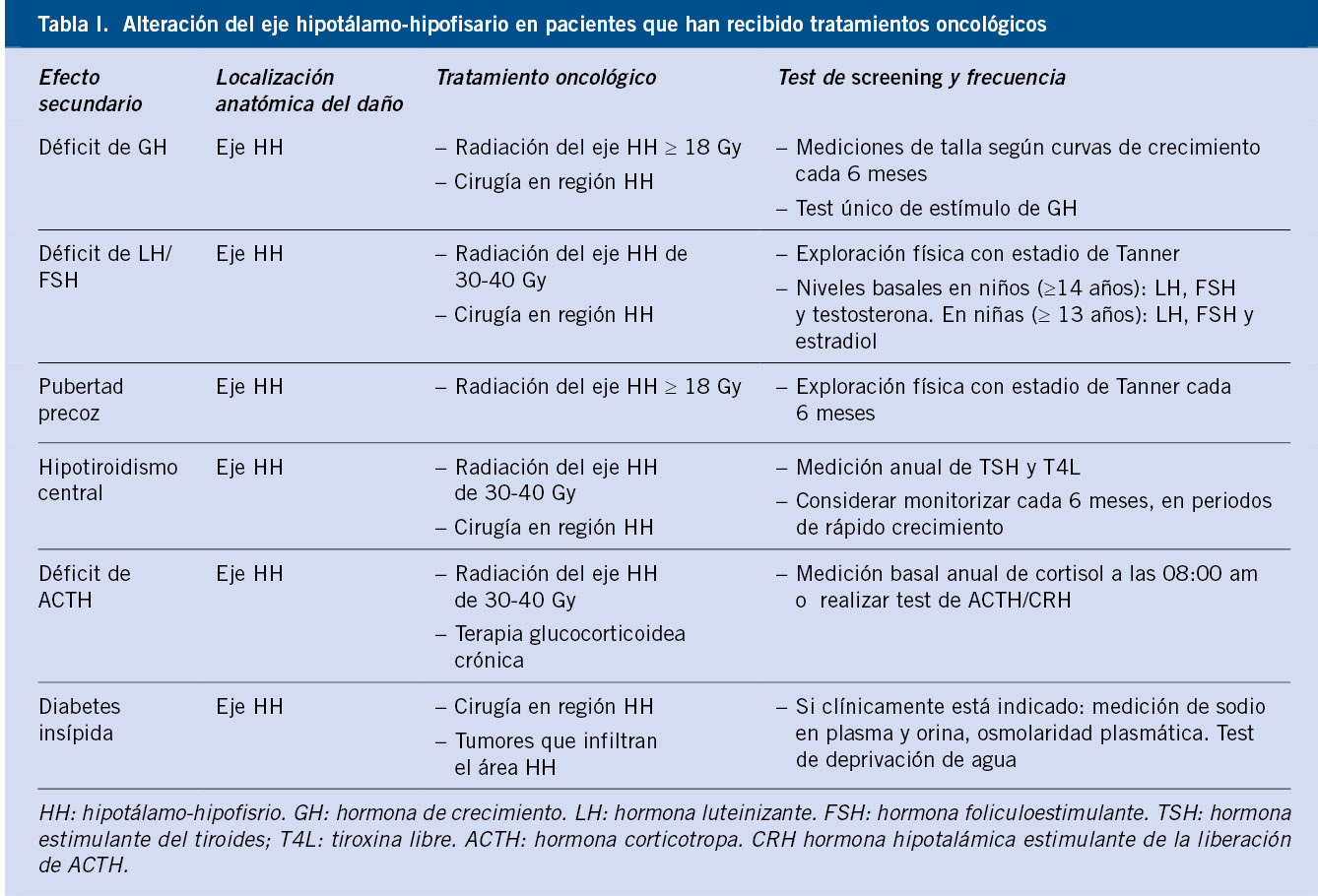
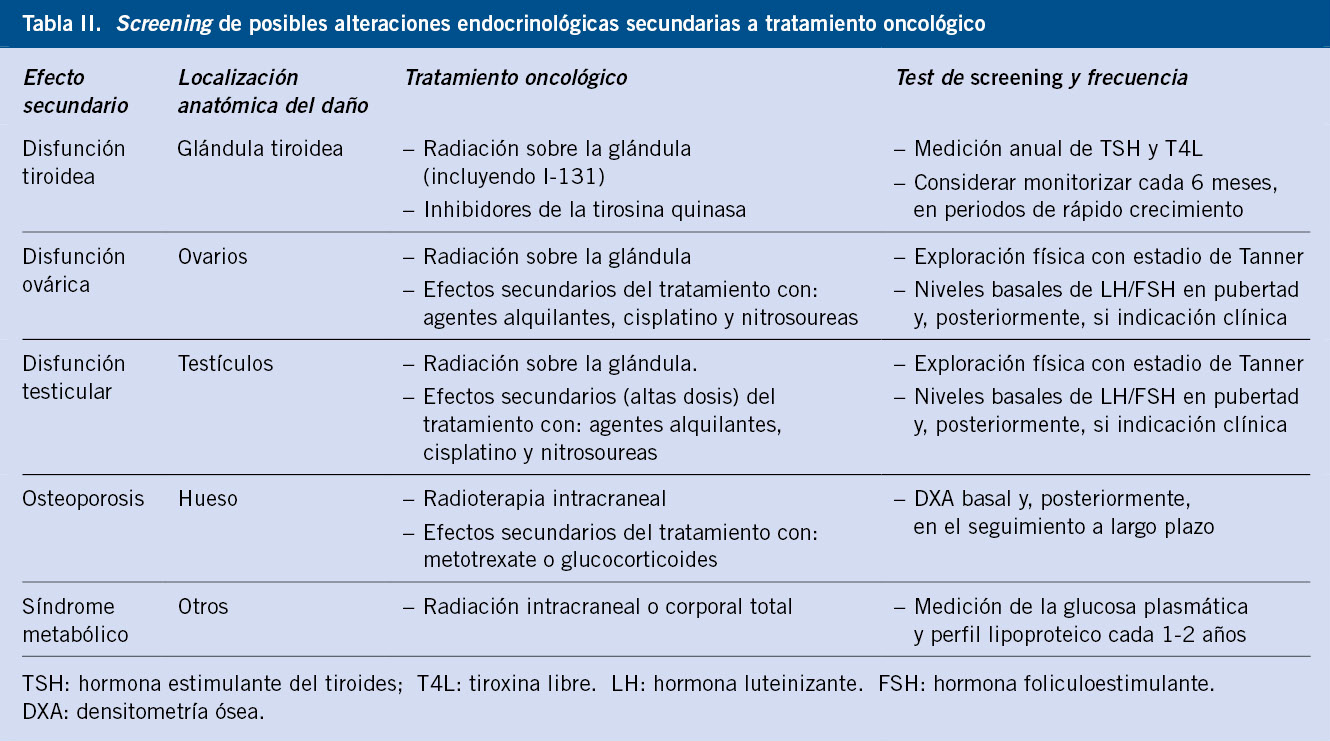
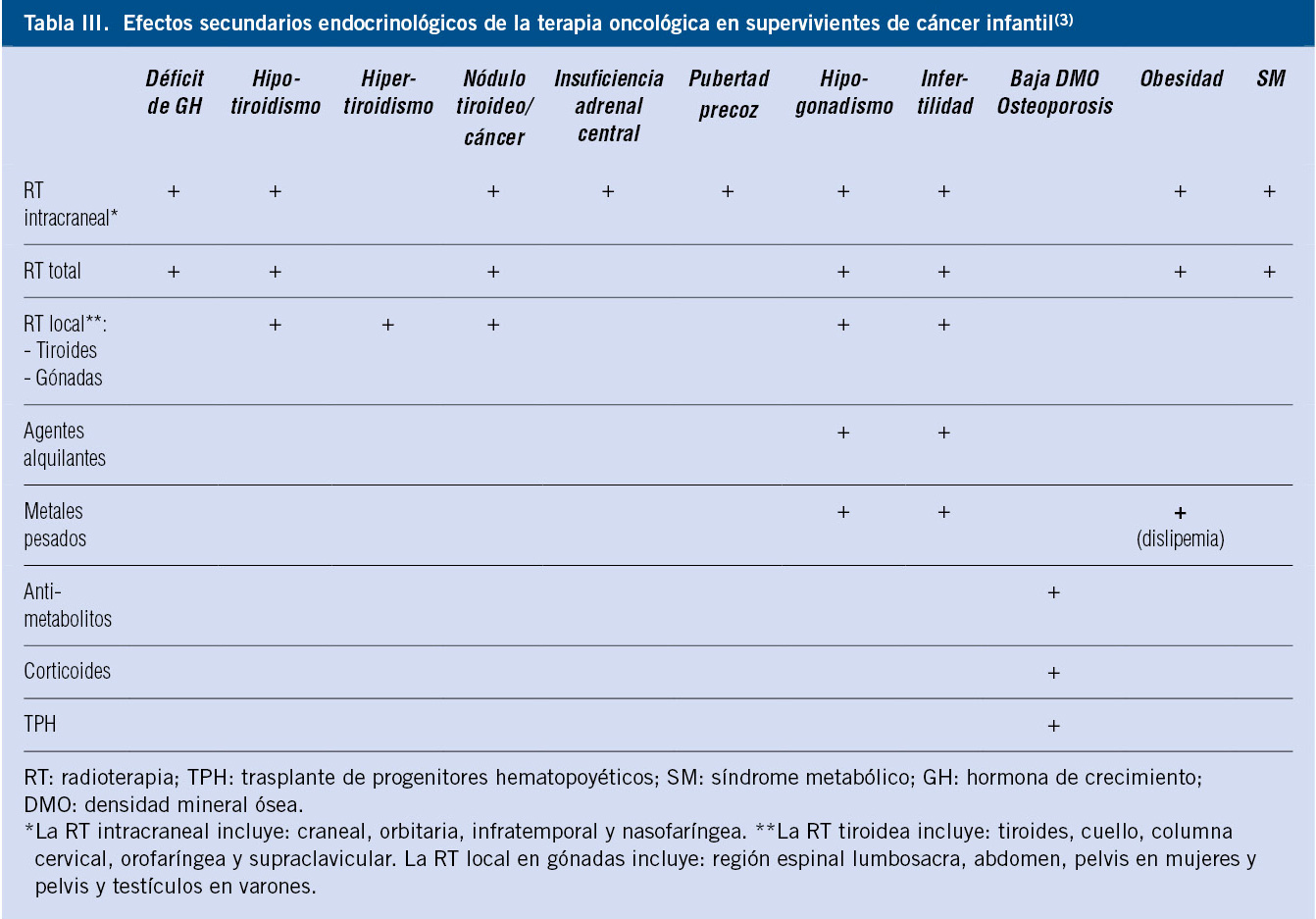
Las curvas para niños/as con síndromes o enfermedades específicas permiten valorar el crecimiento con más precisión (síndrome de Down, Turner, Noonan, Prader-Willi, acondroplasia). Se dispone de curvas de normalidad para un número limitado de patologías o síndromes específicos; si bien, en muchos casos, no tienen el número suficiente ni la metodología necesaria para poder ser utilizadas como estándares de referencia. Las gráficas de crecimiento específicas para síndromes genéticos tienen su utilidad, ya que permiten conocer el patrón de crecimiento específico de la condición a estudio, ayudan a la detección precoz de trastornos de crecimiento adquiridos que determinan una desviación de su crecimiento normal y, en tercer lugar, permiten evaluar los efectos del tratamiento(24). En la tabla I se presentan las referencias bibliográficas de las gráficas de crecimiento de síndromes y patologías específicas.

Estándares de referencia: tipos de estudios
La elaboración de estándares se realiza a través de tres tipos de estudios: transversales, longitudinales y longitudinales mixtos. La selección de la muestra debe incluir todos los estratos socioeconómicos, grupos de edad y sexo y excluir los individuos con patologías que teóricamente pueden afectar al crecimiento. Considerando la aceleración secular, sería deseable realizar estándares de crecimiento cada 15-20 años.
La valoración correcta del patrón de crecimiento exige comparar los datos del sujeto con los estándares obtenidos en una muestra representativa de la población a la que pertenece. La elaboración de estos estándares, que no son más que la distribución de frecuencias de las distintas medidas antropométricas en una determinada población a diferentes edades, puede hacerse utilizando tres métodos que se diferencian en el procedimiento de recogida y seguimiento de la muestra: transversal, longitudinal y mixto.
Los estudios transversales se obtienen efectuando un corte en la población en un momento dado y permiten incluir un elevado número de sujetos para cada grupo de edad y sexo. Son rápidos de realizar, informan sobre la situación actual de la población estudiada, pero no ofrecen información sobre la velocidad de crecimiento. Los estudios transversales son útiles para valorar el crecimiento en las edades en las que no se ha iniciado el desarrollo puberal. Debido a que no todos los niños/as inician el brote puberal a la misma edad, este tipo de estudios no permite evaluar de forma precisa el crecimiento durante la pubertad(1,2,5).
Los estudios longitudinales estudian a una población desde el nacimiento hasta la edad adulta. Estos estudios suministran información de la VC y son indispensables para estudiar con rigor el desarrollo puberal o el rebote adiposo. Cada niño/a tiene su propio tempo madurativo para iniciar la pubertad. El inicio del brote de crecimiento puberal oscila de los 8 a los 13 años en las mujeres y de los 10 a los 15 años en los varones. Esta variabilidad hace que solamente los estudios longitudinales permitan valorar adecuadamente el crecimiento puberal. Este tipo de estudios permite agrupar a los sujetos de acuerdo a la edad de inicio del brote puberal (muy temprano, temprano, medio, tardío, muy tardío) y aporta datos diferenciados para cada grupo en función de su ritmo madurativo. Tienen el inconveniente de que son largos, costosos y puede haber sesgos en la selección y mantenimiento de la muestra a lo largo del estudio. A su vez, si las condiciones socioeconómicas se han modificado a lo largo del periodo de estudio, los datos de los primeros años pueden no ser extrapolables o representativos del momento en el que acaba el estudio(25).
Los estudios longitudinales mixtos realizan el seguimiento de varias cohortes de niños/as de diferentes edades a los que se les sigue longitudinalmente, uniendo al final las curvas resultantes de cada cohorte. La ventaja es que acorta la duración de los estudios necesarios para obtener información sobre la VC.
La metodología para elaborar estándares de referencia incluye una adecuada selección de la muestra que comprenda todos los estratos socioeconómicos, grupos de edad y sexo, y excluir los individuos con patologías que teóricamente pueden afectar al crecimiento. Los datos antropométricos deben obtenerse con precisión con instrumentos debidamente calibrados para la posterior elaboración de tablas y gráficas. Periódicamente, es necesario realizar estudios de crecimiento que, además de su utilidad clínica como estándares de referencia, informen sobre el estado de bienestar de la población. Teniendo en cuenta la aceleración secular, sería deseable realizar estudios transversales cada 15-20 años. Los estudios longitudinales, al ser más costosos y de mayor duración, deberían efectuarse en periodos más amplios y utilizar diseños mixtos con varias cohortes, con el objeto de reducir su duración y limitar el fenómeno de la aceleración secular entre el momento del inicio del estudio y su finalización. Los estudios deben ser realizados a nivel local, con el objeto de que el crecimiento se compare con poblaciones similares en sus condiciones genéticas y ambientales(2,5).
Estudios de crecimiento en España
En España ha habido una fuerte tradición de estudios de crecimiento, tanto longitudinales como transversales. Los estudios longitudinales más relevantes fueron realizados en Bilbao y Zaragoza. El análisis por separado y la comparación de los mismos han permitido comprobar que no existen diferencias, indicando que la población española se comporta, en la actualidad, de manera homogénea a nivel antropométrico. Los estudios de Andalucía, Barcelona, Bilbao, Madrid y Zaragoza han sido analizados conjuntamente, dando lugar a los estudios españoles de crecimiento 2008 y 2010. Se ha producido una aceleración secular y la talla adulta española se sitúa en torno a 177 cm en los varones y 164 cm en las mujeres, similar a la de los países mediterráneos de nuestro entorno. Los estudios transversales son útiles para valorar el crecimiento en las edades en las que todavía no se ha iniciado el desarrollo puberal. Para evaluar el crecimiento durante la pubertad, es necesario disponer de estudios longitudinales donde se clasifique a los niños/as en función del ritmo madurativo. Se distinguen 5 grupos maduradores en función de la edad a la que se inicia el brote de crecimiento puberal (muy temprano, temprano, intermedio, tardío y muy tardío), y esta consideración es especialmente necesaria en la valoración de los trastornos del crecimiento.
En España, ha habido una fuerte tradición de realización de estudios de crecimiento y se han realizado diferentes estudios longitudinales (Bilbao(26), Zaragoza(27), Barcelona(28,29)) y transversales (Bilbao(30,31), Barcelona(32), Andalucía(33), Madrid(34)) en la población caucásica autóctona, que finalizaron entre los años 2000 y 2010. En la tabla II se presentan los estudios de crecimiento más importantes realizados en España, con los parámetros analizados(26,27,29-40). El análisis por separado y la comparación de los datos han permitido comprobar que no existen diferencias de relevancia clínica, por lo que han sido analizados conjuntamente, dando lugar a los estudios españoles de crecimiento 2008 y 2010(37-39,40). Ello indica que la población española se comporta, en la actualidad, de manera homogénea a nivel antropométrico.
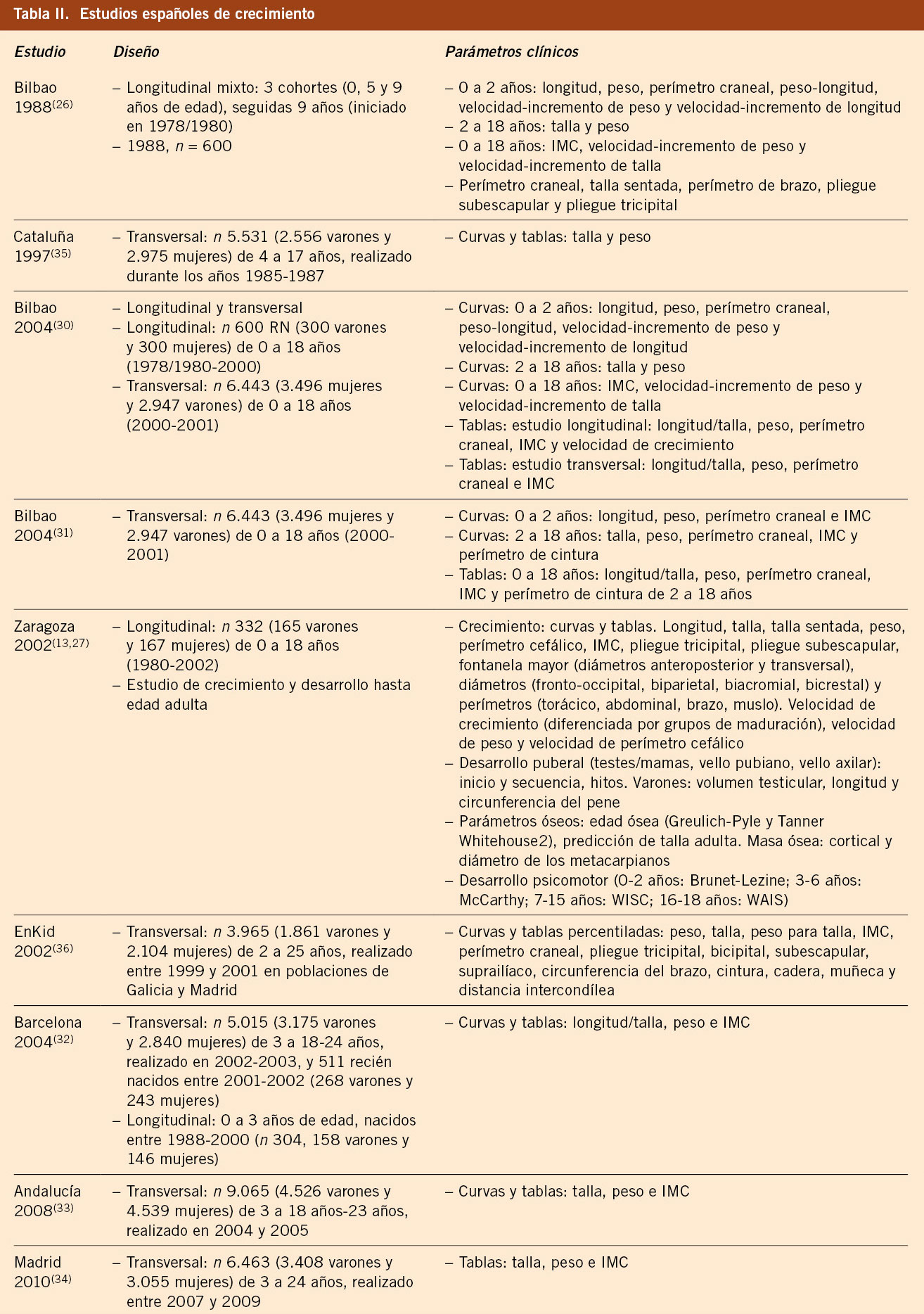
Estudio longitudinal de Bilbao
El estudio longitudinal conocido como Bilbao 1988 se inició en el año 1978-1980, tuvo un diseño longitudinal mixto, seleccionando al azar 3 grupos de 600 niños, con edades de 0, 5 y 9 años, respectivamente, a los que se les siguió durante 9 años. La muestra, en su mayoría, pertenecía a una clase socioeconómica media-baja y la superposición de los grupos durante un año permitió comprobar que no había diferencias significativas entre ellos y, por ello, las gráficas pueden utilizarse como curvas longitudinales puras representativas de la misma población. Este estudio publicado en 1988 incluía gráficas y tablas de crecimiento y tuvo gran difusión(26). Dicho grupo realizó otro estudio, conocido como Bilbao 2002 y publicado en el año 2004, que supone la combinación del estudio longitudinal puro, es decir, la evolución hasta el final del periodo de crecimiento de los niños y niñas nacidas en 1978-1980, y la realización de un estudio transversal durante el año 2000 en una muestra de 6.443 sujetos (3.496 niñas y 2.947 niños), con edades comprendidas entre 0 y 18 años, realizada sobre una muestra amplia de la población de Vizcaya(30,31). El análisis de los resultados de ambos estudios muestra diferencias en la talla adulta en las chicas, en la relación peso/talla y en la morfología del estirón puberal. Es difícil dar una explicación científica de estas diferencias, debido a la distinta metodología seguida en ambos estudios.
Estudio longitudinal de Zaragoza
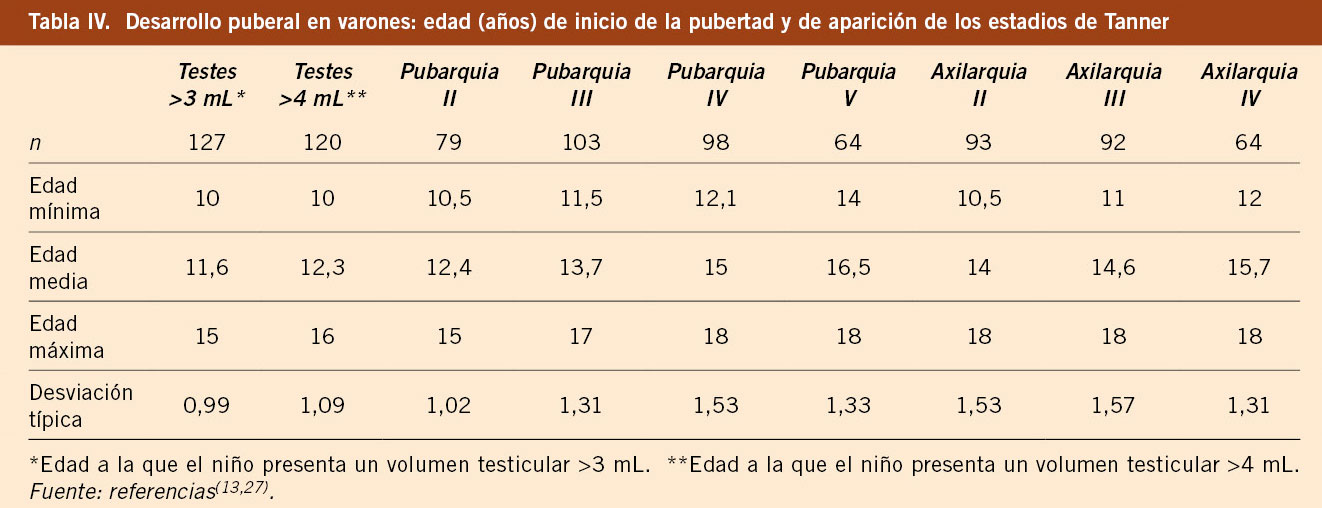
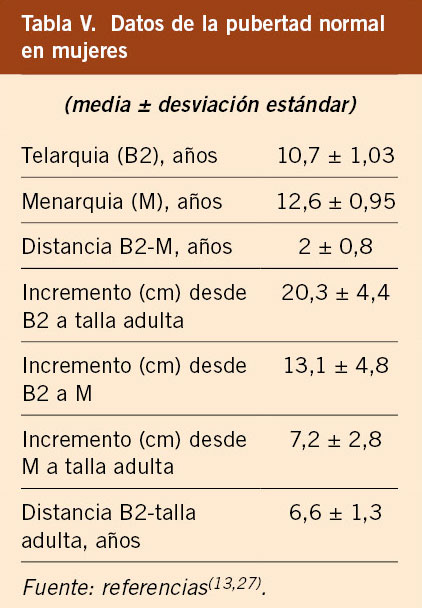
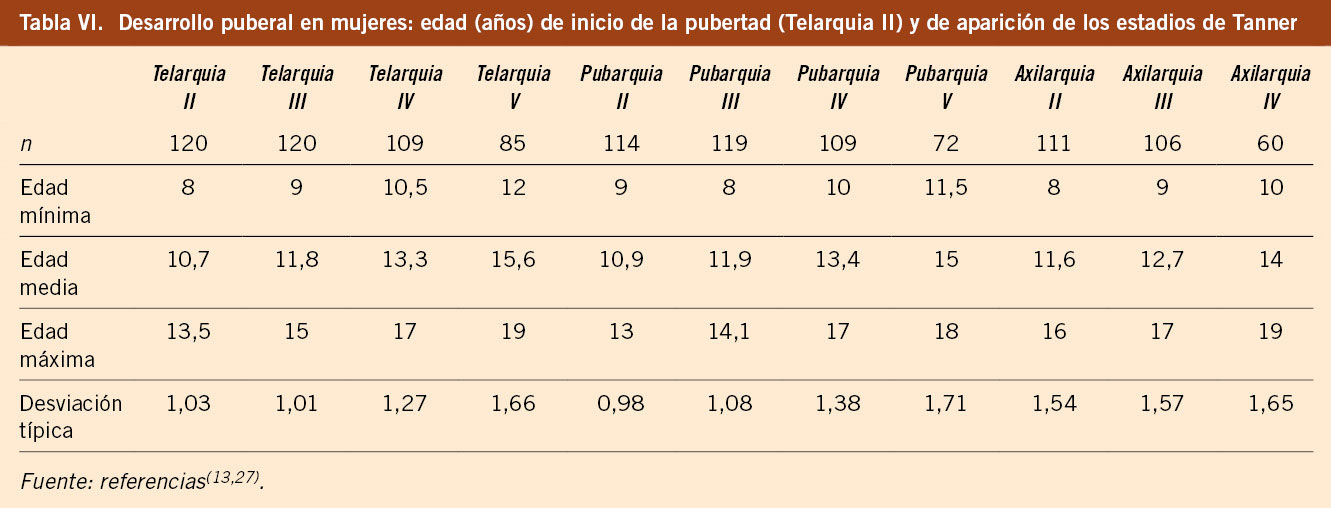
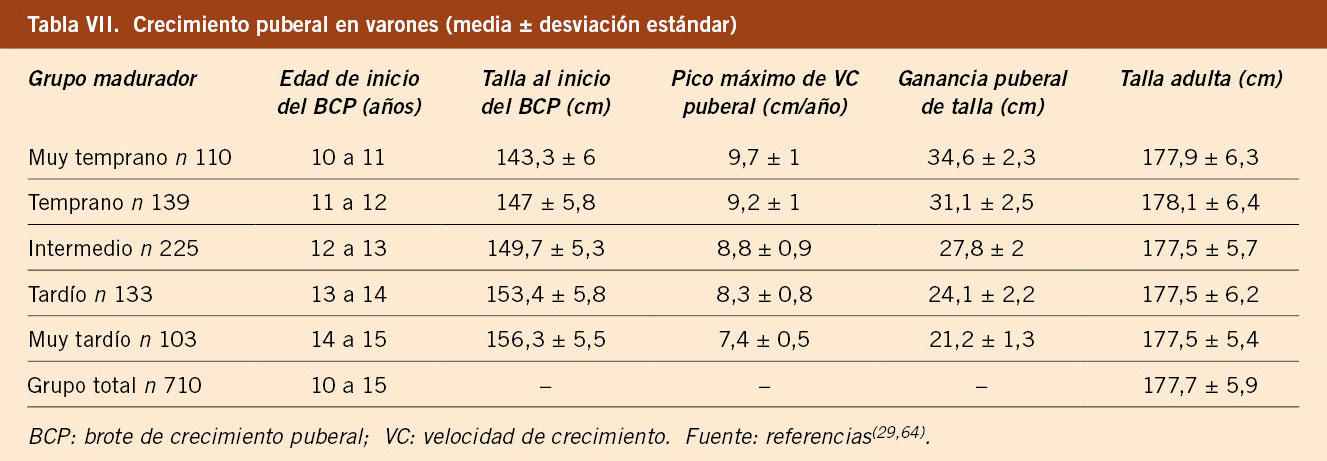
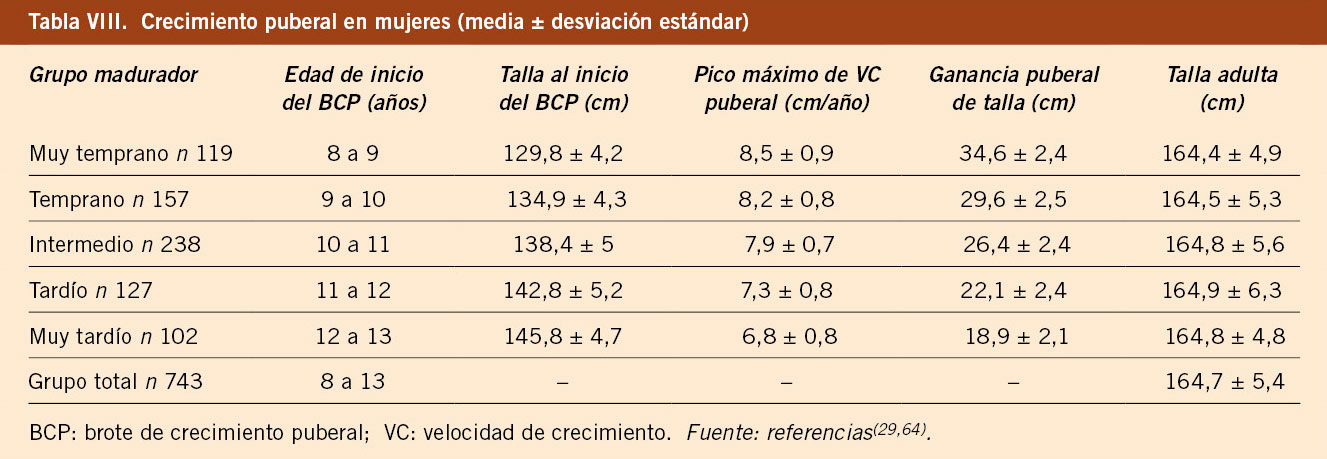
El estudio longitudinal de niños aragoneses tiene una muestra de 332 sujetos (165 varones y 167 mujeres), se inició en el año 1980 y finalizó en el año 2002. Además de valorar parámetros antropométricos de crecimiento, ha elaborado estándares de referencia para edad ósea (métodos de Greulich-Pyle y Tanner-Whitehouse), que ha permitido obtener valores propios de pronóstico de talla adulta, datos de normalidad de densidad mineral ósea, aparición y desarrollo de los caracteres sexuales secundarios (inicio de la pubertad, secuencia del desarrollo puberal, edad de la menarquia, edad de inicio del brote puberal y ganancia de talla durante la pubertad) y datos de normalidad del desarrollo psicomotor e intelectual. En las tablas III, IV, V y VI se presentan los datos de normalidad de la pubertad recogidos en el estudio de Zaragoza en varones y mujeres. El inicio del brote de crecimiento puberal se produce en los varones con un volumen testicular de 4-6 mL y en las mujeres unos 6 meses antes de la aparición del estadio II de Tanner de desarrollo mamario. La edad de inicio puberal muestra una variabilidad de unos 4-5 años entre individuos de condiciones de vida similares (entre 8-13 años en las mujeres y entre 10-14 años en los varones) y este estudio demostró la existencia de cinco patrones de crecimiento puberal(13,27).

Estudio transversal español 2008
En el año 2008 se publicó el estudio transversal español de crecimiento 2008, resultante de la fusión de los estudios transversales realizados en Andalucía, Aragón, Bilbao y Zaragoza. De la fusión de estos estudios nacieron dos estudios complementarios(37).
El primero incluye el crecimiento prenatal, valorando el peso y la longitud de 9.362 recién nacidos (4.884 varones y 4.478 mujeres) de origen caucásico y de gestación única entre 26 y 42 semanas, nacidos en las maternidades de los hospitales Miguel Servet de Zaragoza y Valle Hebrón de Barcelona, entre los años 1999 y 2002(38). Este estudio demuestra el dimorfismo sexual para peso y longitud que es evidente a partir de la semana 30 de edad gestacional, con diferencias significativas en el grupo de RN a término. A su vez, se demuestra una aceleración secular de peso y longitud en el grupo de RN pretérmino respecto a estudios anteriores, hecho que no se observa en los RN a término. Estos datos apoyan la necesidad de disponer de datos actualizados para realizar la clasificación de los RN en función de su antropometría.
Los valores obtenidos son diferentes a los de otros países, por lo que se demuestra la utilidad de disponer de datos nacionales. La heterogeneidad de las poblaciones obstétricas en relación a raza, edad materna, paridad, características antropométricas maternas, estado nutricional y estatus socioeconómico materno pone de relieve las dificultades inherentes que tiene la elaboración de patrones de crecimiento intrauterino normal, sobre todo en recién nacidos pretérmino, en quienes el embarazo no puede considerarse estrictamente normal, al haberse interrumpido prematuramente(5). En este sentido, el estudio transversal español 2008 ha incluido recién nacidos de una misma raza de gestación única y excluyendo gestaciones con riesgo de compromiso del crecimiento prenatal. Este estudio es útil en la clínica diaria para clasificar a los recién nacidos según peso y edad gestacional en: pequeño, adecuado o grande para la edad gestacional. A su vez, son útiles para el seguimiento de peso y de la longitud de los recién nacidos prematuros durante su desarrollo postnatal hasta la edad correspondiente a la 42 semana de su edad gestacional. La actualización periódica de los datos para una determinada población es altamente recomendable(2,41).
Algunos estudios han mostrado que la mejora de la salud materna, relacionada sobre todo con aspectos nutricionales y los hábitos de vida más saludables, ha incrementado progresivamente el tamaño de los fetos y de los recién nacidos a lo largo del tiempo. En los recién nacidos prematuros, la medición del peso al nacimiento para la edad gestacional no se considera tan útil, ya que los recién nacidos prematuros tienen más probabilidad de haber experimentado una restricción del crecimiento prenatal por la propia condición que ha determinado su prematuridad y, por lo tanto, la medición del peso al nacimiento refleja de manera inexacta el crecimiento prenatal de ese feto. Para la valoración del crecimiento prenatal, se utiliza la estimación del peso fetal mediante ecografía. Las curvas de crecimiento fetal permiten identificar casos de retraso de crecimiento intrauterino y existe una buena correlación entre el peso estimado y el peso al nacimiento. La valoración del peso fetal es importante, ya que se ha establecido una correlación entre la presencia de retraso de crecimiento intrauterino y mayor morbimortalidad perinatal. Desde un punto de vista obstétrico, se define como adecuado a la edad gestacional los valores entre el percentil 10 y el percentil 90(42); sin embargo, a nivel pediátrico, en la antropometría realizada postnatalmente, el punto de corte de normalidad se establece entre ± 2 DE para la edad gestacional.
El segundo estudio 2008 lo forma el crecimiento postnatal desde el nacimiento hasta la talla adulta, incluyendo 32.064 sujetos (16.607 varones y 15.457 mujeres) sanos de raza caucásica, de padre de origen español, en cuya muestra participaron individuos de Bilbao, Barcelona, Zaragoza y Andalucía, y que fueron valorados entre los años 2000 y 2004. Este estudio demuestra que no existen diferencias regionales significativas entre las tallas a diferentes edades, incluida la talla adulta(37,39).
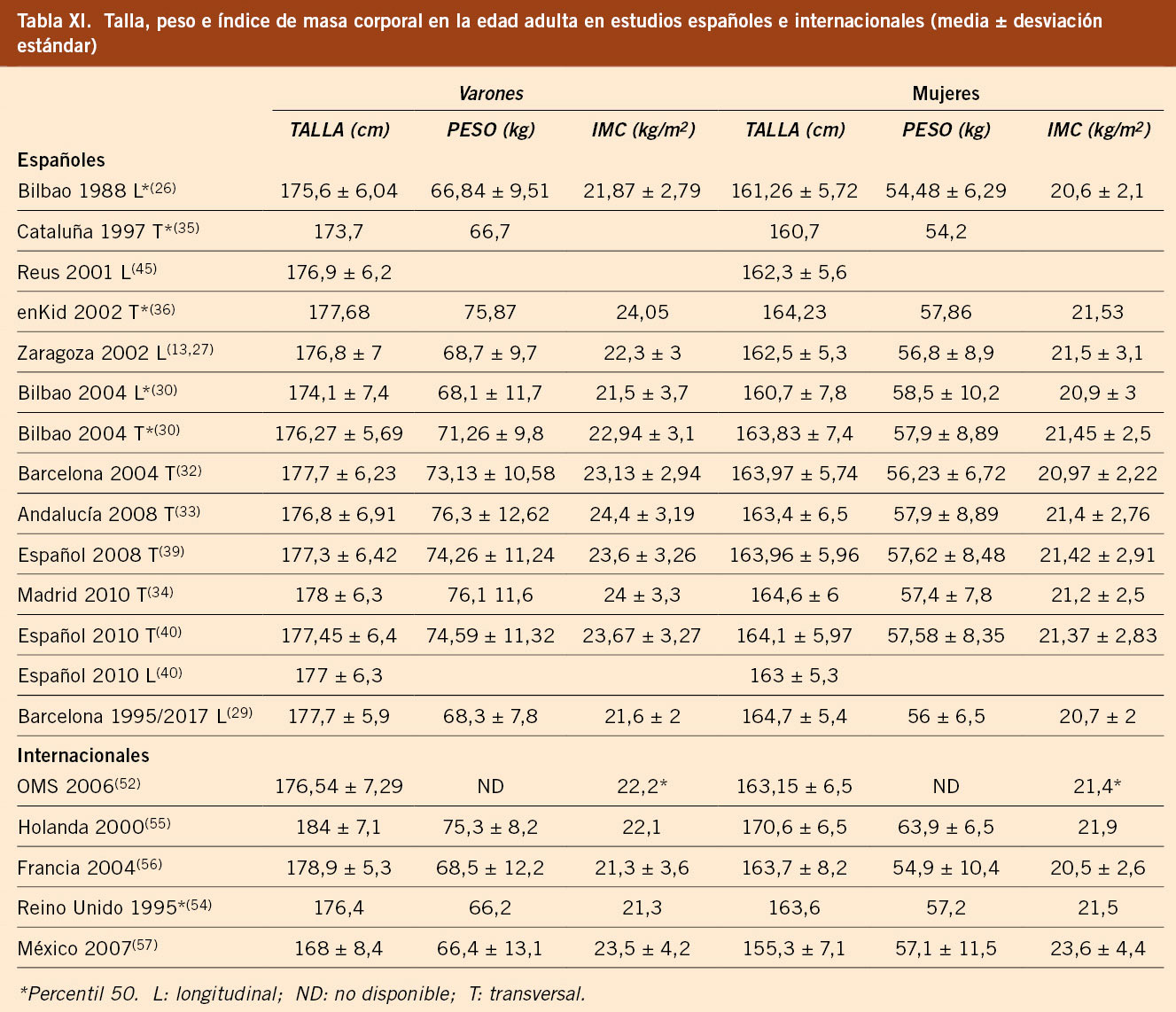
Estudio transversal español 2010
El estudio transversal español de crecimiento 2010 añade la población de Madrid a la muestra del estudio 2008, por lo que se logra una población total de 38.461 sujetos (19.975 varones y 18.486 mujeres), con edades comprendidas entre el nacimiento y los 22 años de edad(40). En este estudio se confirma la aceleración secular del crecimiento comparado con estudios realizados en nuestro país, anteriores a 1988. La talla media adulta en ambos sexos es similar a la establecida en otros estudios realizados en nuestro medio; el percentil 50 se sitúa en torno a 177 cm en varones y a 164 cm en mujeres. Es similar a lo publicado en estudios recientes realizados en países mediterráneos, Reino Unido y Estados Unidos, aunque inferior a la de otros países del centro y norte de Europa, como Holanda, Suecia y Alemania, y superior a la población mexicana bien nutrida. Este estudio refleja la situación de una muestra amplia de la población española, por lo que sus datos son útiles para valorar el crecimiento y la talla adulta de la población española.
Este estudio también demuestra una aceleración secular del IMC en relación a estudios españoles realizados anteriores a 1988, pero únicamente significativos para los valores iguales o superiores al percentil 75. En las mujeres, los valores del percentil 97 del estudio de Bilbao 1988 corresponderían a los valores del percentil 97 (0-5 años) y a los del percentil 95 (5-22 años). En los varones, los valores del percentil 97 del estudio Bilbao 1988 corresponderían a los valores del percentil 95 (0-3 años) y a los del percentil 90 (5-22 años). Esta aceleración del IMC se observa a partir de los 3-5 años de edad en varones y 5-7 años en mujeres y es máxima en ambos sexos durante el desarrollo puberal. Este fenómeno está en relación con la aceleración del ritmo madurativo y con el incremento de sobrepeso en la población. Estos cambios, relacionados con el desarrollo de sobrepeso y obesidad en nuestra población, plantean la cuestión de cuáles son los valores que deben considerarse para definir el sobrepeso y la obesidad en nuestra población actual, teniendo en cuenta la aceleración secular de la talla y los correspondientes incrementos en los valores de peso(40).
Estudio longitudinal español 2010
Los estudios transversales son útiles para valorar el crecimiento en las edades en las que todavía no se ha iniciado el desarrollo puberal y la talla adulta, pero no permiten evaluar el crecimiento durante la pubertad, para lo que es necesario disponer de estudios longitudinales donde se diferencien los niños/as en función del ritmo madurativo. La cronología de la pubertad es extremadamente variable en función del componente genético y ambiental. La edad de inicio del desarrollo puberal y del brote de crecimiento puberal condiciona la intensidad y duración del crecimiento puberal. Cada sujeto tiene su propio tempo o ritmo madurativo para iniciar el desarrollo puberal, y el intervalo de edad en el que se inicia el brote puberal puede oscilar entre los 8 y 13 años en las mujeres y entre los 10 y 15 años en los varones. El brote de crecimiento se puede iniciar a edades tan tempranas como 8-9 años en las mujeres y 10-11 años en los varones o tan tardías como 12-13 años en las mujeres y 14-15 años en los varones; ello obliga a que, en la valoración del crecimiento puberal, sea necesaria la existencia de patrones de referencia diferenciados en función del ritmo de maduración, a diferencia de lo que ocurre en la prepubertad, donde un único patrón es válido para todos los niños/as(43). Ese amplio rango de variabilidad permite agrupar a los sujetos en 5 grupos de maduración en función de la edad a la que se inicia el brote de crecimiento puberal (muy temprano, temprano, intermedio, tardío y muy tardío). En ambos sexos, cada uno de estos grupos tiene diferentes tallas al inicio del brote de crecimiento puberal, diferentes velocidades de crecimiento puberal y diferentes ganancias de talla puberal, es decir, diferentes patrones de crecimiento puberal. Sin embargo, la talla final adulta es la misma, independientemente de la edad de inicio de la pubertad, ya que, si bien los maduradores precoces crecen menos años que los tardíos y tienen menor talla al iniciar la pubertad, presentan un mayor pico de velocidad de crecimiento y mayor ganancia de talla durante la pubertad. Estos datos indican que la edad de inicio del brote puberal no tiene influencia significativa en la talla adulta, tal y como se había sugerido previamente por Tanner y otros autores de nuestro país(44,45).
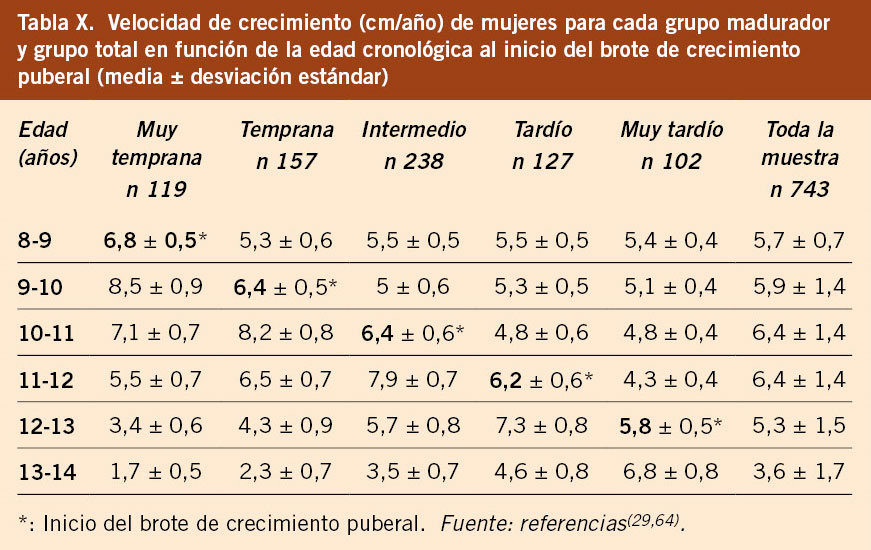
Este estudio, incluido en los estudios integrados de crecimiento 2010, agrupa un total de 540 sujetos (259 varones y 281 mujeres) procedentes de los estudios longitudinales de Barcelona, Bilbao y Zaragoza, realizados en los años 1978-2000, y ha permitido extraer conclusiones relevantes para la valoración del crecimiento durante la pubertad(40). Esta variabilidad, tanto en el inicio (timing) como en la progresión (tempo), es muy importante considerarla. En este sentido, el estudio longitudinal español de crecimiento 1978/2000 aporta información de la media y desviación estándar de la talla para cada intervalo de edad y sexo, desde el nacimiento hasta la talla adulta, de forma separada para cada uno de los 5 grupos de maduración: muy temprano (10-11 años y 8-9 años), temprano (11-12 años y 9-10 años), intermedio (12-13 años y 10-11 años), tardío (13-14 años y 11-12 años) y muy tardío (14-15 años y 12-13 años) en varones y mujeres, respectivamente. También aporta gráficas percentiladas de talla y velocidad de crecimiento, desde el nacimiento hasta talla adulta, de los 3 grupos maduradores más frecuentes: temprano, intermedio y tardío(5,40).
Estudio longitudinal de Barcelona 1995/2017
El estudio longitudinal de crecimiento de Barcelona 1995/2017 incluye 1.453 sujetos de población sana, no obesa ni malnutrida, seguidos desde el nacimiento hasta la edad adulta (743 mujeres y 710 varones). Este estudio confirma los datos previos, con un número de niños/as significativamente superior. Proporciona datos de talla, VC, peso e IMC del grupo total y clasificados en 5 grupos de maduración en función de la edad de inicio del brote puberal y aporta tablas y gráficas de referencia de talla y VC para cada uno de los 5 grupos madurativos. En las tablas VII y VIII se presentan los datos del crecimiento puberal en varones y mujeres, respectivamente, en función del grupo de maduración y como grupo total. En las tablas IX y X se presentan las velocidades de crecimiento en función de la edad cronológica y el grupo de maduración, en varones y mujeres, respectivamente. Estas tablas demuestran que, durante la pubertad, es necesario relacionar la velocidad de crecimiento con el tempo o ritmo madurativo, que depende de la edad de inicio de la pubertad y que se puede categorizar en 5 grupos, y no con la edad cronológica. Los valores de las medias del IMC para la edad en cada sexo son similares a los propuestos por la OMS(5,29).
Otros estudios
En relación con el crecimiento prenatal, se deben mencionar otros estudios realizados en España. Hay que destacar las curvas de crecimiento prenatal de recién nacidos del Hospital de Cruces de Vizcaya, entre 1987 y 1992(46,47). El grupo de Zaragoza realizó un estudio en gestaciones gemelares de peso, longitud y perímetro cefálico entre la 28 y 38 semanas de edad gestacional(48). En España, se disponen de curvas poblacionales de crecimiento en recién nacidos extremadamente prematuros para peso, longitud y perímetro cefálico según edad y sexo, para una edad gestacional ≤28 semanas, provenientes de la base de datos de la Red Nacional SEN 1500(49). Estas gráficas demuestran un dimorfismo sexual desde las 23 semanas de gestación y pueden ser útiles para mejorar la evaluación del crecimiento del prematuro extremo en nuestro país, así como para evaluar el impacto de intervenciones clínicas o de salud pública dirigidas a la optimización del crecimiento fetal.
El fenómeno migratorio, presente en nuestro país, plantea la pregunta de si los datos de los estudios autóctonos en población caucásica son extrapolables a otras poblaciones de origen africano y latinoamericano. El grupo de Barcelona ha realizado un estudio transversal en recién nacidos a término en grupos étnicos de raza no caucásica procedentes de África subsahariana, Marruecos y Sudamérica. Los resultados indican que los valores antropométricos son superiores en los recién nacidos de origen marroquí y sudamericano con respecto a la población local de referencia, mientras que son similares en los nacidos de origen subsahariano. Por ello, este grupo concluye que los patrones antropométricos perinatales de raza caucásica y nacionalidad española no son extrapolables a la hora de evaluar recién nacidos a término de otro grupo étnico(50).
Gráficas de uso en la práctica clínica
La valoración correcta del crecimiento exige comparar los datos del sujeto con estándares obtenidos de una muestra representativa de la población a la que pertenece. Debido a la influencia que tienen la dotación genética y las circunstancias ambientales sobre el crecimiento humano, se recomienda la utilización de estándares nacionales y actualizados. El uso de gráficas no acordes con la población en estudio puede llevar a infradiagnosticar y/o retrasar el diagnóstico de un porcentaje significativo de pacientes con talla baja patológica. La valoración del crecimiento prenatal mediante la longitud y peso al nacimiento se debería realizar mediante las tablas y curvas de referencia del estudio transversal 2008, realizados en recién nacidos de origen caucásico y gestación única. Las gráficas de distancia más adecuadas para la valoración del crecimiento postnatal (talla, peso e IMC) son las del estudio transversal español 2010. En niños con trastornos del crecimiento y en edades puberales, el crecimiento debería valorarse con los patrones propios de cada grupo madurador en lugar de utilizar un único patrón, ya que una incorrecta interpretación del crecimiento puberal puede llevar a realizar diagnósticos erróneos e indicar tratamientos innecesarios. La definición de sobrepeso y obesidad, con los estándares del estudio transversal español 2010, exige una reconsideración de sus puntos de corte.
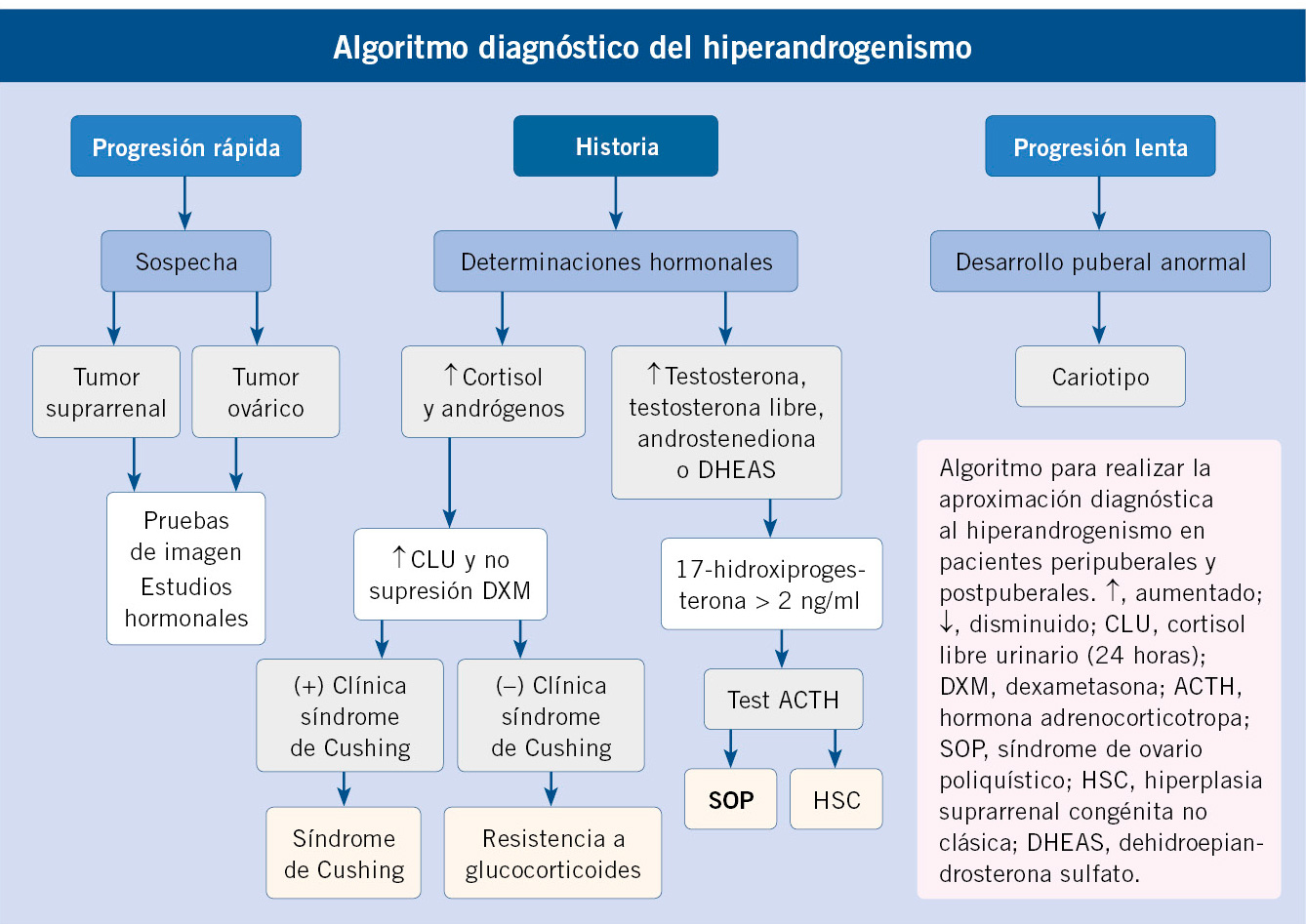
El uso de unas gráficas de crecimiento adecuadas es de enorme importancia en la evaluación de los trastornos del crecimiento para poder identificar niños/as con talla baja patológica elegibles para un potencial tratamiento. La mayoría de los expertos recomienda la utilización de estándares de crecimiento nacionales o locales, debido a la influencia que tienen la dotación genética y las circunstancias ambientales sobre el patrón de crecimiento y el ritmo madurativo. En la figura 1 se presenta un algoritmo de los diferentes estándares de crecimiento para uso en la práctica clínica.
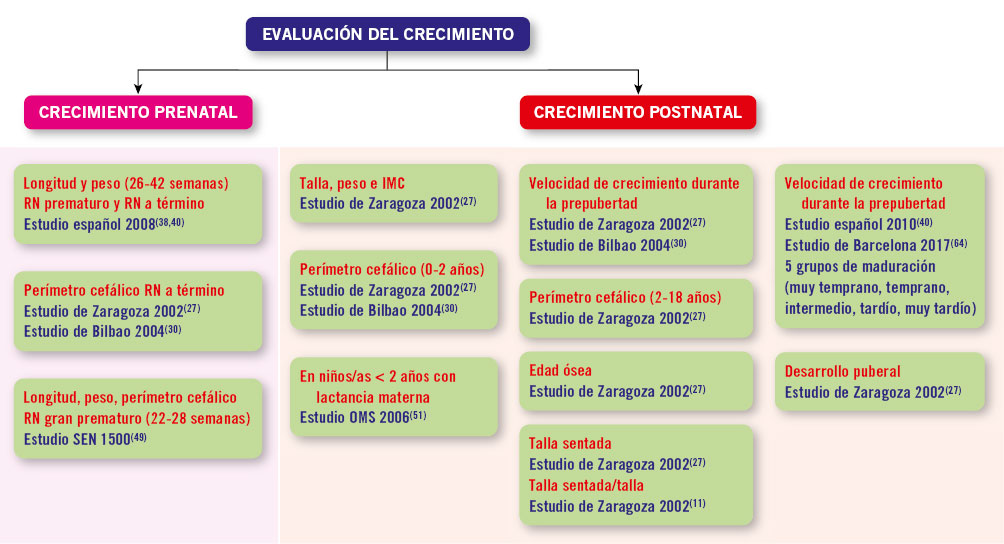
Figura 1. Estándares de crecimiento para uso en la práctica clínica.IMC: índice de masa corporal; RN: recién nacido.
Se ha hipotetizado sobre la conveniencia de utilizar un único estándar o patrón de referencia internacional, dados los movimientos migratorios, pero ello es cuestionado, debido a las diferencias de la talla entre los diferentes países. La OMS realizó un estudio entre 1997 y 2003 en una muestra de 8.440 niños/as, con objeto de elaborar un patrón internacional para la evaluación del crecimiento físico, estado nutricional y el desarrollo motor, desde el nacimiento hasta los 5 años de edad, y combinó un estudio longitudinal desde el nacimiento hasta los 24 meses y otro transversal entre los 18 y 71 meses(51). Fue realizado sobre una muestra de niños/as provenientes de 6 países (Brasil, Gana, India, Noruega, Omán y Estados Unidos) como representación de los cinco continentes. Este estudio publicado, en el año 2006, fue realizado con una orientación novedosa en la forma de construir las gráficas de crecimiento, de manera que reflejen no “cómo crecen”, sino “cómo deberían crecer”. La hipótesis del estudio es que los niños nacidos en diferentes regiones del mundo a los que se les ofrecen unas condiciones de vida óptimas cuentan con un potencial de crecimiento y desarrollo similar, con independencia del país o etnia. Los niños/as que participaron en el estudio fueron criados en condiciones favorables para el crecimiento, como alimentación exclusiva o preferente por lactancia materna en los primeros 4 meses, adecuada nutrición y control de enfermedades, así como ser hijos de madres con hábitos saludables, como no fumar ni durante ni tras el embarazo. Este estudio demuestra que las diferencias en el crecimiento infantil hasta los 5 años dependen más de la nutrición, medio ambiente y atención sociosanitaria que de los factores genéticos o étnicos y, por lo tanto, podrían ser utilizadas para niños de cualquier origen, entorno y tipo de alimentación. Este estudio también presenta factores limitantes, ya que minusvalora las diferencias genéticas entre las distintas etnias, así como la influencia de la nutrición fuera del periodo de lactancia materna. La aportación de este estudio es la disposición de estándares de utilidad internacional que permiten vigilar el crecimiento y desarrollo de los niños en todo el mundo, con independencia de su etnia, nivel socioeconómico y tipo de alimentación; es un instrumento de validez científica para determinar si se satisface el derecho a crecer de todo niño/a. Un patrón internacional que muestra el crecimiento infantil idóneo permite realizar comparaciones entre países para orientar las medidas de promoción de la salud infantil. En muchos países desarrollados existe preocupación por la obesidad en la infancia, pero manejan curvas de crecimiento locales que no descubren el problema hasta después de que el niño haya desarrollado la obesidad. En estos países, el estudio de la OMS puede ser útil para determinar la existencia de problemas de sobrepeso y obesidad antes de que su control o prevención sean demasiado difíciles. La OMS defiende que estos patrones de crecimiento tienen validez mundial y su finalidad es vigilar el crecimiento de todos los niños en todo el mundo, con independencia de su etnia, nivel socioeconómico y tipo de alimentación. Las gráficas de este estudio se completaron hasta los 19 años, con la actualización de datos de estudios previos mediante métodos estadísticos de ajuste(51,52).
Aunque la OMS defiende el uso de unas únicas gráficas de crecimiento universales, diferentes estudios han demostrado diferencias en la talla entre las gráficas de la OMS y las gráficas poblacionales(53). La comparación de los estudios de diferentes países demuestra diferencias en la talla adulta(39,40,54-57). En la tabla XI se presentan la talla adulta, peso e IMC de los principales estudios españoles e internacionales.
La talla del estudio transversal español es superior a la del estudio de la OMS. En un metaanálisis(58) reciente de 55 países, implicando a una población de 11 millones de niños por debajo de 5 años, el 20 % de los valores analizados presentaban un Z-score de la talla 0,5 DE igual o por encima de los valores de la OMS; en general, los niños/as de los estudios europeos tenían valores por encima de 0,5 DE y los de la población del sudeste asiático tenían valores inferiores a -0,5 DE. Un estudio centroeuropeo demostró que, en condiciones patológicas como déficit de GH, síndrome de Turner o talla baja asociada a pequeño para la edad gestacional, un 22 %, 21 % y 32 % de los pacientes, respectivamente, no cumplirían criterios de tratamiento con rhGH, respectivamente, si se utilizan gráficas de la OMS o del CDC (Centers for Disease Control and Prevention), en lugar de los estándares poblacionales(59). Para el diagnóstico del síndrome de Turner antes de los 2 años, el uso de gráficas nacionales se asocia con una mayor sensibilidad (72 %) frente a las gráficas de la OMS (36 %), y lo mismo ocurre para el diagnóstico de la fibrosis quística en la primera infancia, donde el nivel de detección fue mayor al comparar las gráficas del CDC vs. las gráficas de la OMS (26 % vs. 9 %, respectivamente)(60,61). Todas estas observaciones demuestran que el uso de gráficas de la OMS para la evaluación de los trastornos del crecimiento en la primera infancia puede dejar un número significativo de niños/as sin diagnosticar y apoyan la necesidad del uso de gráficas nacionales o poblacionales. La conveniencia del uso de estándares poblacionales y actualizados se pone de manifiesto en un estudio centroeuropeo sobre 3.535 niños/as con enfermedad renal terminal, donde el porcentaje de pacientes con talla baja varía en función del estándar utilizado, siendo del 33 %, 34 %, 40 % y 44 % para los estándares de la OMS, CDC, estándares nacionales no actualizados y estándares nacionales actualizados, respectivamente(62). La aceleración secular del crecimiento y los factores étnicos y ambientales explicarían estas diferencias. Lo ideal es disponer de estándares de referencia actualizados para la población y etnia. El uso de gráficas no apropiadas puede llevar a infradiagnosticar y/o retrasar el diagnóstico de muchos pacientes, con la implicación que ello tiene a nivel del tratamiento y pronóstico de la enfermedad(58,63).
En la clínica, el pediatra debe utilizar gráficas de distancia que representen los valores antropométricos para edad y sexo, así como curvas de velocidad de crecimiento, para su seguimiento. En la valoración del crecimiento y desarrollo se deben utilizar, siempre que sea posible, estándares propios, poblacionales y actualizados, realizados con rigor metodológico. Las consideraciones más relevantes a nivel clínico serían las siguientes(5,7).
• La valoración del crecimiento prenatal mediante la longitud y peso al nacimiento se debería realizar mediante las tablas y curvas de referencia del estudio transversal 2008, realizados en recién nacidos de origen caucásico y gestación única entre 26 y 42 semanas, lo que permite clasificar a los recién nacidos en pequeño (≤2 DE), adecuado (±2 DE) o grande (≥2 DE) para la edad gestacional(37,38).
• Para valorar en un momento determinado la situación de un niño/a y ver si está o no dentro de la normalidad, deben utilizarse las gráficas de distancia, tanto si proceden de un estudio transversal como longitudinal. En la actualidad, las gráficas de distancia más adecuadas para la valoración del crecimiento postnatal (talla, peso e IMC) son las del estudio transversal español 2010 realizado en sujetos sanos, caucásicos de padres españoles desde el nacimiento hasta la edad adulta, que representan a la población actual española, y son las que actualmente se recomiendan(40). Están disponibles en: https://www.aeped.es/noticias/estudios-espanoles-crecimiento-2010. Estas tablas, al carecer de curvas de VC, tienen las dificultades inherentes a cualquier estudio transversal para realizar el seguimiento de un niño. Para realizar el seguimiento individual de un niño/a, hay que utilizar las curvas de VC obtenidas de estudios longitudinales. En este sentido, las tablas del estudio longitudinal Bilbao 2002(30) o las del estudio de Zaragoza 2002(13,27) son las más relevantes para realizar el seguimiento de la VC de un niño de raza caucásica de padres de origen español durante la prepubertad.
• La evaluación del crecimiento (talla y VC) durante la pubertad se debe realizar bien con las gráficas y tablas del estudio longitudinal español 2010, que ha permitido elaborar gráficas en función del tempo madurativo de los niños/as, sea temprano, intermedio o tardío(40) o bien con las gráficas y tablas del estudio longitudinal Barcelona 2017 que aporta información para cada uno de los 5 grupos de maduración (muy temprano, temprano, intermedio, tardío y muy tardío)(29,64). Esta observación tiene importancia clínica, ya que, en los niños alejados del patrón intermedio de desarrollo puberal, es decir, que presentan una maduración/pubertad adelantada o retrasada, situación muy frecuente en la clínica, deben ser valorados con los estándares de referencia acordes a su ritmo madurativo, ya que, si son valorados con los estándares de crecimiento habituales, sus datos de crecimiento serán interpretados de forma errónea, sobre todo para los maduradores muy tempranos y muy tardíos. Esto es especialmente necesario para niños/as con tallas bajas y/o retrasos de crecimiento en edades peripuberales. Es, pues, muy útil poder disponer de tablas y gráficas diferenciadas según el grupo madurador puberal, ya que cada grupo madurador tiene su propio patrón de crecimiento puberal. En las tablas IX y X se observa cómo la velocidad de crecimiento para una misma edad es diferente en función del grupo madurativo. A continuación, se comentan dos situaciones clínicas.
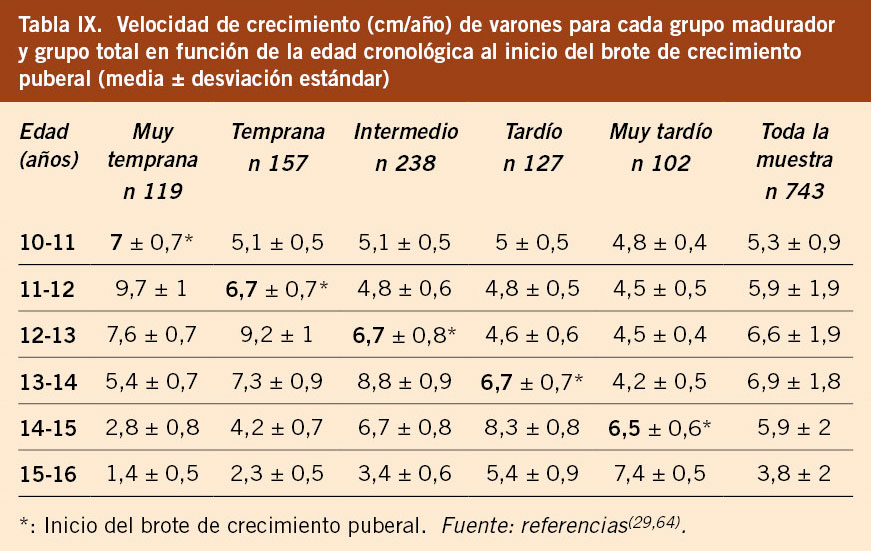
– Si un niño A inicia la pubertad a los 14 años (testes 6 mL), su VC debe ser en ese momento en torno a 5 cm/año y, por lo tanto, le correspondería hacer el pico de máxima velocidad de crecimiento puberal, es decir, el estirón puberal, a los 15 años, alcanzando una VC de en torno a 7,5 cm/año. Si ese mismo niño acude a la consulta un año antes, es decir, a los 13 años, y está creciendo a una velocidad de 3,5-4 cm/año, la familia puede interpretar que es una velocidad de crecimiento disminuida, porque a esa edad la mayoría de los niños están realizando el estirón puberal y se esperaría una velocidad de crecimiento muy superior. Para hacer una correcta valoración, el pediatra debe explorar la pubertad y, al comprobar que está en situación infantil (testes de 2-3 mL), constata que está creciendo a una VC normal, ya que todavía no ha iniciado la pubertad (grupo muy tardío), e informar a la familia que está creciendo como corresponde para su estadio prepuberal. En ese momento, se debe tranquilizar a la familia, informando de la normalidad de la situación y hacer un seguimiento clínico, pero no iniciar pruebas complementarias.
– Igualmente, en el polo opuesto, una niña B ha iniciado la pubertad a los 9,5 años y, como grupo temprano, a esa edad su VC debe situarse en torno a 6 cm/año y hacer el pico de máxima velocidad de crecimiento o estirón puberal en el año siguiente, entre los 10 y los 11 años, alcanzando una VC de 8 cm/año. Si esta niña a los 10,5 años crece a 4,5 cm/año, es evidente que estaría creciendo por debajo de lo que le corresponde por pubertad, por lo que debería ser valorada por el pediatra, quien deberá explicar a la familia que su VC no es normal y que hay que hacer una evaluación para estudiar por qué no está haciendo el estirón puberal.
Ambos casos se sitúan en el rango de la variabilidad normal de la edad de inicio de la pubertad, pero tienen un patrón madurativo muy diferente y su crecimiento debe ser valorado en relación a su estadio de maduración puberal y no en relación a su edad cronológica, para no realizar pruebas innecesarias, en el caso del niño A, o interpretar como normal una situación que no lo es, como el caso de la niña B. Por lo tanto, en el periodo puberal, se debe valorar la VC en relación al tipo de maduración puberal y no en relación a la edad cronológica.
• En la práctica clínica y, sobre todo, al valorar a un niño con talla baja, el crecimiento puberal debería valorarse con los patrones propios de cada grupo madurador en lugar de utilizar un único patrón, ya que una incorrecta interpretación del crecimiento puberal puede llevar a realizar diagnósticos erróneos e indicar tratamientos innecesarios. Este tipo de valoración del crecimiento durante la pubertad en niños/as con retraso de crecimiento asociado se debería realizar por el pediatra endocrino especializado, ya que son decisiones que pueden tener implicaciones diagnósticas y terapéuticas importantes(29,40,64).
• Las gráficas de la OMS serían idóneas para su uso en lactantes alimentados de manera exclusiva o preferentemente con lactancia materna y durante los primeros 2 años de vida, especialmente en lo que se refiere a ganancia ponderal. Ello estaría indicado en casos concretos con lactancia materna y un patrón de crecimiento dudoso, en donde las gráficas de la OMS permiten una mejor interpretación de las ganancias de peso y talla y no sobreestimar en estas circunstancias los retrasos de crecimiento. Su aplicabilidad iría disminuyendo al avanzar en edad, en la medida en que la lactancia materna se va perdiendo como elemento y el componente genético se va haciendo más manifiesto, hecho que ocurre a partir de los 6-12 meses, desapareciendo de esta manera la homogeneidad ambiental y nutricional.
• Un problema que se plantea con frecuencia en la práctica clínica es qué curvas de referencia utilizar en el caso de niños/as inmigrantes o adoptados de países en desarrollo. En un reciente consenso internacional se ha recomendado utilizar gráficas de la OMS para niños/as por debajo de los 5 años de edad, para edades superiores, las gráficas del país de origen, y para los hijos de estos, gráficas del país de adopción(65). En España, los estudios de Barcelona encontraron algunas diferencias entre la población caucásica autóctona y los hijos de inmigrantes de origen magrebí, subsahariano y de América Central y del Sur, pero solamente para recién nacidos a término, diferencias que no se han mantenido en edades posteriores(2,5).
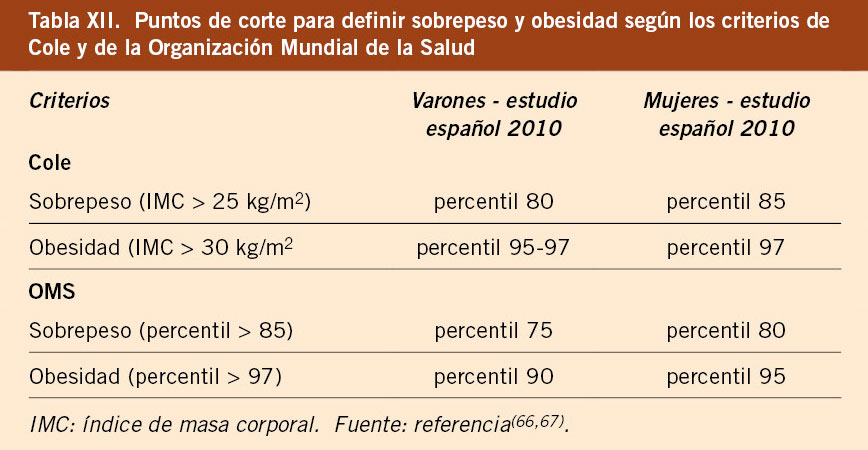
• En la actualidad, la detección precoz del sobrepeso y la obesidad es uno de los retos más importantes en Pediatría. Los estudios transversales españoles 2008 y 2010 han puesto de manifiesto el aumento de su prevalencia con respecto a años anteriores(37,39,40). Estas diferencias en el IMC pueden deberse tanto al incremento del sobrepeso y obesidad en la población española como a la diferente metodología y selección de la muestra. Los valores del IMC del estudio de Bilbao 1988 se encuentran en una posición intermedia entre ambos estudios, el de la OMS y el estudio transversal español 2010. Este argumento es utilizado por aquellos que defienden el uso de las gráficas de la OMS, ya que la muestra de este estudio presentaba menor prevalencia de sobrepeso y obesidad, determinado por la selección de la muestra y la metodología utilizada. Se puede hacer una estimación de la definición de sobrepeso y obesidad usando las gráficas de crecimiento españolas del estudio español 2010, aplicando los criterios antropométricos de Cole, estableciendo como puntos de corte de sobrepeso y obesidad aquellos percentiles que se corresponden con los valores 25 kg/m2 y 30 kg/m2, respectivamente, en la edad adulta. Los valores utilizados para definir sobrepeso y obesidad no son siempre coincidentes y existen diferencias entre los valores propuestos por Cole en el año 2000(66) y los valores propuestos por la OMS en el año 2007(67); por lo tanto, los datos que se obtengan van a variar ampliamente según los criterios utilizados. A su vez, hay que considerar que los estudios de Cole y de la OMS están elaborados con datos procedentes de estudios realizados hace más de 20 años, por lo que no tienen en cuenta la aceleración secular en el inicio del desarrollo puberal y sus datos serían cuestionables en esta época de la vida. En la tabla XII se presentan los puntos de corte para la definición de sobrepeso y obesidad para los valores del estudio español de crecimiento 2010, según los criterios de sobrepeso y obesidad de Cole(66) y de la OMS(67). En la clínica, además de definir la presencia o no de sobrepeso y obesidad, concepto clínico que requiere una adecuada interpretación del IMC, es útil valorar el grado de obesidad, ya que ello se correlaciona con una mayor morbilidad, así como para la valoración de los efectos del tratamiento. En este sentido, el estudio español de crecimiento 2010 permite calcular para cada niño su grado de obesidad, al expresarlo mediante Z-score o desviación estándar de la media.
En resumen, en estos momentos se dispone en nuestro país de estudios transversales actualizados para realizar la antropometría neonatal de los recién nacidos prematuros y a término y el crecimiento postnatal de niños y adolescentes. También se dispone de estudios longitudinales que permiten evaluar el crecimiento y la maduración puberal, diferenciados por grupos maduradores de 1 año de intervalo. En este sentido, estudios transversales y longitudinales muestran su complementariedad. A su vez, se dispone de estudios que permiten la valoración de otros parámetros clínicos complementarios, como perímetro cefálico, segmentos corporales y maduración ósea. La auxología en España ha tenido un desarrollo muy importante en los últimos 35-40 años y ello ha dado como frutos los diferentes estudios de crecimiento. Todo ello ha sido posible gracias al esfuerzo de muchas personas, pero es obligado destacar el liderazgo del grupo de Bilbao (Prof. M. Hernández y la Dra. B. Sobradillo), Barcelona (Prof. A. Carrascosa) y Zaragoza (Dr. A. Ferrández), y sirva este artículo como reconocimiento.
Conflicto de intereses
No hay conflicto de interés en la elaboración del presente manuscrito ni fuente de financiación.
Bibliografía
1. Carrascosa A, Fernández JM, Ferrández A, López-Siguero JP, López D, Sánchez E, et al. Estudios Españoles de Crecimiento 2010. Rev Esp Endocrinol Pediatr. 2011; 2: 59-62. Disponible en: https://doi.org/10.3266/RevEspEndocrinologPediatr.pre2011.Apr.52.
2. Sánchez González E, Carrascosa Lezcano A, Fernández García JM, Ferrández Longás A, López de Lara D, López Siguero JP. Estudios españoles de crecimiento: situación actual, utilidad y recomendaciones de uso. An Pediatr (Barc). 2011; 74: 193.e1-e16.
3. Pozo J. Valoración auxológica del crecimiento. Regreso a las bases. Pediatr Integral. 2011; 6: 590-8. Disponible en: https://www.pediatriaintegral.es/wp-content/uploads/2012/03/Pediatria-Integral-XV-6.pdf.
4. Hermanussen M. Auxology: an update. Horm Res Pediatr. 2010; 74: 153-64.
5. Carrascosa A, Fernández JM, Fernández C, Ferrández A, López-Siguero JP, Sánchez E, et al. Estudios españoles de crecimiento 2008. Nuevos patrones antropométricos. Endocrinol Nutr. 2008; 55: 484-506.
6. Carrascosa A. Aceleración secular de crecimiento en España. Estudios Españoles de Crecimiento 2010. Población autóctona y población inmigrante. Endocrinol Nutr. 2014; 61: 229-33.
7. Argente Oliver J. Curvas de crecimiento españolas. En: Argente Oliver J, Martos Moreno G, Soriano Guillén L, eds. Manual de Endocrinología Pediátrica, 3ª edición. Madrid. Ergon; 2023: p. 455-75.
8. Bogin B, Varela-Silva MI. Leg length, body proportion, and health: a review with a note on beauty. Int J Environ Res Public Health. 2010; 7: 1047-75.
9. Hawkes CP, Mostoufi-Moab S, McCormack SE, Grimberg A, Zemel BS. Sitting height to standing height ratio references charts for children in the United States. J Pediatr. 2020; 226: 221-7.e15. Disponible en: https://doi.org/10.1016/j.jpeds.2020.06.051.
10. Hawkes CP, Mostoufi-Moab S, McCormack SE, Grimberg A, Zemel BS. Leg length and sitting height reference data and charts for children in the United States. Data Brief. 2020: 32: 106131. Disponible en: https://doi.org/10.1016/j.dib.2020.106131.
11. De Arriba Muñoz A, Domínguez Cajal M, Rueda Caballero C, Labarta Aizpún JI, Mayayo Dehesa E, Ferrández Longás A. Sitting/standing height ratio in Spanish children from birth to adulthood. Arch Argent Pediatr. 2013; 111: 309-14.
12. Fredriks AM, Van Buuren S, Ven Heel WJM, Dijkman-Neerincx RHM, Verloove-Vanhorick SP, Wit JM. Nationwide age references for sitting height, leg length and sitting height/height ratio, and their diagnostic value for disproportionate growth disorders. Arch Dis Child. 2005; 90: 807-12.
13. Ferrández A, Baguer L, Labarta JI, Labena C, Mayayo E, Puga B, et al. Estudio longitudinal de niños españoles normales desde el nacimiento hasta la edad adulta. Datos antropométricos, puberales, radiológicos e intelectuales. Edita Fundación Andrea Prader. Zaragoza. 2005.
14. Pozo J, Argente J. Crecimiento: valoración auxológica. En: Argente J, Carrascosa A, Gracias R, Hierro F, eds. Tratado de Endocrinología Pediátrica y de la Adolescencia, 2ª edición. Madrid. Doyma; 2000. p. 177-201.
15. Tanner JM, Goldstein H, Whitehouse RH. Standards for children´s height ages 2-9 years allowing for heights of parents. Arch Dis Child. 1970; 45: 316-23.
16. Van Dommelen P, Schönbeck Y, Van Buuren S. A simple calculation of the target height. Arch Dis Child. 2012; 97: 182.
17. Hermanussen M, Cole TJ. The calculation of target height reconsidered. Horm Res. 2003; 59: 180-3.
18. Scherdel P, Salaün JF, Robberecht-Riquet MN, Reali L, Páll G, Jäger-Roman E, et al. Growth monitoring: a survey of current practices of primary care paediatricians in Europe. PLOS One. 2013; 8: e70871.
19. Groote FK, Oostdijk W, Muinck Keizer-Schrama SMPF, Dekker FW, Verkerk PH, Wit JM. Growth monitoring and diagnostic work-up of short stature: an international inventorization. J Pediatr Endocrinol Metab. 2005; 18: 1031-8.
20. Fayter D, Nixon J, Hartley S, Rithalia A, Butler G, Rudolf M, et al. Effectiveness and cost-effectiveness of height-screening programmes during primary school yars: a systematic review. Arch Dis Child. 2008; 93: 278-84.
21. Scherdel P, Dunkel L, Van Dommelen P, Goulet O, Salün JF, Bauner R, et al. Growth monitoring as an early detection tool: a systematic review. Lancet Diabetes Endocrinol. 2016; 4: 447-56.
22. Stalman SE, Hellinga I, Van Dommelen P, Hennekam RCM, Saari A, Sankilampi U, et al. Application of the Dutch, Finnish and British screening guidelines in a cohort of children with growth failure. Horm Res Pediatr. 2015; 84: 376-82.
23. Savage MO, Backeljauw PF, Calzada R, Cianfarani S, Dunkel L, Koledova E, et al. Early detection, referral, investigation, and diagnosis of children with growth disorders. Horm Res Pediatr. 2016; 85: 325-32.
24. Hall JG, Allanson JE, Gripp KW, Slavotinek AM. Syndrome-specific growth charts. Am J Med Genet Part A. 2012; 158A: 2645-6.
25. Carrascosa A, Mesa J. Estudio Longitudinal de Crecimiento Barcelona 1995-2017. Endocrinol Diabetes Nutr. 2018; 65: 311-3.
26. Hernández M, Castellet J, Narvalza JL, Rincón JM, Ruiz E, Sánchez E, et al. Curvas y tablas de crecimiento. Fundación Faustino Orbegozo. Bilbao. 1988.
27. Ferrández A, Baguer L, Labarta JI, Labena C, Mayayo E, Puga B, et al. Longitudinal study of normal Spanish children from birth to adulthood: anthropometric, pubertal, radiological and intellectual data. Pediatr Endocr Rev. 2005; 2: 423-642.
28. Carrascosa A, Audi L, Bosch-Castañé J, Gussinyé M, Yeste D, Albisu MA, et al. Influencia de la edad de inicio del brote de crecimiento puberal en la talla adulta. Med Clin (Barc). 2008; 130: 645-9.
29. Carrascosa A, Yeste D, Moreno-Galdó A, Gussinyé M, Ferrández A, Clemente M, et al. Crecimiento puberal de 1453 niños sanos según la edad de inicio de la pubertad. Estudio longitudinal de Barcelona. An Pediatr (Barc). 2018; 89: 144-52.
30. Sobradillo B, Aguirre A, Aresti U, Bilbao A, Fernández-Ramos C, Lizárraga A, et al. Curvas y tablas de crecimiento. Estudio longitudinal y transversal. Fundación Faustino Orbegozo. 2004.
31. Fernández C, Lorenzo H, Vrotsou K, Aresti U, Rica I, Sánchez E. Estudio de crecimiento de Bilbao. Curvas y tablas de crecimiento. Estudio transversal. Fundación Faustino Orbegozo. 2011.
32. Carrascosa A, Yeste D, Copil A, Gussinyé M. Aceleración secular del crecimiento. Valores de peso, talla e índice de masa corporal en niños, adolescentes y adultos jóvenes de la población de Barcelona. Med Clin (Barc). 2004; 123: 445-51.
33. López Siguero JP, Fernández García JM, Luna Castillo JD, Moreno Molina JA, Ruiz Cosano C, Jurado Ortiz A. Cross-sectional study of height and weight in the population of Andalucía from age 3 to adulthood. BMC Endocr Disorders. 2008; 8: S1. Disponible en: https://doi.org/10.1186/1472-6823-8-S1-S1.
34. López de Lara D, Santiago P, Tapia M, Rodríguez MD, Gracia R, Carrascosa A. Valoración del peso, talla, IMC en niños, adolescentes y adultos jóvenes de la Comunidad Autónoma de Madrid. Comparación con el estudio español de crecimiento 2008. An Pediatr (Barc). 2010; 73: 305-19.
35. De la Puente ML, Canela J, Álvarez L, Salleras L, Vicens Calvet E. Cross-sectional growth study of the child and adolescent population of Catalonia (Spain). Ann Hum Biol. 1997; 24: 435-52.
36. Serra A, Aranceta J, Pérez C, Moreno B, Tojo R, Delgado A, et al. Dossier de consenso. Curvas de referencia para la tipificación ponderal. Población infantil y juvenil. 2002.
37. Carrascosa A, Fernández JM, Ferrández A, López-Siguero JP, Sánchez E. Estudio transversal español de crecimiento 2008. Barcelona. Pfizer. Ed. Hercu. Barcelona. 2008.
38. Carrascosa Lezcano A, Ferrández Longás A, Yeste Fernández D, García-Dihinx J, Romo Montejo A, Copil Copil A, et al. Estudio transversal español de crecimiento 2008. Parte I: Valores de peso y longitud en recién nacidos de 26-42 semanas de edad gestacional. An Pediatr (Barc). 2008; 68: 544-51.
39. Carrascosa Lezcano A, Fernández García JM, Fernández Ramos C, Ferrández Longás A, López Siguero JP, Sánchez González E, et al. Estudio transversal español de crecimiento 2008. Parte II: Valores de talla, peso e índice de masa corporal desde el nacimiento a la talla adulta. An Pediatr (Barc). 2008; 68: 552-69.
40. Carrascosa A, Fernández García JM, Ferrández Longás A, López-Siguero JP, Sánchez González E. Estudios españoles de crecimiento 2010. 2010.
41. Kramer MS, Morin I, Yang H, Platt RW, Usher R, McNamara H, et al. Why are babies getting bigger? Temporal trends in fetal growth and its determinants. J Pediatr. 2002; 141: 538-42.
42. Grantz KL. Fetal growth curves: is there a universal reference? Obstet Gynecol Clin North Am. 2021; 48: 281-96.
43. Ferrández A, Carrascosa A, Audi L, Baguer L, Rueda C, Bosch-Castané J, et al. Longitudinal puberty growth according to age at pubertal growth spurt onset: data from a Spanish study including 458 children (223 boys and 235 girls). J Pediatr Endocrinol Metab. 2009; 22: 715-26.
44. Tanner JM, Davies PSW. Clinical longitudinal standards for height and height velocity for North American children. J Pediatr. 1985; 107: 4535-52.
45. Vizmanos B, Marti-Henneberg C, Civillé R, Moreno A, Fernández-Ballart J. Age of pubertal onset affects the intensity and duration of pubertal growth peak but no final height. Am J Hum Biol. 2001; 13: 409-16.
46. Delgado Beltrán P, Melchor Marcos JC, Rodríguez-Alarcón Gómez J, Linares Uribe A, Fernández-Llebrez del Rey L, Barbazán Cortés MJ, et al. Curvas de desarrollo fetal de los recién nacidos en el Hospital de Cruces (Vizcaya). I. Peso. An Esp Pediatr. 1996; 44: 50-4.
47. Delgado Beltrán P, Melchor Marcos JC, Rodríguez-Alarcón Gómez J, Linares Uribe A, Fernández-Llebrez del Rey L, Barbazán Cortés MJ, et al. Curvas de desarrollo fetal de los recién nacidos en el Hospital de Cruces (Vizcaya). II. Longitud, perímetro cefálico e índice ponderal. An Esp Pediatr. 1996; 44: 54-9.
48. García Dihinx J, Romo Montejo A, Ferrández-Longás A. Estándares neonatales. Hospital Infantil Miguel Servet. Zaragoza. 2001. En: Patrones de crecimiento y desarrollo en España. Atlas de Gráficas y Tablas. Eds. Carrascosa A, Delgado Beltrán A, Ferrández Longás A, García Dihinx J, Hernández Rodríguez M, Romo A, A, Sobradillo B. Ergon. Madrid. 2004; 21-48.
49. García-Muñoz F, García-Alix Pérez A, Figueras Aloy J, Saavedra Santana P y Grupo español SEN 1500. Nuevas curvas poblacionales de crecimiento en recién nacidos extremadamente prematuros españoles. An Pediatr (Barc). 2014; 81: 107-14.
50. Copil A, Yeste D, Teixidó R, Macia J, Santana S, Almar J, et al. Patrones antropométricos de los recién nacidos a término de grupos étnicos de raza no caucásica procedentes de África subsahariana, Marruecos y Sudamérica nacidos en Cataluña. An Pediatr (Barc). 2006; 65: 454-60.
51. WHO Multicentre Growth Reference Study Group, Onis MD. WHO Child Growth Standards based on length/height, weight and age. Acta Pediatr. 2006; 95: 76-85.
52. World Health Organization 2008. The WHO child growth standards. Disponible en: https://www.who.int/childgrowth/en/.
53. Centers for Diseases Control. 2000 CDC growth charts: United States. Disponible en: http://www.cdc.gov/growthcharts/.
54. Freeman JV, Cole TJ, Chinn S, Jones PRM, White EM, Preece MA. Cross sectional stature and weight reference curves for the UK. Arch Dis Child. 1995; 73: 17-24.
55. Fredriks AM, Buuren SV, Burgmeijer RJF. Continuing positive secular growth change in the Netherlands 1955-1997. Pediatr Res. 2000; 47: 316-23.
56. Deheeger M, Rolland-Cachera MF. Etude longitudinale de la croissance d´enfants parisiens suivis de l´age de 10 mois a 18 ans. Arch Pediatr. 2004; 11: 1130-44.
57. Del Río Navarro BE, Velázquez Monroy O, Santos Preciado JI, Lara Esqueda A, Berber A, Lorendo Abdala A, et al. Mexican anthropometric percentiles for ages 10-18. Eur J Clin Nutr. 2007; 61: 963-75.
58. Natale V, Rajagopalan A. Worldwide variation in human growth and the World Health Organization growth standards: a systematic review. BMJ Open. 2014; 4: e003735. Disponible en: https://doi.org/10.1136/bmjopen-2013-003735.
59. Christesen HT, Pedersen BT, Pournara E, Petit IO, Juliusson PB. Short stature: comparison of WHO and National Growth Standards / References for height. PLOS One. 2016. Disponible en: https://doi.org/10.1371/journal.pone.0157277.
60. Saari A, Sankilampi U, Dunkel L. Multiethnic WHO growth charts may not be optimal in the screening of disorders affecting height: Turner syndrome as a model. JAMA Pediatr. 2013; 167: 194-5.
61. Machogu E, Cao Y, Miller T, Simpson P, Levy H, Quintero D, et al. Comparison of WHO and CDC growth charts in predicting pulmonary outcomes in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2015; 60: 378-83.
62. Bonthuis M, Van Stralen KJ, Verrina E, Edefonti A, Molchanava E, Hokken-Koelega ACS, et al. Use of national and international growth charts for studying height in European children: development of up-to-date European height-for-age charts. PLOS One. 2012; 7: e42506. Disponible en: https://doi.org/10.1371/journal.pone.0042506.
63. Patel R, Dave C, Agarwal N, Mendpara H, Shukla R, Bajpai A. Predictive value of IAP 2015, IAP 2007 and WHO Growth Charts in identifying pathological short stature. Indian Pediatr. 2021; 15: 149-51.
64. Carrascosa A. Millennials´ Growth. Estudio longitudinal de crecimiento. Barcelona 1995-2017. Disponible en: https://doi.org/10.3266/Pulso.ed.MillennialsGrowth.2018.ESP.
65. Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Pediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008; 93: 4210-7.
66. Cole TJ, Bellizzi MC, Flegal KM, Dietz WH. Establishing a standard definition for child overweight and obesity worldwide: international survey. BMJ. 2000; 320: 1240-3.
67. De Oliveira MH, Dos Santos Pereira, Sousa Melo D, Cumpian Silva J, Lisboa Conde W. Accuracy of international growth charts to assess nutritional status in children and adolescents: a systematic review. Rev Paul Pediatr. 2022; 40: e2021016.
Bibliografía comentada
– Sánchez González E, Carrascosa Lezcano A, Fernández García JM, Ferrández Longás A, López de Lara D, López Siguero JP. Estudios españoles de crecimiento: situación actual, utilidad y recomendaciones de uso. An Pediatr (Barc). 2011; 74: 193.e1-e16.
Artículo excelente de revisión sobre los estudios españoles de crecimiento donde, además, se realiza una comparación con el estudio de la OMS y otros estudios internacionales. Es de obligada lectura, ya que aborda de manera muy acertada los estudios realizados, el valor que aporta cada uno de ellos y ofrece conocimientos sobre auxología y metodología que son muy necesarios para el clínico. Revisa también el fenómeno de la aceleración secular y cómo se deberían realizar los estudios de crecimiento en el futuro.
– Carrascosa A, Fernández JM, Fernández C, Ferrández A, López-Siguero JP, Sánchez E, et al. Estudios españoles de crecimiento 2008. Nuevos patrones antropométricos. Endocrinol Nutr. 2008; 55: 484-506.
Artículo de revisión sobre los estudios de crecimiento españoles realizados en el 2008. Presenta el estudio transversal realizado en recién nacidos entre 26-42 semanas y el estudio transversal de crecimiento postnatal hasta talla adulta. Demuestra la no existencia de diferencias entre las regiones de España y la aceleración secular del IMC, por lo que plantea la necesidad de reconsiderar los puntos de corte que se deben usar para definir sobrepeso y obesidad. Además de ello, realiza una muy acertada comparación de dicho estudio con otros estudios nacionales e internacionales, tanto longitudinales como transversales. Artículo muy bien redactado, lectura fácil y muy docente, con tablas y figuras muy ilustrativas. Es una revisión de obligada lectura.
– Scherdel P, Dunkel L, Van Dommelen P, Goulet O, Salün JF, Bauner R, et al. Growth monitoring as an early detection tool: a systematic review. Lancet Diabetes Endocrinol. 2016; 4: 447-56.
Excelente revisión sobre las guías y protocolos de decisión clínica que existen en la literatura para distinguir un crecimiento normal de un crecimiento patológico y expone las dificultades que hay para identificar auxológicamente una talla baja patológica. Pone de manifiesto la existencia de una variabilidad importante en la práctica clínica y presenta los criterios auxológicos que se utilizan con más frecuencia. También revisa el tema de las gráficas de crecimiento que se utilizan y sus limitaciones.
– Carrascosa Lezcano A, Ferrández Longás A, Yeste Fernández D, García-Dihinx J, Romo Montejo A, Copil Copil A, et al. Estudio transversal español de crecimiento 2008. Parte I: Valores de peso y longitud en recién nacidos de 26-42 semanas de edad gestacional. An Pediatr (Barc). 2008; 68: 544-51.
Artículo original que presenta el estudio transversal 2008 para la valoración del peso y longitud al nacer en 9.362 recién nacidos de gestación única, entre 26-42 semanas de raza caucásica. Se demuestra la existencia de un dimorfismo sexual y se presentan los datos en forma de tablas y gráficas. La discusión es muy formativa.
– Carrascosa Lezcano A, Fernández García JM, Fernández Ramos C, Ferrández Longás A, López Siguero JP, Sánchez González E, et al. Estudio transversal español de crecimiento 2008. Parte II: Valores de talla, peso e índice de masa corporal desde el nacimiento a la talla adulta. An Pediatr (Barc). 2008; 68: 552-69.
Artículo original que presenta el estudio transversal 2008 de crecimiento postnatal (longitud/talla, peso, IMC), desde el nacimiento hasta talla adulta, con la integración de los estudios de Andalucía, Barcelona, Bilbao y Zaragoza, con una muestra total de 32.064 sujetos. Con respecto a estudios españoles previos, se demuestra una aceleración secular de talla y peso, con un incremento del IMC en los percentiles superiores. Este estudio demuestra la necesidad de actualizar periódicamente los estándares de referencia utilizados en la práctica diaria. La discusión es amplia y compara los datos del estudio español con otros estudios internacionales.
– Carrascosa A, Fernández García JM, Ferrández Longás A, López-Siguero JP, Sánchez González E. Estudios españoles de crecimiento 2010. 2010.
Es una monografía muy útil para el clínico y para tenerla en la consulta, ya que presenta, de manera muy clara, las tablas y las gráficas del estudio transversal 2010 de crecimiento postnatal (longitud/talla, peso, IMC), de nacimiento a talla adulta, que integra el estudio 2008 y el estudio de la Comunidad de Madrid, lo que supone un total de 38.461 sujetos. Igualmente, presenta los datos del estudio longitudinal 2010 y la diferenciación del crecimiento puberal en 5 grupos madurativos (muy temprano, temprano, intermedio, tardío y muy tardío). En esta monografía también se presentan las tablas y las gráficas del estudio 2008 de peso y longitud al nacimiento. Estos estudios son los estándares de referencia actualmente recomendados.
– Ferrández A, Carrascosa A, Audi L, Baguer L, Rueda C, Bosch-Castané J, et al. Longitudinal puberty growth according to age at pubertal growth spurt onset: data from a Spanish study including 458 children (223 boys and 235 girls). J Pediatr Endocrinol Metab. 2009; 22: 715-26.
Artículo original de los grupos de Zaragoza y Barcelona, que presenta el estudio longitudinal de 458 niños sanos desde el nacimiento a talla adulta. La edad de inicio del brote de crecimiento puberal oscila en las mujeres entre 8 y 13 años y en los varones entre 10 y 15 años, de modo que divide a la población en estudio en 5 grupos madurativos con intervalos de un año. Se demuestra que el pico de máxima velocidad de crecimiento y la ganancia de talla durante la pubertad varía entre cada grupo, pero no así la talla final alcanzada. Se presentan tablas de talla y velocidad de crecimiento para cada grupo madurativo, y demuestra la necesidad de que, durante la pubertad, el crecimiento sea valorado considerando el tipo de maduración del niño/a. Este estudio sirvió de base para la realización de ulteriores estudios longitudinales durante la pubertad, como el estudio longitudinal español 1978/2000 y el de Barcelona 1995/2017.
– Carrascosa A. Millennials´ Growth. Estudio longitudinal de crecimiento. Barcelona 1995-2017. Disponible en: https://doi.org/10.3266/Pulso.ed.MillennialsGrowth.2018.ESP.
Monografía muy completa y útil para el clínico. Presenta los datos del estudio longitudinal de crecimiento de Barcelona en edad puberal con 1.453 sujetos. Presenta tablas y gráficas de los 5 grupos madurativos (muy temprano, temprano, intermedio, tardío y muy tardío). Este estudio tiene la fuerza del número de niños/as seguidos, que es muy elevado, por lo que cada grupo tiene una muestra superior a 100, y el hecho de haber descartado para el estudio a los niños/as con obesidad o malnutrición.
Gráficas de crecimiento en España. Cuál y cómo utilizarlas
1. Señale qué ESTÁNDARES se recomiendan, en la actualidad, para realizar una valoración de la antropometría neonatal en un RN de 37 semanas:
a. Estándares transversales del estudio de Bilbao 1988.
b. Estándares transversales del estudio integrado español 2008.
c. Estándares longitudinales del estudio integrado español 2010.
d. Estándares longitudinales del estudio de Zaragoza 2002.
e. Estándares transversales del estudio de Barcelona 2004.
2. En relación a la interpretación de la velocidad de crecimiento en un niño/a antes de iniciar la pubertad, señale a partir de qué PERCENTIL se puede considerar patológica cuando el periodo de observación es prolongado:
a. Percentil 1.
b. Percentil 3.
c. Percentil 10.
d. Percentil 25.
e. Percentil 35.
3. El cálculo del pronóstico de talla adulta de un niño/a a la edad de 10 años se realiza habitualmente mediante el método de Bayley-Pinneau. Señale qué PARÁMETROS necesita disponer para realizar dicho cálculo:
a. Talla y peso.
b. Talla y talla media parental.
c. Talla y talla genética.
d. Talla y estadio puberal.
e. Talla y edad ósea.
4. Señale qué ESTÁNDARES se recomiendan, en la actualidad, para realizar una valoración de la talla y peso de un niño de 8,5 años nacido en Bilbao y que reside en Andalucía:
a. Estándares transversales del estudio integrado español 2010.
b. Estándares transversales del estudio de Bilbao 2004.
c. Estándares longitudinales del estudio de Bilbao 2004.
d. Estándares longitudinales del estudio integrado español 2010.
e. Estándares transversales del estudio de Andalucía 2008.
5. Señale la respuesta INCORRECTA en relación al estudio integrado español 2010:
a. Los datos del estudio demuestran una aceleración secular de la talla.
b. Los datos del estudio demuestran una aceleración secular del índice de masa corporal.
c. Es un estudio que integra los estudios de crecimiento de Andalucía, Barcelona, Bilbao, Madrid y Zaragoza.
d. Los datos del estudio demuestran una aceleración secular de la pubertad.
e. El estudio integrado español 2010 es una continuación del estudio integrado español 2008.
6. Una niña de 12 años acude a tu consulta porque la madre está preocupada por su crecimiento. Tiene una talla en el percentil 3 y ha crecido 4 cm en el último año. No hay antecedentes personales de interés y es una niña sana y sociable. En la exploración destaca que no ha iniciado la pubertad (Tanner I). En relación a la valoración de la velocidad de crecimiento, indique en qué grupo de MADURACIÓN PUBERAL la incluirías:
a. Grupo de maduración muy tardío.
b. Grupo de maduración tardío.
c. Grupo de maduración intermedio.
d. Grupo de maduración temprano.
e. No es necesario considerar el tipo de maduración puberal.
7. Un niño de 12,5 años tiene un índice de masa corporal de 23,5 kg/cm2 que corresponde al percentil 85 en los estándares del estudio español 2010. Señale la respuesta CORRECTA:
a. Presenta obesidad leve.
b. Presenta normopeso.
c. Presenta sobrepeso.
d. Exploraría la velocidad de crecimiento antes de hacer una valoración.
e. Esperaría a que llegara a la edad adulta para hacer una valoración.
8. Acude a tu consulta un niño de 12 años con talla inferior al percentil 3, peso en el percentil 10 y testes infantiles. Tienes la sospecha de que presenta una talla baja desproporcionada por acortamiento de las extremidades. Indique qué PARÁMETRO confirmará su sospecha clínica:
a. Cociente talla sentada/talla en el percentil 50.
b. Cociente talla sentada/talla a +0,5 DE de la media.
c. Cociente talla sentada/talla superior al percentil 90.
d. Cociente talla sentada/talla a -0,5 DE de la media.
e. Cociente talla sentada/talla inferior al percentil 3.
Respuestas correctas:
Pregunta 1 – Respuesta correcta: b.
Los estándares más adecuados son los provenientes del estudio español de crecimiento 2008, que agrupan 9.362 recién nacidos. Este estudio aporta tablas de normalidad desde la semana 26 a la semana 42 para longitud y peso. El resto de estudios aporta datos de peso y longitud al nacimiento, pero tiene una muestra significativamente inferior y no distingue por edades gestacionales. La valoración de la antropometría neonatal exige conocer el peso y la longitud y la edad gestacional para poder decir si un recién nacido es pequeño, adecuado o grande para la edad gestacional.
Pregunta 2 – Respuesta correcta: d.
A diferencia de las gráficas de distancia para talla, peso o IMC, donde el límite de la normalidad lo marca el percentil 3, en las gráficas de velocidad de crecimiento, el límite de la normalidad lo marca el percentil 25, ya que se ha observado que si un niño/a crece ≤ al percentil 25 durante un periodo de 2-3 años, se va cayendo progresivamente de su carril. Por ello, en un periodo prolongado de observación, una velocidad de crecimiento ≤ al percentil 25 se debe considerar como patológica.
Pregunta 3 – Respuesta correcta: e.
El método de Bayley-Pinneau se basa en el porcentaje de talla adulta alcanzada a una edad ósea determinada y existen unas tablas que indican qué porcentaje corresponde a cada edad ósea, en función de si está adelantada, retrasada o acorde a su edad cronológica. Por tanto, para poder hacer el cálculo del pronóstico de talla adulta, en un momento dado, necesita saber la talla y la edad ósea. El estadio puberal no es necesario para hacer este cálculo. La talla genética y la talla media parental son conceptos diferentes. La talla media parental es la media de la talla de los progenitores y la talla genética es la talla media parental con la aplicación de un factor de corrección en función del sexo.
Pregunta 4 – Respuesta correcta: a.
El estudio español 2010 supone la integración de los estudios de Andalucía, Barcelona, Bilbao, Madrid y Zaragoza, con una muestra total de 38.461 sujetos. Los estudios integrados de crecimiento españoles, realizados en los años 2008 y 2010, demostraron que no hay diferencias en las tallas en función de la región, por lo que se puede considerar que la población española se comporta de manera homogénea a nivel antropométrico. Por ello, estos estándares son aplicables en toda España, independientemente del lugar de nacimiento y el lugar de residencia.
Pregunta 5 – Respuesta correcta: d.
El estudio de crecimiento español 2010 es continuidad del estudio 2008 (que agrupa los estudios de Andalucía, Barcelona, Bilbao y Zaragoza) y que añade los datos de la Comunidad de Madrid. Este estudio demuestra una aceleración secular de la talla y del IMC, pero este estudio no ha valorado el desarrollo puberal, por lo que no demuestra la existencia de una aceleración del inicio de la pubertad. Únicamente, se explica que la aceleración secular del IMC está en relación al incremento del peso y a la posible aceleración del ritmo madurativo, pero el estudio no ha valorado este fenómeno directamente, ya que no analiza el desarrollo puberal. El único estudio longitudinal que ha valorado el inicio y desarrollo puberal en la población española es el estudio de Zaragoza.
Pregunta 6 – Respuesta correcta: a.
La valoración del crecimiento en la edad puberal se debe hacer con las tablas y gráficas de referencia del estudio longitudinal español 2010 o de Barcelona 2017. Ello es especialmente necesario en un niño/a con talla en percentiles bajos o que consulta por retraso de crecimiento. Estos estudios permiten clasificar al niño/a en función de su ritmo de maduración puberal. Una niña con 12 años que no ha iniciado la pubertad debe ser clasificada en el grupo de maduración puberal muy tardío, que son las niñas que inician la pubertad muy tarde y, por lo tanto, la edad de inicio del brote puberal es igualmente muy tardía, entre los 12 y 13 años. Una velocidad de crecimiento de 4 cm/año en una niña sana de 12 años en estadio Tanner I es normal.
Pregunta 7 – Respuesta correcta: c.
El estudio de crecimiento español 2010 demostró una aceleración secular del IMC y puso de manifiesto la necesidad de redefinir los puntos de corte para hablar de sobrepeso y de obesidad. Debido a dicha aceleración y si se utilizan dichos estándares de referencia, el punto de corte de sobrepeso está en el percentil 80 y, si el IMC está entre el percentil 80 y 97, se habla de sobrepeso y, si está por encima del percentil 97, se habla de obesidad. La valoración del IMC es independiente de la velocidad de crecimiento y no hay que esperar a la edad adulta para clasificar a un niño en normopeso, sobrepeso u obesidad. Este niño con IMC en el percentil 85 en los estándares del estudio español 2010 presenta sobrepeso.
Pregunta 8 – Respuesta correcta: c.
El cociente talla sentada/talla se utiliza a nivel clínico para valorar la proporcionalidad de los segmentos corporales. Un cociente por encima de la normalidad indicaría una cortedad del segmento inferior de las extremidades inferiores, y un cociente por debajo de la normalidad indicaría lo contrario, un segmento inferior elevado. El estudio longitudinal de Zaragoza ha establecido los valores de la normalidad de dicho cociente desde el nacimiento a talla adulta en tablas y gráficas. A nivel de gráficas de crecimiento, si el cociente está por encima del percentil 90, ya avalaría la sospecha clínica. Este cociente se sitúa en torno a 0,65 en el nacimiento y desciende hasta 0,5 al iniciar la pubertad. A nivel clínico, se considera que si el cociente se sitúa por encima de 0,55, ya indica que está en los límites altos de la normalidad.













































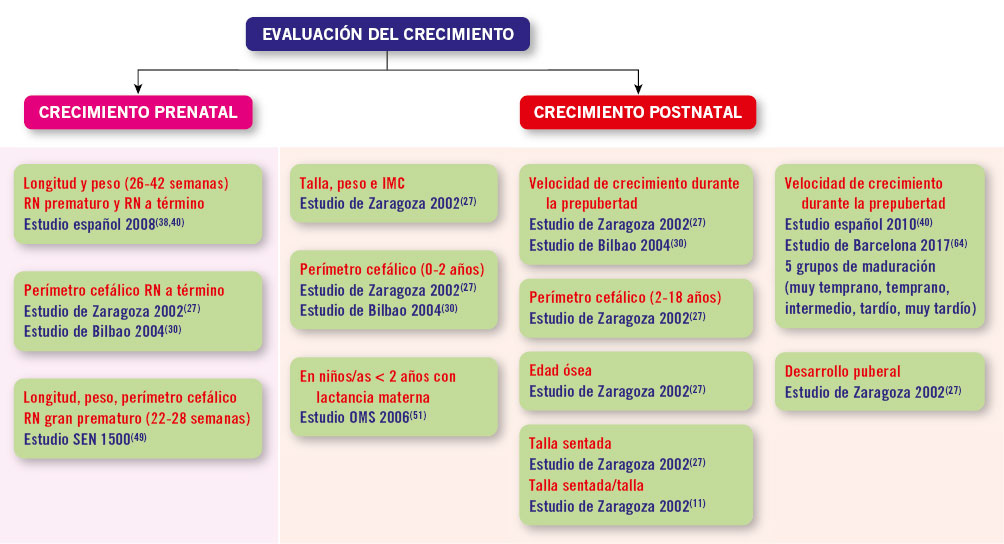









































































 La dieta cetogénica es un tipo de dieta con una proporción baja de hidratos de carbono, alta de lípidos y adecuada de proteínas, con el objetivo de inducir un estado cetósico.
La dieta cetogénica es un tipo de dieta con una proporción baja de hidratos de carbono, alta de lípidos y adecuada de proteínas, con el objetivo de inducir un estado cetósico.
 Además, es de elección en dos metabolopatías: el déficit de GLUT1 y el déficit de piruvato deshidrogenasa. En ambas entidades, la glucosa no puede ser empleada para generar energía, por lo que se necesitan otros sustratos energéticos.
Además, es de elección en dos metabolopatías: el déficit de GLUT1 y el déficit de piruvato deshidrogenasa. En ambas entidades, la glucosa no puede ser empleada para generar energía, por lo que se necesitan otros sustratos energéticos. Por otra parte, está contraindicada en pacientes con metabolopatías que impidan una correcta síntesis o degradación de los cuerpos cetónicos, y en los trastornos de la oxidación mitocondrial de los ácidos grasos.
Por otra parte, está contraindicada en pacientes con metabolopatías que impidan una correcta síntesis o degradación de los cuerpos cetónicos, y en los trastornos de la oxidación mitocondrial de los ácidos grasos. Existen varios tipos de dieta cetogénica en función de la proporción de lípidos respecto al resto de macronutrientes. Las más empleadas son, en orden decreciente de proporción de grasas: la dieta cetogénica a clásica con ratios 4:1 o 3:1, la dieta modificada de Atkins y la dieta de bajo índice glucémico.
Existen varios tipos de dieta cetogénica en función de la proporción de lípidos respecto al resto de macronutrientes. Las más empleadas son, en orden decreciente de proporción de grasas: la dieta cetogénica a clásica con ratios 4:1 o 3:1, la dieta modificada de Atkins y la dieta de bajo índice glucémico. El inicio de la dieta cetogénica debe ser valorado por un equipo interdisciplinar, y normalmente precisa de 1-2 semanas para su instauración completa una vez iniciada, a fin de evitar efectos adversos digestivos (vómitos o alteraciones del ritmo gastrointestinal) y/o metabólicos (hipoglucemias o hipercetonemias sintomáticas).
El inicio de la dieta cetogénica debe ser valorado por un equipo interdisciplinar, y normalmente precisa de 1-2 semanas para su instauración completa una vez iniciada, a fin de evitar efectos adversos digestivos (vómitos o alteraciones del ritmo gastrointestinal) y/o metabólicos (hipoglucemias o hipercetonemias sintomáticas). Al principio, requiere de un control estrecho diario de cuerpos cetónicos y glucosa capilar, que se puede llevar a cabo en domicilio.
Al principio, requiere de un control estrecho diario de cuerpos cetónicos y glucosa capilar, que se puede llevar a cabo en domicilio. Dichos controles se pueden espaciar tras los primeros meses, volviendo a intensificar su control en procesos intercurrentes (fiebre o procesos gastrointestinales con vómitos y diarrea).
Dichos controles se pueden espaciar tras los primeros meses, volviendo a intensificar su control en procesos intercurrentes (fiebre o procesos gastrointestinales con vómitos y diarrea). Si un paciente con dieta cetogénica requiere antitérmicos, analgésicos, antibióticos u otros fármacos de forma aguda, se debe revisar su compatibilidad con la dieta (riesgo de pérdida de eficacia). En general, las formulaciones tipo jarabe contienen una alta proporción de hidratos de carbono.
Si un paciente con dieta cetogénica requiere antitérmicos, analgésicos, antibióticos u otros fármacos de forma aguda, se debe revisar su compatibilidad con la dieta (riesgo de pérdida de eficacia). En general, las formulaciones tipo jarabe contienen una alta proporción de hidratos de carbono. Ante un paciente con dieta cetogénica, el pediatra de Atención Primaria debe conocer su indicación, el tipo, los controles a realizar y los rangos de glucemia y cetonemia necesarios en cada paciente, así como saber identificar aquellas situaciones que puedan descompensar estos niveles y su manejo inicial, remitiendo a urgencias pediátricas en caso de descompensación aguda.
Ante un paciente con dieta cetogénica, el pediatra de Atención Primaria debe conocer su indicación, el tipo, los controles a realizar y los rangos de glucemia y cetonemia necesarios en cada paciente, así como saber identificar aquellas situaciones que puedan descompensar estos niveles y su manejo inicial, remitiendo a urgencias pediátricas en caso de descompensación aguda.