 |
| Temas de FC |
M. Güemes*, B. Corredor Andrés**, M.T. Muñoz Calvo***
*Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús. Madrid. **Sección de Endocrinología. Hospital Virgen de la Salud. Toledo. ***Unidad de Endocrinología. Servicio de Pediatría. Hospital Ruber Internacional. Madrid
| Resumen
Desde el periodo neonatal y a lo largo de infancia y adolescencia, una función tiroidea normal es imprescindible para un adecuado neurodesarrollo y crecimiento. Las alteraciones tiroideas son comunes, por lo que su detección a través del cribado neonatal y, posteriormente, por las manifestaciones clínicas, junto con hallazgos analíticos y radiológicos, permitirá su pronta detección y tratamiento pertinente. Se describe de forma práctica la presentación, evaluación y terapia de las alteraciones tiroideas más frecuentes en la infancia y adolescencia. |
| Abstract
An adequate neurodevelopment and growth rely on a normal thyroid function from the neonatal period, throughout childhood and adolescence. Thyroid disorders are common, thus its detection by means of the neonatal Guthrie screening test and later on by clinical manifestations, along with analytical and imaging findings, will enable a prompt detection and suitable treatment. |
Palabras clave: Hipotiroidismo; Tiroiditis; Hipertiroidismo; Nódulo tiroideo.
Key words: Hypothyroidism; Thyroiditis; Hyperthyroidism; Thyroid nodule.
Pediatr Integral 2020; XXIV (5): 248 – 257
Patología tiroidea en la infancia y la adolescencia
Introducción
Las hormonas tiroideas presentan efectos pleiotrópicos en múltiples sistemas del organismo; de ahí, que la mayoría de las alteraciones tiroideas se manifiesten como clínica multiorgánica. A continuación, se expondrá la patología tiroidea característica del periodo infantojuvenil, realizando especial mención a las alteraciones más frecuentes.
Hipotiroidismo
El hipotiroidismo indica insuficiencia bioquímica de hormonas tiroideas, que puede ser debida a diferentes etiologías que alteran su producción a nivel: primario (glándula tiroidea), secundario (hipofisario) o terciario (hipotalámico), y quedan recogidas en la tabla I.
Hipotiroidismo congénito (HC)
Es la causa más frecuente de las alteraciones endocrinas del recién nacido (RN). Su incidencia es de 1/2.000 RN vivos. El HC tiene especial importancia en el niño, debido a que las hormonas tiroideas son imprescindibles para el desarrollo cerebral, representando la causa de retraso mental prevenible más frecuente(1).
Etiología
La causa más frecuente es la disgenesia tiroidea (75-85%), correspondiendo la mayoría de los casos a ectopias tiroideas, siendo la localización sublingual la más habitual.
Las dishormonogénesis representan el 10-20%, siendo un grupo heterogéneo de errores congénitos que consisten en bloqueo total o parcial de la síntesis de hormonas tiroideas. El HC de origen central es el menos frecuente. Existen formas de HC transitorio donde la función tiroidea se normaliza en tiempo variable, siendo necesaria, en algunas circunstancias, el tratamiento sustitutivo(2) (Tabla I).

Manifestaciones clínicas
La exploración inicial del RN con HC suele ser normal o presentar signos sutiles, incluso en los hipotiroidismos graves(3) (Tabla II).
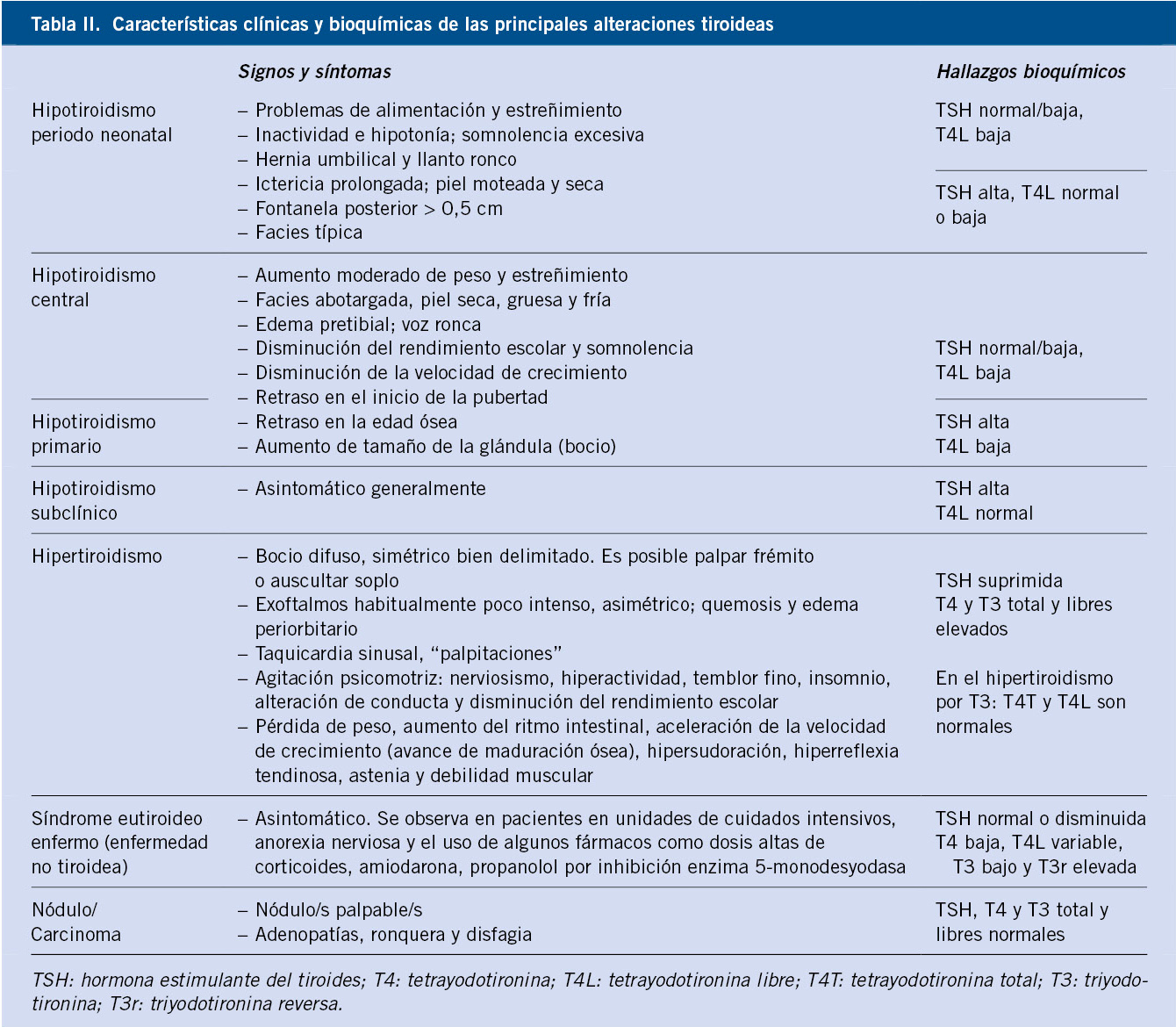
Diagnóstico por cribado neonatal
El cribado neonatal está diseñado para el diagnóstico precoz de HC, para evitar el daño cerebral y permitir así el desarrollo neurológico y físico normal.
La detección se lleva a cabo a través de la medición de TSH (hormona estimulante del tiroides) a las 48 horas de vida, evitando su aumento fisiológico inicial. En algunas comunidades autónomas (País Vasco, Navarra y Cantabria), se analiza de forma conjunta TSH y T4L (tetrayodotironina libre), permitiendo así la detección de hipotiroidismo de origen central. Según la Sociedad Española de Endocrinología Pediátrica(1), el punto de corte de TSH, por encima del cual existe sospecha de presentar la enfermedad es de 7-10 UI/ml (Fig. 1).

Figura 1. Algoritmo de actuación para la detección precoz de hipotiroidismo congénito (modificado de cita 1). TSH: hormona estimulante del tiroides. TG: tiroglobulina.
Ac. antitiroideos: anticuerpos antitiroglobulina, antiperoxidasa y antirreceptor de TSH.
RN: recién nacido. *Yoduria: en casos con sospecha de déficit o exceso de yodo.
Existen situaciones especiales donde el RN puede presentar una elevación tardía de la TSH o que precisa su confirmación, recomendándose la realización de muestras seriadas de TSH a las 2 y 4 semanas de vida(1,4).
Pruebas complementarias
Para la confirmación y orientación diagnóstica, se realizará un estudio en: sangre venosa de: T4L, TSH, TG (tiroglogulina), anticuerpos antitiroideos y yoduria (cuando se sospecha deficiencia o exceso de yodo).
La gammagrafía tiroidea es imprescindible para el diagnóstico etiológico. La administración del isótopo I123 o Tc99 por vía parenteral resulta en la captación y localización de la glándula tiroidea. La ecografía tiroidea puede realizarse de forma diferida. Esta tiene especial interés en el caso de detectar una ausencia de captación en la gammagrafía con TG detectable (“falsas agenesias” por la presencia de tiroides in situ visible por la ecografía).
En todos los casos de HC permanente, se debe descartar la presencia de malformaciones asociadas, en especial, a nivel cardiaco. La relación entre HC e hipoacusia está bien establecida; recomendándose un estudio de audición en la infancia. La evaluación inicial por Neuropediatría es de gran utilidad, junto con un seguimiento estrecho de los hitos del desarrollo(1,4).
Tratamiento
El tratamiento debe iniciarse lo antes posible, en el momento del estudio de confirmación y no debe retrasarse a esperar realizar el diagnóstico.
El objetivo es iniciarlo lo más precoz posible, antes de los 15 días de vida. Se trata con levotiroxina sódica sintética por vía oral, en dosis única diaria, preferiblemente en ayunas unos 30 minutos antes de la toma de alimento y siempre a la misma hora. Se administra en forma de comprimidos triturados. No se recomienda usar fórmulas magistrales por su concentración errática, ni el uso de genéricos por su biodisponibilidad variable. La dosis recomendada en el RN es entre 10-15 µg/kg/día.
Seguimiento: realizar una segunda valoración de T4L y TSH a los 15 días (se deberá observar una normalización de T4L con descenso de TSH) y después cada 2 semanas hasta normalización de TSH. Posteriormente: mensualmente, los primeros 6 meses; bimensual, hasta los 12 meses de vida; y cada 3-4 meses hasta los 3 años de edad. Si se produce una modificación en la dosis, se efectuará un control a las 4-6 semanas. Los objetivos son normalizar y elevar el nivel de T4L en el rango superior de la normalidad y normalizar la TSH. A partir de los 3 años, se programará la reevaluación de la función tiroidea en tiroides eutópicos sin alteraciones genéticas confirmadas(1,4).
Hipotiroidismo adquirido
El hipotiroidismo adquirido primario autoinmune es la alteración tiroidea más común en la infancia y adolescencia, siendo más frecuente (como toda la patología tiroidea) en el sexo femenino (4:1), con una prevalencia aproximada de 1,13:1.000 en menores de 18 años(5).
Etiología
La causa más frecuente del hipotiroidismo adquirido, excluida la deficiencia de yodo, es la tiroiditis crónica autoinmune. En aproximadamente el 50% de los casos, existen antecedentes familiares de enfermedad tiroidea autoinmune(6).
Manifestaciones clínicas
Los principales síntomas y signos quedan incluidos en la tabla II. Destacar que, ante un niño cuya velocidad de crecimiento y percentil de talla disminuyan sin causa conocida, se debe considerar el diagnóstico de hipotiroidismo(5).
Estudios complementarios
En el hipotiroidismo primario, los valores de T4 libre están disminuidos, mientras que los de TSH aparecen elevados. Valores de TSH entre 5-10 mU/L deberían ser repetidos antes de iniciar tratamiento; pues en el 70% de los casos, serán normales en la siguiente determinación. La determinación de los anticuerpos antitiroideos [antitiroglobulina (antiTG) y antiperoxidasa (antiTPO)] es de gran interés para confirmar el carácter autoinmune(6). Por el contrario, en el hipotiroidismo hipofisario o hipotalámico, los valores de TSH son bajos o inapropiadamente normales, para valores de T4 disminuídos(5,7).
Entre las técnicas de imagen, la ecografía es el método de elección para valorar el tamaño del tiroides. La radiografía de la mano indicará un retraso en la maduración ósea(8).
Tratamiento
Consiste en la administración de levotiroxina sódica sintética, con la dosis necesaria para mantener los valores de T4 libre plasmáticos en rango normal-alto y los de TSH en rango normal(5,7,8).
Según la edad, las dosis sustitutivas oscilan entre: 10-15 mcg/kg/día, en menores de 3 meses; 4-7 mcg/kg/día, hasta los 3 años; 3-4 mcg/kg/día, de los 3 a los 10 años; y en mayores de 10 años, en torno a 2 mcg/kg/día(8). Deben realizarse determinaciones analíticas cada 4-6 semanas después de administrar o modificar una dosis, para comprobar la normalización de la función tiroidea. Una vez alcanzado el estado eutiroideo, deben efectuarse controles periódicos cada 6 meses. En niños obesos, es frecuente encontrar valores moderadamente elevados de TSH (hipertirotropinemia leve), que no precisan tratamiento con hormona tiroidea y se normalizan tras la pérdida ponderal(8).
Hipotiroidismo subclínico
Su prevalencia está comprendida entre 2-9,5% y consiste en un cuadro clínico de insuficiencia tiroidea leve, caracterizado por valores de TSH por encima de los límites superiores de la normalidad, junto con concentraciones de T4 libre dentro del rango de la normalidad(9). La progresión a hipotiroidismo es más frecuente en adolescentes con tiroiditis autoinmune, siendo incluso en este grupo la progresión de solo un 31% a los 4 años, mientras que el 34% revierte a eutiroidismo(7-9). Existe controversia sobre cuando tratar el hipotiroidismo subclínico, aunque la recomendación actual es tratar cuando los valores de TSH son superiores a 10 mU/L o si están sintomáticos(9).
Tiroiditis
Son procesos que cursan con destrucción de la estructura normal del folículo tiroideo. Según su etiología, se clasifican en diferentes tipos, recogidos en la tabla III.

La tiroiditis crónica autoinmune o de Hashimoto constituye la afectación tiroidea más frecuente durante la infancia y la adolescencia, así como la causa más común de bocio y de hipotiroidismo adquirido en regiones sin carencia de yodo(6,7,8).
Hipertiroidismo
Es el exceso de síntesis y liberación mantenida de hormonas tiroideas por la glándula tiroides. Tirotoxicosis, hace referencia a las repercusiones clínicas multiorgánicas y analíticas derivadas de la exposición de los tejidos al exceso de hormonas tiroideas. Es un trastorno poco frecuente en Pediatría, con una incidencia en torno al 3/100.000 y pico de edad entre los 11 y 15 años(8). En la tabla IV, se presenta la etiología del hipertiroidismo.
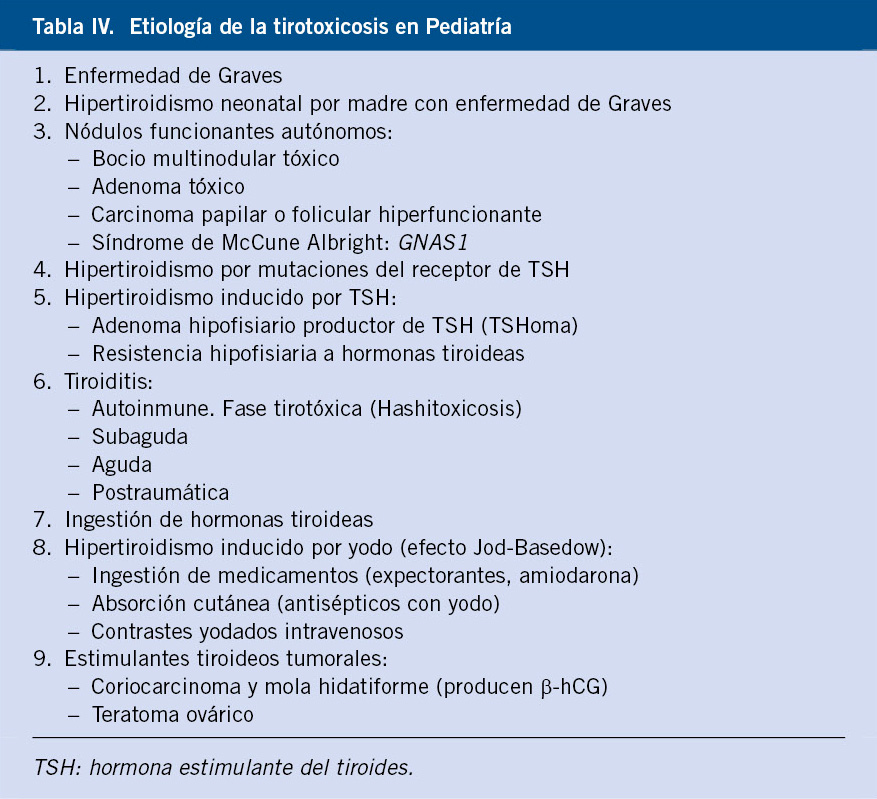
En más del 90% de los casos, el hipertiroidismo en la edad pediátrica es debido a enfermedad de Graves, seguido de la fase hipertiroidea de una tiroiditis crónica autoinmune (Hashitoxicosis) y del nódulo tiroideo hiperfuncionante(7).
Enfermedad de Graves (EG)
La EG es una afección de mecanismo autoinmune, que implica la producción de autoanticuerpos contra el receptor de TSH (TSH-R o TSH-RAb). Es posible detectar también anticuerpos antiTPO, antiTG y contra el cotransportador yodosodio de la membrana de las células foliculares del tiroides, pero estos tienen poca influencia en la patogenia de la enfermedad(10). Es más frecuente en el sexo femenino en sujetos puberales, mientras que en prepúberes, la prevalencia parece ser similar en ambos sexos.
Etiología
La susceptibilidad a la enfermedad depende de: factores ambientales (estrés, tabaquismo, estrógenos e infecciones [Y. enterocolítica]) y genéticos (susceptibilidad aumentada en pacientes caucásicos HLA-DR3 y HLA-B8, así como mutaciones en el gen TSH-R (receptor de TSH) y en el gen de la subunidad α de la proteína G, que regula la transducción del mensaje de la TSH al interior de la célula)(7,10). El 60% de los pacientes tiene antecedentes familiares de enfermedad tiroidea autoinmune.
Manifestaciones clínicas
La EG se caracteriza por la presencia de hipertiroidismo, bocio (en >95% de los casos) y oftalmopatía (en el 50% de ellos)(8). Los síntomas quedan recogidos en la tabla II.
Estudios complementarios
Los niveles circulantes de T4 y T3 (triyodotironina) total y libres elevados y, los de TSH suprimidos confirman el diagnóstico. Frecuentemente, se incrementan más los niveles de T3 que los de T4, e incluso, a veces, presentan niveles normales de T4 (hipertiroidismo por T3). La tasa de anticuerpos TSI o TSAb (thyroid stimulating antibodies), aunque no siempre positivos al comienzo del cuadro clínico, tienden a disminuir con el tratamiento, a pesar de que su utilidad como marcador de remisión de la enfermedad no está totalmente establecida(10). Los anticuerpos antiTPO y antiTG son detectables en la mayoría de los pacientes, aunque con títulos inferiores a los de las tiroiditis autoinmunes. Si los TSI son negativos y la sospecha de EG es alta, pueden determinarse los anticuerpos inhibidores de la unión de la TSH a su receptor (TBII), que suelen ser positivos en casi el 100% de los pacientes. La ecografía tiroidea es útil para descartar nódulo/s o confirmar la presencia de bocio, que muestra un tiroides aumentado de tamaño con ecogenicidad homogénea y con flujos vasculares elevados en el Doppler. La realización de una gammagrafía tiroidea I123 o I131 queda reservada para casos de bocio multinodular o con sospecha de adenoma tóxico(7,8,10).
Tratamiento
Hay tres modalidades terapéuticas, sin haber demostrado superioridad una sobre otra; de ahí, la controversia sobre el tratamiento idóneo de la EG.
Estarían indicados: en primer lugar, los fármacos antitiroideos; en segundo lugar, en caso de falta de remisión, se recomendaría tratamiento definitivo con I131, salvo si el bocio es grande o el paciente presenta oftalmopatía severa, en cuyo caso, es recomendable la tercera alternativa, el tratamiento quirúrgico(10-12).
1. Fármacos antitiroideos. Es la forma de terapia inicial más frecuentemente utilizada en Pediatría, sobre todo, en Europa. Los fármacos antitiroideos (tionamidas) son metimazol y carbimazol. El propiltiouracilo no está, actualmente, recomendado en Pediatría por sus potenciales graves efectos secundarios (necrosis hepática fulminante), estando reservado solo para el primer trimestre de gestación. Provocan la disminución de síntesis de hormonas tiroideas, además de poseer un efecto inmunosupresor, disminuyendo los niveles de TSI(10).
La dosis inicial de tratamiento recomendada de metimazol o carbimazol es de 0,5-1 mg/kg/día, en tres dosis. Hasta que se consiga el estado eutiroideo puede requerirse la administración de betabloqueantes (propanolol o atenolol), al objeto de controlar los síntomas pseudo-adrenérgicos (sobre todo, si frecuencia cardiaca >100 lpm en reposo), a dosis de 0,5-1 mg/kg/día, vía oral, repartidos cada 8 horas (dosis máxima de 2 a 4 mg/kg/día para el propanolol y de 2 mg/kg/día para el atenolol). Tras iniciar antitiroideos, se suele alcanzar eutiroidismo clínico y analítico en 4-6 semanas, momento en el que se reducirá la dosis a la mitad o a dos terceras partes, en una sola dosis al día. Se realizará el seguimiento del paciente cada 2-3 meses y, en cada una de estas revisiones, se valorará la función tiroidea con la determinación de T3, T4 total y libre, y de TSH. Durante el tratamiento, el tamaño de la glándula suele reducirse de un 30 a un 50%. La duración del tratamiento con antitiroideos permanece controvertida(10-12), habitualmente se mantiene de 2 a 4 años, hasta alcanzar la remisión, que viene definida por la presencia de eutiroidismo bioquímico que se mantiene por un tiempo superior al año, después de la retirada del fármaco. En la EG en Pediatría, el índice de remisión con fármacos antitiroideos tras tratamiento prolongado es bajo, entre un 30 y un 40%. Obtenida la remisión, la recidiva se produce entre el 3-47% de los adolescentes. El porcentaje de recaídas es más alto cuanto más corto ha sido el periodo de tratamiento(8).
Los efectos tóxicos de los fármacos antitiroideos se presentan entre el 17 y el 30% de los casos, en las distintas series. En general, en la edad pediátrica, los efectos adversos son leves y no requieren retirada del tratamiento (neutropenia transitoria, exantema papular urticariano, náuseas, cefaleas, artralgias y caída del cabello)(11). La agranulocitosis, generalmente acompañada de fiebre y odinofagia, así como la hepatitis severa y fallo hepático, son raras, pero precisan del cese inmediato de la medicación.
2. Tratamiento con radioyodo. Es un tratamiento eficaz (índice de curación superior al 90%), fácil de realizar, de bajo coste y aparentemente seguro(7). Las indicaciones para la administración de I131 son:
• Ineficacia de los fármacos antitiroideos para inducir la remisión permanente de la enfermedad.
• Necesidad de utilización de dosis altas de antitiroideos de manera crónica.
• Reacciones de hipersensibilidad moderadas-severas de los fármacos.
• Olvido de la medicación.
Es mejor individualizar la dosis según el tamaño de la glándula tiroidea y su capacidad de captar radioyodo, siendo recomendable una única dosis de 220-275 mcCi/g de tejido tiroideo; pudiendo ser necesaria dar una nueva dosis de dos a seis meses, más tarde. Se debe descartar embarazo previo a su administración.
Los efectos secundarios del tratamiento con I131 incluyen: hipotiroidismo (eventualmente el 90% de los niños), recurrencia del hipertiroidismo, alteración de la función paratiroidea, tiroiditis inducida por radiación e incremento del riesgo de desarrollar adenomas benignos de tiroides(12). Estudios de seguimiento de más de 20 años, realizados en amplias poblaciones de niños y adolescentes, tratados con altas dosis de I131, no demuestran mayor riesgo de cáncer de tiroides(7,8,10,12).
3. Tratamiento quirúrgico. La tiroidectomía total/subtotal bilateral es el método de elección en aquellos adolescentes que:
• No sigan el tratamiento con fármacos antitiroideos o tengan reacciones tóxicas a los mismos.
• Presenten una oftalmopatía grave.
• Tengan un bocio difuso grande.
La preparación de la cirugía requiere tratamiento antitiroideo, al menos, 1-2 meses y, en algunos casos, para disminuir la vascularización glandular se emplea yodo lugol 10 días antes (18 gotas al día, divididas en 3 tomas). Los efectos secundarios incluyen: hipotiroidismo permanente (la mayoría), recurrencia de EG a partir del tejido tiroideo residual (3%), hipoparatiroidismo y lesión del nervio laríngeo recurrente(10,12).
Independientemente del tratamiento recibido, una vez completado, se debe realizar control de función tiroidea semestral hasta el fin del crecimiento y, posteriormente, anual de por vida(11).
Nódulo tiroideo
Ni los datos de la anamnesis ni los signos físicos exploratorios del paciente aportan información concluyente acerca de la malignidad o no del nódulo tiroideo (NT).
Los NT son masas localizadas en el tiroides, de consistencia distinta al resto de la glándula. Pueden ser múltiples o únicos y estar asociados o no a un bocio difuso. En la población pediátrica, la incidencia es aproximadamente del 0,2 al 1,5%, siendo más frecuentes en el sexo femenino que en el masculino (6:1) y aumentando linealmente con la edad(13).
Etiología
Entre los factores inductores de la formación de NT, el más documentado es la radiación externa (radioterapia craneal, cervical o zona superior del tórax), déficit de yodo y factores hormonales: la TSH y los factores reguladores del crecimiento de las células tiroideas (EGF, TGF-β, PDGF, FGF)(8,13).
Hay que descartar patología del tiroides en: los antecedentes familiares, la enfermedad tiroidea previa del paciente, los síndromes de neoplasia endocrina múltiple y la exposición previa a radiación.
Manifestaciones clínicas
Los NT pueden ser descubiertos incidentalmente durante una exploración médica y cursar de forma asintomática o acompañarse de sintomatología, dependiendo de la funcionalidad del NT. Los datos sugestivos de benignidad y malignidad quedan recogidos en la tabla V.
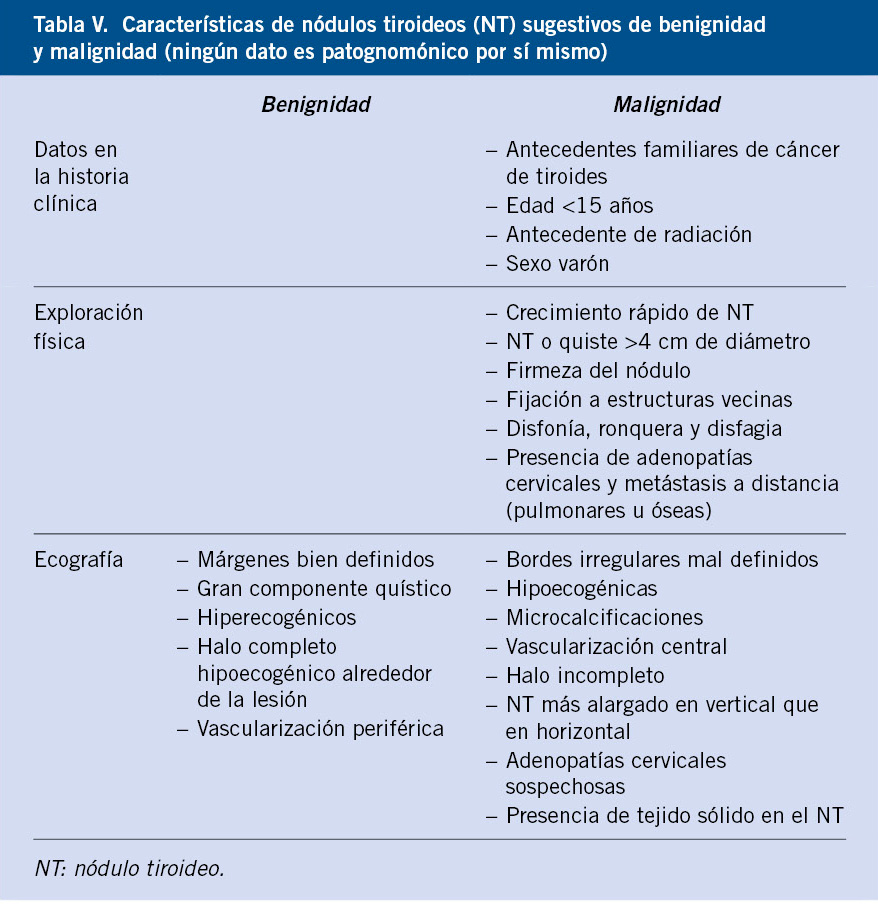
El signo que tiene mayor valor es la velocidad de crecimiento del nódulo, ya que en general, los tumores malignos aparecen de modo brusco y tienen un crecimiento rápido(13).
Estudios complementarios
La determinación de T3 libre, T4 libre, TSH y anticuerpos antiTG, antiTPO y antiTSI ayuda a establecer el grado de funcionalidad del tiroides. Generalmente, el cáncer de tiroides no produce alteración de la función tiroidea, pero no se excluye su presencia ante nódulos encontrados en la EG o en la tiroiditis autoinmune. La determinación de tiroglobulina no ayuda al diagnóstico de precisión de un NT, ya que puede estar elevada también en situaciones benignas, como las tiroiditis. Los niveles de calcitonina, que son útiles como marcador tumoral en familiares asintomáticos portadores de mutación RET, se encuentran elevados en el carcinoma medular(13).
La ecografía Doppler de alta resolución detecta NT a partir de 1 mm de diámetro, a la vez que descubre cambios en las estructuras vecinas. Es un método rápido e inocuo que, además de aportar información sobre la consistencia del NT (sólido, quístico o mixto) y su localización dentro o fuera de la glándula, permite guiar la punción-aspiración con aguja fina (PAAF) del mismo y, en última instancia, controlar su tamaño durante el tratamiento con levotiroxina(14). La clasificación Thyroid Imaging Reporting and Data System (TIRADS) estratifica a los NT en riesgo, según sus características ecográficas(15). Los patrones ecográficos que orientan hacia la benignidad o malignidad de un NT quedan recogidos en la tabla V.
La punción-aspiración con aguja fina (PAAF) guiada por ecografía debe realizarse de forma sistemática en todos los NT mayores de 6 mm (por debajo no es posible). Es una técnica segura, de fácil realización, indolora, de bajo coste y alta fiabilidad, con una exactitud cercana al 95%, en manos de un citopatólogo experto(13).
La limitación más importante de esta técnica es la escasa especificidad para diferenciar los tumores foliculares benignos de los malignos(14). Según el resultado de la citología, se distinguen 6 categorías en el Bethesda System for Reporting Thyroid Cytopathology (BSRTC) (Tabla VI)(15).

La gammagrafía con I123 o Tc99 solo está indicada si la TSH está suprimida o la calcitonina elevada. Su utilización permite detectar la presencia de nódulos, siempre que tengan un tamaño superior a 1 cm de diámetro, así como su funcionalidad. Los NT se pueden clasificar gammagráficamente en:
• Hipofuncionante o frío (mayor riesgo de malignidad).
• Hiperfuncionante o caliente (menor riesgo de malignidad).
• Isocaptante o indeterminado.
Tratamiento
La actitud terapéutica ante un NT queda representada en el algoritmo de la figura 2, donde el resultado citológico de la PAAF, juega un papel fundamental en la toma de decisiones(13,16).
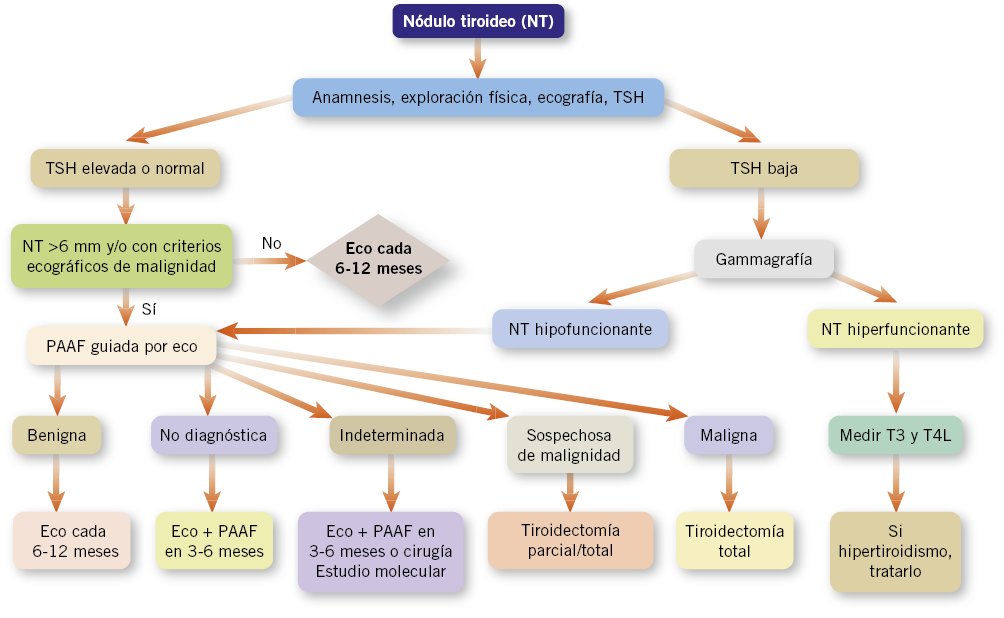
Figura 2. Esquema de actitud a seguir ante nódulos tiroideos. TSH: hormona estimulante del tiroides; PAAF: punción-aspiración con aguja fina; T4: tetrayodotironina; T3: triyodotironina; (figura modificada de cita 16).
Carcinoma de tiroides
Representa solo el 0,6-1,6% de los tumores malignos en la infancia, pero su incidencia mundial está en aumento.
Etiología
Los factores etiológicos son los mismos que los mencionados para el NT. Además, el 70% de los carcinomas diferenciados presentan mutaciones activadoras en la vía del cAMP, en concreto en los genes RET, NTRK1, BRAF y RAS. También se ha encontrado relacionado con mutaciones en TP53: gen supresor APC, gen supresor PTEN, así como con el síndrome DICER1(16).
Tipos de tumores malignos
La clasificación histológica realizada por la OMS describe 4 tipos principales de tumores malignos de origen epitelial: papilar (el más común), folicular (el segundo más común), medular y anaplásico.
Los denominados tumores diferenciados, el papilar y el folicular, derivan ambos de las células foliculares y suponen el 89% de los tumores epiteliales malignos y es frecuente que presenten metástasis al diagnóstico.
El tumor medular deriva de las células parafoliculares C y se han descrito formas de carácter familiar, y una no familiar. Entre las formas familiares asociadas a otras alteraciones endocrinas, se encuentra el carcinoma medular asociado a neoplasia endocrina múltiple tipo 2 (MEN 2) por mutaciones en el protooncogén RET (10q11.2)(17).
Respecto a las manifestaciones clínicas y estudios complementarios, sirve lo mencionado previamente en el apartado de NT. Además, solo existen marcadores tumorales específicos del carcinoma medular, que son la calcitonina y el antígeno carcinoembrionario (CEA). En la ecografía, además de determinar las características del NT, se valorará la extensión tumoral a ganglios linfáticos y cuerdas vocales. La PAAF, guiada por ecografía, debe realizarse de forma sistemática(18).
Tratamiento
Se realizará tiroidectomía total (excepcionalmente subtotal en los tumores diferenciados) y vaciamiento ganglionar cervical más o menos extenso(16,17).
Posteriormente, se realizará un rastreo de tejido tiroideo con I123 o I131 y, en la mayoría de los casos, se administrará radioablación con I131, con el objeto de eliminar el resto de tejido tiroideo y las posibles metástasis. Para carcinomas refractarios y metástasis, se puede emplear quimioterapia o radioterapia. Para las formas familiares de carcinoma medular, debe practicarse la tiroidectomía profiláctica (a diferentes edades según el riesgo), cuando se detecte la mutación del protooncogén RET, puesto que la concordancia entre la presencia de la enfermedad y el estado de portador de la mutación es del 100%.
Función del pediatra de atención primaria
La anamnesis dirigida, exploración física y función tiroidea realizadas por el pediatra con un alto índice de sospecha de patología tiroidea, acelerará el proceso diagnóstico y terapéutico de las alteraciones de esta glándula.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Rodríguez Sánchez A, Chueca Guindulain MJ, Alija Merillas M, Ares Segura S, Moreno Navarro JC, Rodríguez Arnao MD, et al. Diagnóstico y seguimiento de los pacientes con hipotiroidismo congénito diagnosticados por cribado neonatal. An Pediatr. 2019; 90: 1-250.
2. Peters C, van Trotsenburg ASP, Schoenmakers N. DIAGNOSIS OF ENDOCRINE DISEASE: Congenital hypothyroidism: update and perspectives. European Journal of Endocrinology. 2018: 179; 297-317.
3. Wassner AJ. Congenital Hypothyroidism. Clinics in Perinatology. W.B. Saunders. 2018; 45; 1-18.
4.** Léger J, Olivieri A, Donaldson M, Torresani T, Krude H, Van Vliet G, et al. European society for paediatric endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. Horm Res Paediatr. 2014; 99: 363-84.
5. LaFranchi S. Acquired hypothyroidism in childhood and adolescence. UpToDate. Última actualización: 2018 (acceso en febrero de 2020). Disponible en www.uptodate.com.
6. Pasala P, Francis GL. Autoimmune thyroid diseases in children. Expert Rev Endocrinol Metab. 2017; 12: 129-42.
7. Hanley P, Lord K, Bauer AJ. Thyroid Disorders in Children and Adolescents: A Review. JAMA Pediatr. 2016; 170: 1008-19.
8. Segni M. Disorders of the Thyroid Gland in Infancy, Childhood and Adolescence. Endotext. Thyroid Disease Manager. Última actualización: 2017 (acceso en febrero de 2020). Disponible en www.endotext.org.
9. Catli G, Abaci A, Büyükgebiz A, Bober E. Subclinical hypothyroidism in childhood and adolescense. J Pediatr Endocrinol Metab. 2014; 27: 1049-57.
10.** Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, et al. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid. 2016; 26: 1343-421.
11. Minamitani K, Sato H, Ohye H, Harada S, Arisaka O. Guidelines for the treatment of childhood-onset Graves’ disease in Japan, 2016. Clin Pediatr Endocrinol. 2017; 26: 29-62.
12. De Luca F, Valenzise M. Controversies in the pharmacological treatment of Graves’ disease in children, Expert Review of Clinical Pharmacology. 2018; 11: 1113-21.
13. Burman KD, Wartofsky L. Thyroid Nodules. New England Journal of Medicine. 2015; 373: 2347-56.
14. Triana G, Valencia S, Morillo AJ. Pediatric American Thyroid Association Guidelines Are Not Completely Applicable to Pediatric Nodule Evaluation. Radiology. 2018; 289: 881-2.
15. Singaporewalla RM, Hwee J, Lang TU, Desai V. Clinico-pathological Correlation of Thyroid Nodule Ultrasound and Cytology Using the TIRADS and Bethesda Classifications. World Journal of Surgery. 2017; 41: 1807-11.
16. Francis GL, Waguespack SG, Bauer AJ, Angelos P, Benvenga S, Cerutti JM, et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2015; 25: 716-59.
17. Paulson VA. Thyroid cancer in the pediatric population. Genes. 2019; 10: 723.
18. Agrawal VR, Jodon G, Mushtaq R, Bowles DW. Update on multikinase inhibitor therapy for differentiated thyroid cancer. Drugs Today. 2018; 54: 535.
Bibliografía recomendada
– Hanley P, Lord K, Bauer AJ. Thyroid Disorders in Children and Adolescents: A Review. JAMA Pediatr. 2016; 170: 1008-19.
Revisión práctica y holística de fisiopatología tiroidea pediátrica que incluye ilustraciones para una correcta exploración tiroidea.
– Francis GL, Waguespack SG, Bauer AJ, Angelos P, Benvenga S, Cerutti JM, et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2015; 25: 716-59.
Esta es la primera guía específicamente pediátrica y la más reciente sobre el manejo de nódulos tiroideos y carcinoma tiroideo en esta población.
| Caso clínico |
|
Motivo de consulta: niña de 10 años y 6 meses, remitida por su pediatra al detectar aumento del tamaño de la glándula tiroidea. Clínicamente asintomática. Rendimiento escolar normal. Antecedentes familiares: sin interés. Antecedentes personales: alopecia areata desde hace tres años. En seguimiento en la consulta de Gastroenterología por anticuerpos antitransglutaminasa levemente positivos, pero sin cumplir criterios de enfermedad celiaca. Sin otros antecedentes de interés. Exploración física: Buen estado general, piel y cabello normales. Antropometría: peso: 33 kg (p25, -0,6DE) y talla: 143 cm (p50, +1DE). IMC: 16,1 kg/m2 (-0,7DE). TA: 105/60 mmHg. FC: 65 l/m. Talla genética: 163 ± 5 cm (P50-75). Bocio grado 1B (palpable y visible cuando el cuello está extendido), de consistencia normal, sin nódulos. Desarrollo puberal en estadio I de Tanner (M1,P1,Aa). Resto de la exploración normal. Pruebas complementarias: hemograma y bioquímica normales. Función tiroidea: TSH 3,28 mcUI/ml (0,36-5,0), T4L 1,23 ng/dl (0,63-1,4), anticuerpos antitiroglobulina 1.198 UI/ml (<37) y antiperoxidasa >600 UI/ml (<37). Edad ósea acorde a la edad cronológica.
|
