 |
| Temas de FC |
I. Rica Echevarría*, A. Vela Desojo**, G. Grau Bolado***
*Jefa de Sección de Endocrinología Pediátrica. CIBERDEM. CIBERER. Hospital Universitario Cruces. Bizkaia. **Médico Adjunto. Sección Endocrinología Pediátrica. CIBERDEM. CIBERER. Hospital Universitario Cruces. Bizkaia. ***Médico Adjunto. Sección Endocrinología Pediátrica. Hospital Universitario Cruces. Bizkaia
| Resumen
Las alteraciones del metabolismo fosfocálcico son relativamente frecuentes en Pediatría. Son muchas las hormonas implicadas en dicho metabolismo. Los recientes avances en el campo de la genética están ayudando a mejorar el diagnóstico etiológico de estas patologías. La hipocalcemia neonatal es frecuente, generalmente transitoria y multifactorial. La causa más frecuente de hipocalcemia permanente en el niño es el hipoparatiroidismo. La hipercalcemia es menos frecuente y aparece a una mayor edad. La clínica de las alteraciones del metabolismo fosfocálcico suele ser larvada y los tratamientos son eficaces. Mantener la sospecha diagnóstica adecuada en escenarios clínicos sugestivos y hacer un diagnóstico precoz de estas entidades es una prioridad que, además, puede evitar complicaciones graves derivadas de las mismas. Por lo anteriormente mencionado: el conocimiento del diagnóstico, etiopatogenia y tratamiento de esta patología es fundamental. |
| Abstract
Calcium and phosphate metabolism disorders are relatively common in pediatrics. There are many hormones involved in the regulation of its homeostasis. Recent advances in genetics are aiding to ascertain the etiological diagnosis of these pathologies. Neonatal hypocalcaemia is common, usually transient and multifactorial. |
Palabras clave: Hipocalcemia; Hipercalcemia; Vitamina D; Paratohormona (PTH); Tetania; Hipoparatiroidismo; Hiperparatiroidismo; MEN; Pseudohipoparatiroidismo; CaSR.
Key words: Hypocalcemia; Hypercalcemia; Vitamin D; Parathyroid hormone (PTH); Tetany; Hypoparathyroidism; Hyperparathyroidism; MEN; Pseudohypoparathyroidism; CaSR.
Pediatr Integral 2020; XXIV(5): 268 – 275
Metabolismo fosfocálcico
Introducción
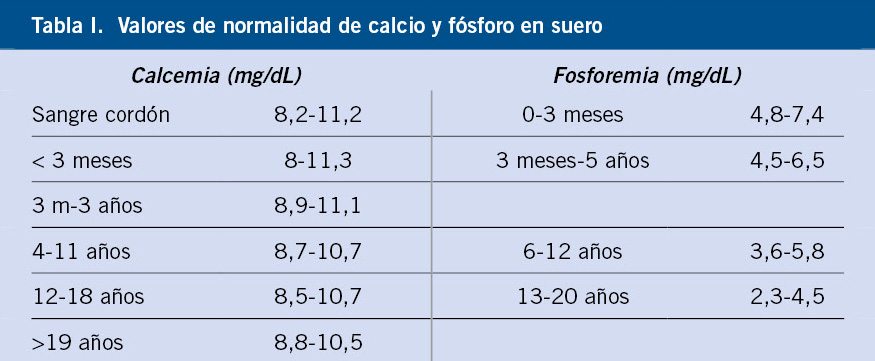
El calcio (Ca) y el fósforo (P) suponen el 65% del peso del hueso, siendo este el principal reservorio de Ca. El Ca, además, es un cofactor imprescindible en distintos procesos enzimáticos y el P está ampliamente distribuido por los tejidos, formando parte de: ácidos nucleicos, fosfolípidos, enzimas, proteína G, ATP y segundos mensajeros. Ambos cationes se obtienen fundamentalmente con la alimentación. Los niveles de Ca y P en sangre deben mantenerse en un rango estrecho y lo hacen gracias a un complejo mecanismo regulador, siendo fundamentales la paratohormona (PTH) y la vitamina D. La PTH favorece la reabsorción activa de Ca y reduce la del P. El calcitriol [1,25(OH)2D3], que es la forma activa de la vitamina D, favorece tanto la absorción de Ca como la de P. La calcemia considerada normal varía dependiendo de los laboratorios y de la edad del paciente, siendo más preciso referirnos al Ca iónico. Los valores normales de P permanecen más contantes a lo largo de la vida (Tabla I).
Fisiología de la homeostasis del calcio y fósforo
La homeostasis del Ca tiene lugar en tres órganos diana: intestino, hueso y riñón. El 99% del Ca corporal está fijado al hueso y un 1% circula en sangre (50% en forma libre o activa, Ca iónico). La absorción de Ca se produce de forma activa y pasiva a nivel intestinal. El glomérulo renal filtra aproximadamente el 50% del Ca iónico y, posteriormente, se reabsorbe un 85% a nivel tubular, mediante un proceso pasivo.
La vitamina D se genera en la piel por la acción de la luz solar, posteriormente a nivel hepático se transforma en 25 hidroxivitamina D3 [25(OH)D3] y, finalmente en el riñón, mediante la 1 alfa hidroxilasa, pasa a su forma activa, la 1,25(OH)2D3 o calcitriol. Esta enzima renal es activada por la PTH y regulada por los niveles de P (la depleción de P estimula su formación y el aumento de P la disminuye). La 1,25(OH)2D3 estimula la absorción intestinal de Ca y, en menor medida, de P(1). La PTH favorece: la salida de Ca del hueso, su absorción intestinal y disminuye su excreción urinaria. La secreción de PTH es regulada por el receptor sensor del Ca (CaSR) localizado en paratiroides y túbulo renal(2).
El FGF23 actúa a nivel renal, regulando el manejo tubular del P y el metabolismo de la vitamina D. Ejerce una función fosfatúrica y protege al organismo de los excesivos niveles de [1,25(OH)2D3], mediante la inhibición de su producción renal y el aumento de su catabolismo. El exceso de FGF23 produciría: hipofosfatemia, alteración de la vitamina D, defectos de crecimiento y osteomalacia/raquitismo, y su defecto daría como resultado hiperfosfatemia y calcificaciones en tejidos blandos.
Las hormonas principales que regulan el manejo renal de P son: la PTH y el FGF23. La PTH aumenta los niveles de P al estimular la resorción ósea y la síntesis de [1,25(OH)2D3](1), frena la absorción intestinal de fósforo. El FGF23, como ya se ha mencionado, reduce el P sérico.
Alteraciones del calcio
Hipocalcemia
La determinación de Ca iónico es fundamental en el estudio de la hipocalcemia. En el periodo neonatal suele ser transitoria, mientras que en niños mayores, la causa más frecuente es el hipoparatiroidismo. El tratamiento es sencillo, pero requiere un exquisito control.
Se considera hipocalcemia, de forma general, a la concentración de Ca total < 8,5 mg/dl o de Ca iónico < 4,4 mg/dl. El periodo neonatal es el momento más frecuente de riesgo de hipocalcemia relacionada con factores transitorios de estrés perinatal (Tabla II).

En niños mayores, las causas de hipocalcemia son múltiples, muchas de origen genético. Actualmente, gracias al avance de las técnicas de diagnóstico molecular, la hipocalcemia idiopática debe ser un diagnóstico de exclusión (Tabla III).

Los síntomas de la hipocalcemia se deben a un aumento de la excitabilidad neuromuscular y son variables, dependiendo de la edad del paciente y de la rapidez de su instauración. La crisis de tetania es la manifestación habitual en los casos agudos e incluye: parestesias, temblores, hiperreflexia, calambres, signo de Chevostek, signo de Trousseau y convulsiones. Si la hipocalcemia es crónica, la clínica es muy variada: afectación ectodérmica (sequedad de la piel, fragilidad ungueal, alteración del esmalte), ocular (edema de papila, cataratas subcapsulares), pérdida de memoria, alteraciones psiquiátricas, alteración del ritmo cardiaco con QT alargado.
El enfoque diagnóstico inicial de la hipocalcemia debe incluir: la determinación en sangre de Ca total e iónico, fósforo, magnesio, PTHi y 25(OH)D3, así como la determinación en orina del cociente calcio/creatinina (Tabla IV).
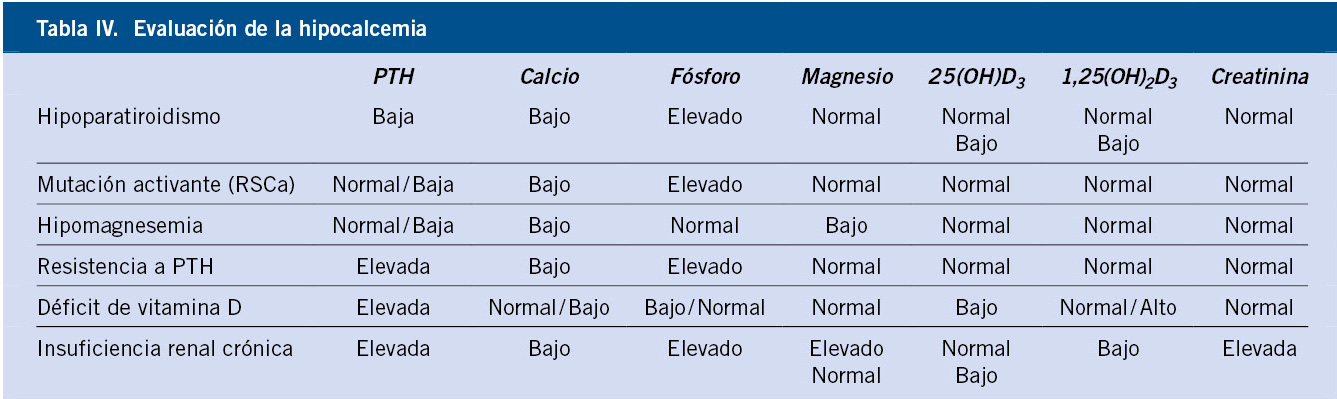
Posteriormente, en función de la sospecha diagnóstica, se debe completar el estudio con la determinación de 1,25(OH)2D3 y con la realización de estudios genéticos y/o de imagen(3).
Clasificación, clínica y fisiopatología de la hipocalcemia
Hipocalcemia neonatal
Durante el embarazo, el Ca es transferido activamente de la madre al feto. Tras el nacimiento, estos niveles descienden de forma brusca y alcanzan la normalidad a las dos semanas de edad. La hipocalcemia neonatal es frecuente y muchas veces transitoria (Tabla II). Puede ser precoz, si aparece en las primeras 72 horas, favorecida por: prematuridad, asfixia, diabetes o preeclampsia maternas, o tardía, desencadenada por una hipercalcemia materna o exceso de P en la alimentación. Debemos considerar otras etiologías en función de las características clínicas acompañantes y de su evolución.
Hipocalcemia neonatal permanente, lactante y niño mayor
Hipocalcemia secundaria a hipoparatiroidismo
En el hipoparatiroidismo(4,5) existe una disminución de la función de la PTH que puede deberse a: una ausencia/hipoplasia paratiroidea, una disminución de la síntesis/secreción de la PTH, una alteración del sensor receptor del Ca (CaSR) o a una resistencia del receptor de la PTH (Tabla III).
• Niveles bajos de PTH. Generalmente se asocian a causas genéticas(6). La Deleción del 22q11.2 destaca por su frecuencia e incluye: un cuadro sindrómico variable con aplasia/hipoplasia de timo, defectos cardiacos, disfunción velo-palatina, fenotipo peculiar e hipoparatiroidismo(7).
Mutaciones activantes del CaSR. Familiar (AD) o esporádica. Se produce un aumento en el umbral de calcio para la respuesta de la PTH, siendo esta insuficiente a pesar de hipocalcemia, con hipercalciuria relativa o absoluta.
• Niveles altos de PTH (Pseudohipoparatiroisdismo). Grupo heterogéneo de enfermedades producidas por una resistencia a la acción de la PTH, que actualmente se denomina iPPSD (inactivating PTH/PTHr Signalling Disorder)(8). Se caracterizan por: hipocalcemia, hiperfosfatemia, niveles elevados de PTH y niveles normales de 25(OH)D3. En función del fenotipo y alteración bioquímica, se dividen en subtipos, siendo los más frecuentes el Ia y el Ib, ambos producidos por alteración en la subunidad α de la proteína G codificada por el gen GNAS(9,10).
• Hipomagnesemia. Los niveles de Mg por debajo de 1 mg/dl condicionan de forma reversible resistencia y disminución en la síntesis de PTH.
Hipocalcemias secundarias al déficit de vitamina D
El raquitismo carencial es la causa más frecuente. La deficiencia de vitamina D(1,11) debe considerarse ante: una falta grave de exposición solar, una disminución de su ingesta y/o absorción, un aumento del catabolismo hepático de vitamina D o una disminución de su síntesis endógena. En la etapa neonatal, debe descartarse en hijos de madres con deficiencia grave de vitamina D. Los raquitismos hereditarios no carenciales (resistentes) son excepcionales. Las razas de piel oscura tienen mayor predisposición a este déficit y hemos de tenerlas en cuenta dada la población inmigrante en el momento actual en España.
Hipocalcemia autoinmune
La enfermedad poliglandular tipo 1 por mutación en el gen AIRE (21q22.3) es una enfermedad autosómica recesiva que puede causar un hipoparatiroidismo por destrucción de la glándula. Se puede asociar a candidiasis mucocutánea, insuficiencia suprarrenal, hipotiroidismo, insuficiencia ovárica o hepatitis autoinmune.
Otras causas de hipocalcemia adquirida
Tiroidectomía, paratiroidectomía, enfermedades de depósito (hemocromatosis o enfermedad de Wilson), infecciones, síndrome de hueso hambriento, osteopetrosis o hiperfosfatemia.
Tratamiento de la hipocalcemia
• Crisis de hipocalcemia: es una urgencia hospitalaria que se trata por vía intravenosa lenta, con monitorización electrocardiográfica (gluconato cálcico 1-2 ml/kg, en solución al 10%, diluido al medio con solución salina o glucosada). Puede ser preciso repetir la dosis y, en cuanto se consigan niveles estables de calcemia, se iniciará vía oral.
• Mantenimiento: Ca por vía oral (20 mg/kg/día), para mantener la calcemia normo-baja y evitar el riesgo de nefrocalcinosis. Se aconseja la restricción de los alimentos ricos en fósforo (lácteos y huevos) para evitar hiperfosforemia. En función de la etiología, se requerirá 25(OH)D3 o 1,25(OH)2D3(3,12).
Hipercalcemia
La hipercalcemia suele ser un hallazgo casual en la edad pediátrica. La PTH determina la orientación diagnóstica. Solo las hipercalcemias sintomáticas o severas precisan tratamiento urgente.
La hipercalcemia se sospecha ante una concentración plasmática de Ca total > 10,5 mg/dL o de Ca iónico > 5,2 mg/dL. Este hecho debe confirmarse en una segunda determinación, incluyendo el Ca iónico o valorando la calcemia corregida. La hipercalcemia en Pediatría es menos frecuente que en adultos, pero su relevancia clínica suele ser mayor. El hiperparatiroidismo primario (HPP) es la causa más frecuente de hipercalcemia franca y mantenida en la infancia. Otras situaciones frecuentes son la hipercalcemia hipocalciúrica familiar (HHF) y la inmovilización prolongada(13-15).
Clasificación, clínica y fisiopatología de la hipercalcemia
La severidad clínica de la hipercalcemia dependerá del nivel del Ca y de la velocidad de instauración de la misma. Se clasifican en: leve (Ca <12 mg/dl), moderada (12-14 mg/dl) y severa (Ca>14 mg/dl). La hipercalcemia puede ser un hallazgo incidental; pero, en ocasiones, se refieren síntomas como: letargia, hipotonía, anorexia, estancamiento ponderal, poliuria y polidipsia, vómitos, dolor abdominal y estreñimiento. En los casos de larga evolución y/o severos, nos podemos encontrar ante: fallo renal (por hipercalciuria y nefrolitiasis), pancreatitis (por depósitos de Ca y activación de tripsinógeno a nivel pancreático), arritmias (por acortamiento del QT), deterioro del nivel de conciencia y/o convulsiones. En el adolescente puede originar un cuadro psiquiátrico (ansiedad, depresión y/o psicosis). La calcemia y la severidad de los síntomas pueden orientar hacía una etiología (Tabla V).

A la hora de plantear una correcta orientación diagnóstica, es de gran utilidad clasificar la hipercalcemia en función de los niveles de PTH (Fig. 1).

Figura 1. Algoritmo diagnóstico de la hipercalcemia. HHF: hipercalcemia hipercalciúrica familiar. HPPF: hiperparatiroidismo primario familiar. IRC: insuficiencia renal crónica. PTH: paratohormona. PTHrP: péptido relacionado con la PTH.
Hipercalcemia PTH dependiente
Hiperparatiroidismo primario (HPP)
• Esporádico:
- En el recién nacido o en el lactante: en hijos de madres con hipocalcemia gestacional o en neonatos con una depleción de P (inadecuada suplementación).
- En el niño mayor o en el adolescente: el adenoma paratiroideo es la causa más frecuente de HPP en este grupo de edad (65% casos). Generalmente existe afectación de órganos diana ya al diagnóstico: quistes pardos y/o nefrocalcinosis.
• Genético:
- No sindrómico. La etiología más frecuente es la hipercalcemia hipocalciúrica familiar (HHF) que afecta, sobre todo, a niños y adolescentes. Es un cuadro benigno de herencia autosómica dominante originado por mutaciones inactivantes en heterocigosis del CaSR. que condicionan una hiposensibilidad relativa al Ca. Se caracteriza por: una elevación crónica de la calcemia, PTH elevada o inapropiadamente normal y calciuria baja. El diagnóstico diferencial se plantea con el HPP familiar (HPPF), lo cual supone un reto diagnóstico en ausencia de otros tumores.
Mutaciones con pérdida de función severa del CaSR (en homozigosis u heterozigosis si alteración grave del receptor o si asocian deficiencia de vitamina D), originan una hipercalcemia grave en el recién nacido o lactante, incluso con compromiso vital(2).
- Sindrómico: la causa más frecuente es la neoplasia endocrina múltiple (MEN), de herencia autosómica dominante, por lo que suele haber antecedentes familiares. Existen varios subtipos y es, en el MEN tipo 1, donde el hiperparatiroidismo constituye la 1ª manifestación de la enfermedad en el 90% de los casos, estando la hipercalcemia presente en la adolescencia. Se debe a mutaciones en el gen codificante de la menina y se caracteriza por la presencia de: tumores paratiroideos (90%), tumores pancreáticos (40%), hipofisarios (30%) y adrenocorticales (40%)(17,18). Los MEN 2 y 3 están causados por mutaciones en el protooncogén RET y la prevalencia de tumores paratiroideos es menor.
Otra etiología de tumores de paratiroides, menos frecuente, es el síndrome HPP-tumor de mandíbula, de herencia autosómica dominante que asocia tumores fibro-óseos de maxilar y mandíbula.
Hiperparatiroidismo terciario o adquirido
Es secundario a una insuficiencia renal crónica (IRC) o al tratamiento de raquitismos hipofosfatémicos.
Hipercalcemia PTH independiente
Asociada a altos niveles de vitamina D
Por exceso de vitamina D: una ingesta accidental o por prescripción incorrecta de cantidades excesivas de vitamina D (o sus metabolitos) en el niño, puede originar una hipercalcemia. Si esto acontece en una madre gestante, puede verse en el neonato. Los niveles de 25(OH)D3 estarán elevados, los de calcitriol normales y la PTH estará suprimida.
La hipercalcemia idiopática infantil se caracteriza por: hipercalcemia, hipercalciuria y nefrocalcinosis, generalmente resueltas antes del año de vida. Se ha relacionado con alteraciones en la reabsorción renal de fosfato determinantes de un descenso de FGF23 y un aumento de 1,25(OH)2D3, y con mutaciones inactivantes de CYP24A1 (enzima responsable del catabolismo de la 1,1,25(OH)2D3.
La necrosis grasa del recién nacido es una paniculitis de causa desconocida, más frecuente en neonatos post-término y relacionada con estrés perinatal, en la que existe un aumento de 1,25(OH)2D3 en el tejido de granulación cutáneo, con el consiguiente aumento de absorción de Ca en el intestino.
Existen linfomas y disgerminomas ováricos, así como algunas enfermedades inflamatorias y granulomatosas con actividad 1α-hidroxilasa que causan una hipercalcemia.
No asociada a los metabolitos de vitamina D
• Inmovilización. Origina con frecuencia hipercalciuria en niños/adolescentes y también puede causar hipercalcemia.
• Tóxicos y drogas. Existen fármacos que producen hipercalcemia por diferentes motivos, incluyendo: tiazidas, ácido retinoico, litio, tamoxifeno o teofilina. Una administración excesiva de carbonato cálcico o bicarbonato sódico, y una intoxicación de vitamina A, también pueden originarla.
• Hipercalcemia maligna. Aparece en menos del 1% de los niños con cáncer asociado a tumores: hematológicos, neurológicos, hepáticos, disgerminomas o rabdomiosarcomas. Se debe a una osteolisis directa producida por las células tumorales o a la síntesis tumoral de factores con actividad osteclástica, destacando el PTHrP (péptido homólogo a PTH), que es capaz de activar el receptor de la PTH(19).
• Otras causas. Entre otras causas infrecuentes de hipercalcemia, se incluyen: aporte excesivo de Ca (nutrición parenteral), algunas endocrinopatías (feocromocitoma, Addison, acromegalia, patología tiroidea), tubulopatías (acidosis tubular renal distal y síndrome de Bartter), el síndrome de Williams-Beuren, la trisomía 21, la condrodisplasia metafisaria de Jansen, el síndrome IMAGe, y algunos errores innatos del metabolismo (hipofosfatasia, deficiencia congénita de lactasa, intolerancia a disacáridos, mucopolisacaridosis tipo I y el síndrome del pañal azul).
Tratamiento de la hipercalcemia
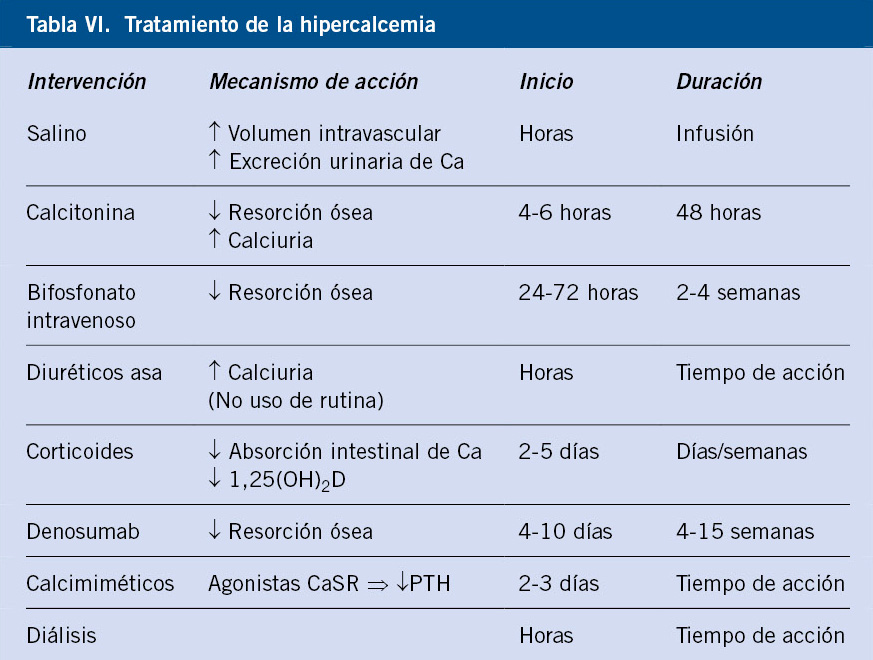
La hipercalcemia leve y asintomática no precisará tratamiento inmediato. En la crónica moderada y asintomática, la actitud inicial puede ser expectante, pero en las situaciones restantes, el tratamiento será urgente(16). La hiperhidratación junto a la administración de calcitonina y/o bifosfonatos son las medidas de elección, tal y como se representa en la tabla VI.
Alteraciones del fósforo
La hipofosforemia de define por una concentración de fósforo sérica < 2,5 mg/dl, y se considera grave si es < 1,5 mg/dl. Es una entidad rara en el niño sano y su orientación diagnóstica estará determinada por la capacidad de reabsorción tubular de fósforo (Fig. 2).
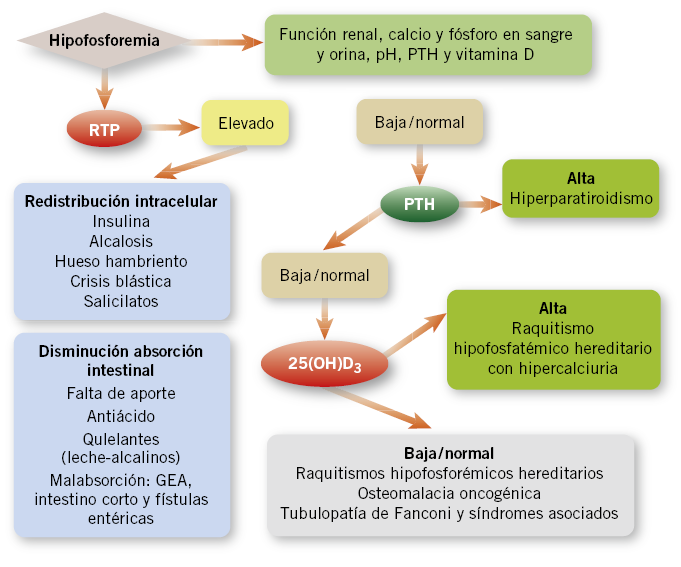
Figura 2. Algoritmo de diagnóstico de la hipofosforemia. PTH: paratohormona. RTP: reabsorción tubular de fosfatos.
Solo en los casos graves, aparece clínica relacionada con el descenso del 2-3 difosfoglicerato del hematíe que aumentará su afinidad por el oxígeno, reduciendo el ATP intracelular. Los síntomas serán fundamentalmente neurológicos (encefalopatía) y osteomusculares (debilidad, mialgias, rabdomiolisis e insuficiencia cardíaca y respiratoria)(20). En los casos graves, el tratamiento será hospitalario e intravenoso con una dosis de choque de 0,1-0,2 mmol/kg/dosis de fósforo elemento (1 mmol = 2 mEq = 31 mg P elemento) durante 6 horas (infusión máxima 0,2 mmol/kg/hora).
La hiperfosforemia se define por una concentración de fósforo sérica > 6,5 mg/dl. Es rara, salvo en niños con insuficiencia renal crónica (70% casos), en los que conlleva la aparición de un hiperparatiroidismo secundario y osteodistrofia renal. En el resto de las situaciones suele ser asintomática, aunque si el producto Ca-P séricos supera 60, existe riesgo de hipocalcemia y calcificaciones metastásicas por precipitación de las sales de P cálcico en tejidos blandos(20). El hallazgo de una hiperfosforemia en un niño previamente sano, obliga a descartar una enfermedad tumoral o metastásica (Fig. 3).

Figura 3. Algoritmo de diagnóstico de la hiperfosforemia. PTH: paratohormona. RTP: reabsorción tubular de fosfatos.
El papel del pediatra de Atención Primaria es relevante en varios aspectos: conocer las situaciones de riesgo que pueden facilitar una alteración del metabolismo fosfocálcico, llevar a cabo una correcta prevención del raquitismo carencial en los primeros meses de la vida e identificar, con celeridad, los síntomas muchas veces inespecíficos, que ponen de manifiesto este tipo de patología.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Clifford J. Rosen M.D. Vitamin D Insufficiency. N Engl J Med. 2011; 364: 248-54.
2. Hannan FM, Thakker RV. Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. 2013; 27: 359-71.
3. Cooper MS, Gittoes NJ. Diagnosis and Management of hipocalcemia, BMJ. 2008; 336: 1298-302.
4. Vela A, Pérez de Nanclares G, Grau G, Aguayo A, Rodríguez A, Rica I. Hipoparatiroidismo. Rev Esp Endocrinol Pediatr. 2013; 4.
5. Shoback D. Clinical practice. Hypoparathyroidsm. N Engl J Med. 2008; 359: 391-403.
6.*** Gordon RJ, Levine MA. Genetic Disorders of Parathyroid Development and Function. Endocrinol Metab Clin North Am. 2018; 47: 809-23.
7. Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical Guidelines for managing patients with 22q11.2 Deletion Syndrome. J Pediatrics. 2011; 159: 332-9.
8. Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L, et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. 2016; 175: 1-17.
9.** Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, Lisardi A, et al. Diagnosis and management of pseudohypoparathyroidism and related disorder: first international Consensus Statement. Nature. 2018; 14: 476-500.
10. Martos-Moreno GA. Lecumberri B, Pérez de Nanclares G. Implicaciones en Pediatría del primer consenso internacional para el diagnóstico y asistencia a pacientes con pseudohipoparatiroidismo y enfermedades relacionadas. An Pediatr (Barc). 2019; 90: 125.e1-125.e12.
11. Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, et al. Global Consensus Recommendation on Prevention and Management of Nutricional Rickets. Horm Res Paediatr. 2016; 85: 83-106.
12. Bilezikian JP, Brandi ML, Cusano NE, Mannstadt M, Rejnmark L, Rizzoli R, et al. Management of Hypoparathyroidism: Present and Future. J Clin Endocrinol Metab. 2016; 101: 2313-24.
13. Shane E, Dinaz I. Hypercalcemia: Pathogenesis, clinical manifestations, differencial diagnosis, and management. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 6th ed. American Society of Bone and Mineral Research. 2006. p. 179.
14.** Stokes VJ, Nielsen MF, Hannan FM, Thakker RV. Hypercalcemic Disorders in Children. J Bone Miner Res. 2017; 32: 2157-70.
15. Lietman SA, Germain-Lee LE, Levine MA. Hypercalcemia in children and adolescents. Curr Opin Pediatr. 2010; 22: 508-15.
16.** Davies JH, Shaw NJ. Investigation and management of hypercalcemia in children. Arch Dis Child. 2012; 97: 533-8.
17. Bilezikian JP. Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2018; 103: 3993-4004.
18. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical Practice Guidelines for Multiple Endocrine Neoplasia Type 1 (MEN1). J Clin Endocrinol Metab. 2012; 97: 2990-3011.
19. Wysolmerski JJ. Parthyroid Hormone-Related: An Update. J Clin Endocrinol Metab. 2012; 97: 2947-56.
20.*** Guerrero Fernández J, González Casado I. Manual de Diagnóstico y Terapéutica en Endocrinología Pediátrica v.1.0. 2018. ISBN 978-84-171194-44-4.
Bibliografía recomendada
– Shoback D. Clinical practice. Hypoparathyroidsm. N Engl J Med. 2008; 359: 391-403.
Un artículo clásico, pero completísima revisión para entender el hipoparatiroidismo.
– Gordon RJ, Levine MA. Genetic Disorders of Parathyroid Development and Function. Endocrinol Metab Clin North Am. 2018; 47: 809-23.
Completísima revisión del hipoparatiroidismo, incidiendo en los diferentes genes implicados en la enfermedad.
– Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, Lisardi A, et al. Diagnosis and management of pseudohypoparathyroidism and related disorder: first international Consensus Statement. Nature. 2018; 14: 476-500.
Actualización, tanto clínica como genética del pseudohipoparatiroidismo.
– Stokes VJ, Nielsen MF, Hannan FM, Thakker RV. Hypercalcemic Disorders in Children. J Bone Miner Res. 2017; 32: 2157-70.
Reciente revisión de la fisiopatología, etiología y enfoque diagnóstico de la hipercalcemia en el niño.
– Davies JH, Shaw NJ. Investigation and management of hypercalcemia in children. Arch Dis Child. 2012; 97: 533-8.
Sencilla y breve revisión del enfoque diagnóstico y tratamiento de la hipercalcemia en el niño.
– Guerrero Fernández J, González Casado I. Manual de Diagnóstico y terapéutica en Endocrinología Pediátrica v.1.0. 2018. ISBN 978-84-171194-44-4.
Excelente manual de diagnóstico y tratamiento que facilita el manejo de las principales patologías del metabolismo fosfocálcico en la edad pediátrica.
| Caso clínico 1 |
|
Anamnesis: niña de 1 mes y 10 días de vida, que ingresa desde urgencias por sospecha de convulsiones. Refieren que desde hace 3 días tiene episodios frecuentes y autolimitados (segundos de duración) consistentes en rubefacción facial con desviación de cabeza y mirada hacia lado derecho. La niña está normal entre los episodios. Los padres comentan que tiene un tono muscular aumentado desde el nacimiento, como única sintomatología asociada. Antecedentes familiares: ambos padres viven sanos; ausencia de consanguinidad. Antecedentes personales: embarazo bien controlado: gemelar bicorial biamniótico. Madre hiperemesis gravídica. Parto pretérmino (34 semanas), vaginal instrumental (fórceps). 1er gemelo. PN: 2.030 g (P 25). LN: 43 cm (P 25). Apgar: 8/10. Ingreso neonatal del 4º al 7º día de vida por hiperbilirrubinemia (fototerapia). Recibe lactancia mixta con fórmula de inicio (fórmula para prematuros previamente). No se han iniciado suplementos vitamínicos. Exploración física: peso: 3,180 kg (P 3). Longitud: 50,3 cm (P 3-10). FC: 145 lpm. FR: 26 rpm. TA: 102/57 mm Hg. Sat.: O2 100%. Fenotipo normal. Color ligeramente ictérico. Hidratación mucocutánea normal. ACP: normal. Pulsos simétricos. Abdomen: normal. SNC: hipertonía generalizada con tendencia a la flexión, con resto de exploración negativa. Genitales femeninos prepuberales normales. Analítica en urgencias: gasometría venosa: pH: 7,30; pCO2: 47 mm Hg; Bicarbonato: 23 mmol/L; EB: -2,8 mmol/L. Hematimetría normal. Bioquímica: glucosa: 66 mg/dL; urea: 14 mg/dL; creatinina: 0,28 mg/dL; sodio: 136 mEq/l; potasio: 4,8 mEq/L; cloro: 101 mEq/L; GPT: 9 U/L; Ca: 5,3 mg/dL; Ca iónico: 0,50 mmol/L; fósforo: 9,2 mg/dL. Mg: 2,30 mg/dl. Proteínas totales: 5,4 g/dl. Ante la hipocalcemia grave, ingresa en UCIP para tratamiento.
|
| Caso clínico 2 |
|
Anamnesis: varón de 1 mes y 11 días de vida, que consulta en urgencias por haber presentado 10-15 episodios breves, autolimitados, de sacudidas de extremidades con lateralización de la cabeza en las últimas 20 horas. El niño está normal entre los episodios y no asocia ninguna otra sintomatología. Antecedentes familiares: hermana gemela que ingresó 24 horas (caso 1). Antecedentes personales: presentación de nalgas. 2º gemelo. PN 1.650 g. (P 3). Longitud: 42,1 cm (P 3-10). Apgar: 8/10. Ingreso en Unidad Neonatal hasta el 7º día de vida por hiperbilirrubinemia (fototerapia). Resto compartido con caso 1. Exploración física: peso: 3,300 kg (P 3). Longitud: 50,4 cm (P 3). FC: 167 lpm. FR: 32 rpm. TA: 85/55 mm Hg. Sat.: O2 100%. Fenotipo normal. Color ligeramente ictérico. Bien hidratado. Fontanela anterior normotensa. ACP: normal. Pulsos simétricos. Abdomen: normal. Hipertonía generalizada con resto de exploración neurológica normal. Testes 2 ml en bolsa. Pene infantil normal. Analítica en urgencias: gasometría venosa: pH 7,29; pCO2: 16 mm Hg; Bicarbonato: 8 mmol/L; EB: 15 mmol/L. Hematimetría normal. Bioquímica: glucosa: 66 mg/dL; urea: 18 mg/dL; creatinina: 0,19 mg/dL; sodio: 136 mEq/l; potasio: 4,8 mEq/L; cloro: 101 mEq/L; GPT: 10 U/L; calcio: 5,4 mg/dL; calcio iónico: 0,59 mmol/L; fósforo: 8,5 mg/dL. Mg: 1,8 mg/dl. Proteínas totales: 5,4 g/dl. Ante la hipocalcemia grave, ingresa en UCIP para tratamiento. Diagnóstico diferencial de hipocalcemia en un lactante Las causas más frecuentes de hipocalcemia en un lactante de 1 mes son: el hipoparatiroidismo, el pseudohipoparatiroidismo, el raquitismo o la hipocalcemia neonatal tardía. Dado que se trata de 2 hermanos gemelos de diferente sexo, a priori lo más razonable es pensar que su hipocalcemia pudiera deberse a un problema intrauterino. En este sentido, se re-historió a la madre y se consultaron los resultados de las analíticas que se le habían realizado durante el embarazo. Evidenciamos en 2 ocasiones, una hipercalcemia leve con hipofosforemia, que el ginecólogo no había valorado (calcio: 11,4 mg/dl con fósforo: 1,2 mg/dl; calcemia de control: 10,8 mg/dl). Ante este hecho, la primera sospecha diagnóstica es una hipocalcemia secundaria a un hiperparatiroidismo materno, y completamos los estudios con determinación del metabolismo fosfocálcico a los niños y su madre: • Analítica de la madre: calcio: 10,3 mg/dl; fósforo: 3 mg/dl; magnesio: 2,11 mg/dl. PTH: 173 pg/ml y 25-hidroxi vitamina D < 7 ng/ml. • Analítica de la niña (24 h de ingreso): calcio: 5,7 mg/dl; fósforo: 7,4 mg/dl; PTH: 58 pg/ml y 25-hidroxi vitamina D: 10 ng/ml. • Analítica del niño (24 h de ingreso): calcio: 5,6 mg/dl; fósforo: 6,5 mg/dl; PTH: 116 pg/ml y 25-hidroxi vitamina D: 9,4 ng/ml. Evolución Los niños estuvieron 9 días ingresados en la UCIP, recibiendo calcio intravenoso a dosis altas y vitamina D. Tras iniciar la terapia, no volvieron a tener episodios de clonías y la hipertonía fue mejorando progresivamente. El 7º día se cambió a terapia con Ca oral y pasaron a planta de hospitalización, donde estuvieron ingresados durante 2 semanas. Evolución analítica de los niños: • Caso 1. A los 7 días: Ca: 8,9 mg/dl; fósforo: 7,4 mg/dl; PTH: 43 pg/ml. Control a las 2 semanas: Ca: 10,5 mg/dl; fósforo: 5,3 mg/dl; 25-hidroxi vitamina D: 34 ng/ml. • Caso 2. A los 7 días: Ca: 7,8 mg/dl; fósforo: 5,1 mg/dl; PTH: 43 pg/ml y 25-hidroxi vitamina D: 13 ng/ml. Control a las 2 semanas: Ca: 10,7 mg/dl; fósforo: 6,8 mg/dl; 25-hidroxi vitamina D: 22 ng/ml. Los niños fueron controlados en consultas externas de endocrinología pediátrica. De forma progresiva, se fueron bajando las dosis de Ca y calcitriol, hasta poder suspender ambas terapias a los 8-9 meses de vida. Se confirmó que posteriormente mantenían valores analíticos normales. Fueron dados de alta a los 18 meses con peso/longitud en P 50 y exploración física completa rigurosamente normal. La madre fue referida al servicio de endocrinología. Inicialmente, indicación de tratamiento con colecalciferol. Tras normalizar la vitamina D y objetivarse la persistencia de una elevación de la PTH, se solicitó gammagrafía-MIBI que confirmó la presencia de un adenoma paratiroideo izquierdo. Se realizó una paratiroidectomía inferior izquierda, con normalización de la PTH intraoperatoria. En resumen, interpretamos el caso de estos dos lactantes, como una hipocalcemia sintomática derivada de un hipoparatiroidismo funcional transitorio, debido fundamentalmente a un hiperparatiroidismo materno al que se asociaron algunos otros factores de riesgo de hipocalcemia, incluyendo: la prematuridad y los niveles bajos de vitamina D.
|
