|
| Temas de FC |
J.I. Labarta Aizpún*,1,2, M. Ferrer Lozano**,1,2, A. de Arriba Muñoz***,1,2, M. Vara Callau**,1,2
*Jefe de Servicio de Pediatría. Profesor Titular de Pediatría. Universidad de Zaragoza. **Facultativo Especialista de Pediatría. ***Facultativo Especialista de Pediatría. Profesor Asociado de Pediatría. Universidad de Zaragoza. 1Unidad de Endocrinología Pediátrica. Hospital Universitario Miguel Servet. 2Instituto de Investigación Sanitaria de Aragón. Zaragoza
| Resumen
El retraso puberal es una situación muy frecuente que se define como ausencia de inicio de la pubertad a los 13 años en las mujeres y a los 14 años en los varones. El concepto de retraso puberal también incluye la falta de progresión o pubertad incompleta y el infantilismo o ausencia de pubertad. La afectación psicológica puede ser importante y ello es el motivo principal de consulta. Las causas de retraso puberal son múltiples, pero la más frecuente es el retraso constitucional del crecimiento y desarrollo (RCCD). Las enfermedades crónicas, en función de la severidad y duración, pueden causar también retraso puberal. El hipogonadismo puede ser por fallo hipotálamo-hipofisario (hipogonadotropo) o por fallo gonadal primario (hipergonadotropo). El diagnóstico diferencial entre el hiopogonadismo hipogonadotropo y el retraso constitucional puede ser difícil y exige una valoración especializada. El RCCD no suele requerir tratamiento salvo que exista una afectación psicológica importante. El tratamiento del hipogonadismo depende de la etiología y tiene como objetivos fundamentales: inducir el desarrollo puberal y preservar la fertilidad. El pediatra de Atención Primaria debe ser capaz de identificar un retraso puberal y diferenciar si se trata de un retraso constitucional o de un posible hipogonadismo, en cuyo caso deberá remitir el paciente a una Unidad de Endocrinología Pediátrica. |
| Abstract
Delayed puberty is a very common condition and it is defined as the absence of onset of puberty at 13 years of chronological age in girls and 14 years in boys. It also includes the lack of normal progression or incomplete pubertal development and infantilism. Delayed puberty may cause psychological distress. There are different causes of delayed puberty but the most frequent is the normal variant constitutional delay of growth and puberty. Chronic diseases, depending on severity and duration, may also produce delayed puberty. Hypogonadism might be either due to abnormal hypothalamic – pituitary function (hypogonadotropic) or to primary gonadal failure (hypergonadotropic). It is difficult to differentiate between constitutional delay of growth and puberty and hypogonadotropic hypogonadism and this usually requires a specialized consideration. Constitutional delay of growth and puberty does not usually require medical treatment unless there is an important psychological distress. Treatment of hypogonadism depends on its etiology and the main objectives are to induce puberty and preserve fertility. General pediatricians should be able to recognize a pubertal delay and a normal variant, constitutional delay of puberty, and refer the patient to a specialized Pediatric Endocrine Unit when hypogonadism is suspected. |
Palabras clave: Retraso puberal; Retraso constitucional del crecimiento y desarrollo; Hipogonadismo.
Key words: Delayed puberty; Constitutional delay of growth and puberty; Hypogonadism.
Pediatr Integral 2020; XXIV (4): 191 – 207
Retraso puberal
Introducción
La cronología de la pubertad, tanto en su inicio como en su progresión, es muy variable en función de factores genéticos y ambientales, y es necesario conocerla antes de poder hablar de un trastorno patológico de la pubertad.
La pubertad es la fase del desarrollo humano que lleva a la adquisición de la madurez sexual y a la capacidad reproductiva, y supone la transición de la infancia a la edad adulta. En este periodo: se desarrollan los caracteres sexuales secundarios, se alcanza la fertilidad, se desarrollan cambios en la composición corporal y en las proporciones corporales, tiene lugar una aceleración del crecimiento lineal, se alcanza la talla adulta y se producen cambios psicológicos y emocionales propios de la adolescencia.
Este proceso requiere un eje hipotálamo-hipófiso-gonadal (HHG) intacto, tanto a nivel funcional como anatómico. El fenómeno final que pone en marcha la pubertad es el aumento, tanto en número como en amplitud de los picos de secreción de hormona liberadora de gonadotropinas (GnRH) por parte de las neuronas hipotalámicas productoras de GnRH y ello conlleva el aumento en la secreción de gonadotropinas y, consecuentemente, de esteroides sexuales. El eje HHG es ya activo durante la época fetal y continúa su actividad en la primera infancia. En el varón, los niveles de LH se incrementan dramáticamente tras el nacimiento y, posteriormente, van disminuyendo hasta el 6º mes; por el contrario, en la mujer, predominan los niveles de FSH, que permanecen elevados durante los primeros 24 meses de vida postnatal. Posteriormente y hasta el inicio de la pubertad, los niveles de LH y FSH son bajos, pero detectables; si bien, se ha demostrado que se mantiene la pulsatilidad de la secreción de gonadotropinas con niveles moderadamente más elevados durante la noche que por el día(1).
El inicio de la pubertad es el resultado final de la interacción de determinantes genéticos y de un gran número de factores reguladores que incluyen elementos endógenos y señales ambientales. La reactivación de la actividad pulsátil de GnRH es el elemento clave en el inicio de la pubertad. Este evento parece estar iniciado por dos mecanismos complementarios como son la pérdida del tono inhibitorio transináptico y la activación de los estímulos favorecedores, que provienen tanto de señales transinápticas como de la glia(2). En el momento actual, el descubrimiento del sistema kispeptina/receptor de kispeptina ha contribuido a mejorar el conocimiento de la regulación de la secreción de GnRH. Las kispeptinas son secretadas por neuronas hipotalámicas y son esenciales en la regulación de la síntesis y liberación de GnRH. A su vez, la síntesis de este péptido se ha mostrado sensible a los niveles de esteroides gonadales y al estado nutricional y metabólico. Ello ha hecho que las kispeptinas sean un factor primordial en la neurorregulación de la pubertad.
La cronología de la pubertad es extremadamente variable en función del componente genético (racial y familiar) y ambiental (condiciones intrauterinas, estado nutricional, ambiente afectivo, enfermedades crónicas y disruptores endocrinos). Esta variabilidad, tanto en el inicio (timing) como en la progresión (tempo), es muy importante considerarla antes de poder hablar de un trastorno patológico de la pubertad. Desde un punto de vista clínico, el inicio de la pubertad lo marca la aparición de los caracteres sexuales secundarios, botón mamario en la mujer y testes de 4 mL en el varón. El Estudio Longitudinal Andrea Prader aporta datos de normalidad en cuanto al inicio y a la secuencia del desarrollo puberal(3), que sigue una secuencia predecible y que está categorizada los estadios por Tanner. En dicho estudio, el primer signo puberal de las mujeres es la aparición de la telarquia (II) que ocurre a una edad media de 10,7 años, seguida 2,5 meses más tarde por el inicio del vello pubiano, un año más tarde por la axilarquia y la menarquia ocurre a los 12,6 años. Los varones alcanzan un volumen testicular igual o mayor a 4 mL a una edad media de 12,3 años, la pubarquia se inicia inmediatamente después y la axilarquia aparece casi 2 años más tarde. La edad de inicio puberal muestra una variabilidad de unos 4-5 años entre individuos de la misma población y condiciones de vida similares (entre 8-13 años en las mujeres y entre 9-14 años en los varones). Trabajos recientes, como el estudio longitudinal español de crecimiento(4), dividen el crecimiento en 5 grupos diferentes según la forma de madurar y se demuestra que todos ellos, en ambos sexos, pese a comenzar la pubertad a edades diferentes y tener una duración muy distinta, alcanzan una talla adulta similar y acorde a su genética. Esta variabilidad parece estar determinada por factores genéticos (50-80% de la variabilidad) y ambientales.
Es un hecho bien conocido, como la edad de inicio de la pubertad se ha adelantado en los últimos siglos con mayor consistencia en las mujeres que en los varones(5); si bien, esta tendencia se habría detenido en las últimas décadas. Esta aceleración secular se ha relacionado con una mejoría en las condiciones de vida. Otras variables implicadas han sido: mayor prevalencia de obesidad / sobrepeso, inactividad física, cambios en los hábitos dietéticos y el posible efecto de los disruptores ambientales. Los factores genéticos no se podrían descartar, ya que este proceso ocurre con mayor intensidad en unas poblaciones que en otras.
Concepto y clasificación
El concepto de retraso puberal incluye no solo el retraso en su inicio, sino también su falta de progresión o pubertad detenida, y el infantilismo sexual o ausencia de pubertad. Las causas son múltiples y se clasifican en tres categorías: retraso constitucional del crecimiento y desarrollo, hipogonadismo hipogonadotropo e hipogonadismo hipergonadotropo.
El retraso puberal es una situación frecuente en la práctica asistencial. Es importante su correcta identificación, ya que puede conllevar repercusiones psicosociales que son habitualmente el motivo de consulta. La falta de desarrollo puberal y el retraso de crecimiento, que habitualmente le acompaña, son motivo de daño psicológico en forma de menor autoestima e introversión, que puede conllevar fracaso escolar y problemas sociales.
Las alteraciones que cursan con retraso puberal pueden tener diversas formas de presentación y no existe un consenso claro en su definición. Se entiende por pubertad retrasada a la ausencia de cualquier signo de pubertad a la edad en que la ha iniciado el 97% de la población general de la misma área geográfica, es decir, a una edad cronológica superior a 2-2,5 desviaciones estándar respecto a la media de la población de referencia. Desde el punto de vista práctico, se considera que un varón tiene un retraso puberal, cuando no ha alcanzado un volumen testicular de 4 mL a los 14 años de edad cronológica y, en una niña, cuando no ha iniciado el desarrollo mamario a la edad de 13 años. Se habla de pubertad no progresiva, pubertad detenida o pubertad incompleta cuando la pubertad, iniciada tardíamente o no, presenta una ausencia de progresión de los caracteres sexuales durante dos años o cuando transcurren más de 5 años entre los primeros signos de pubertad y el desarrollo genital completo en el varón y la menarquia en la mujer, en la que el término amenorrea primaria indica la ausencia de la menarquia a los 15 años de edad(6,7). Se han publicado recientemente nomogramas que evalúan la progresión de la pubertad, expresándolo en desviación estándar por año, permitiendo identificar una progresión anormal en forma de precocidad o retraso. Se entiende por amenorrea secundaria la ausencia de menstruaciones tras la existencia de sangrado uterino previo durante 6 meses o tras un período de tiempo igual a tres ciclos previos regulares. Por último, se habla de ausencia de pubertad o infantilismo sexual, cuando la pubertad no llega a iniciarse.
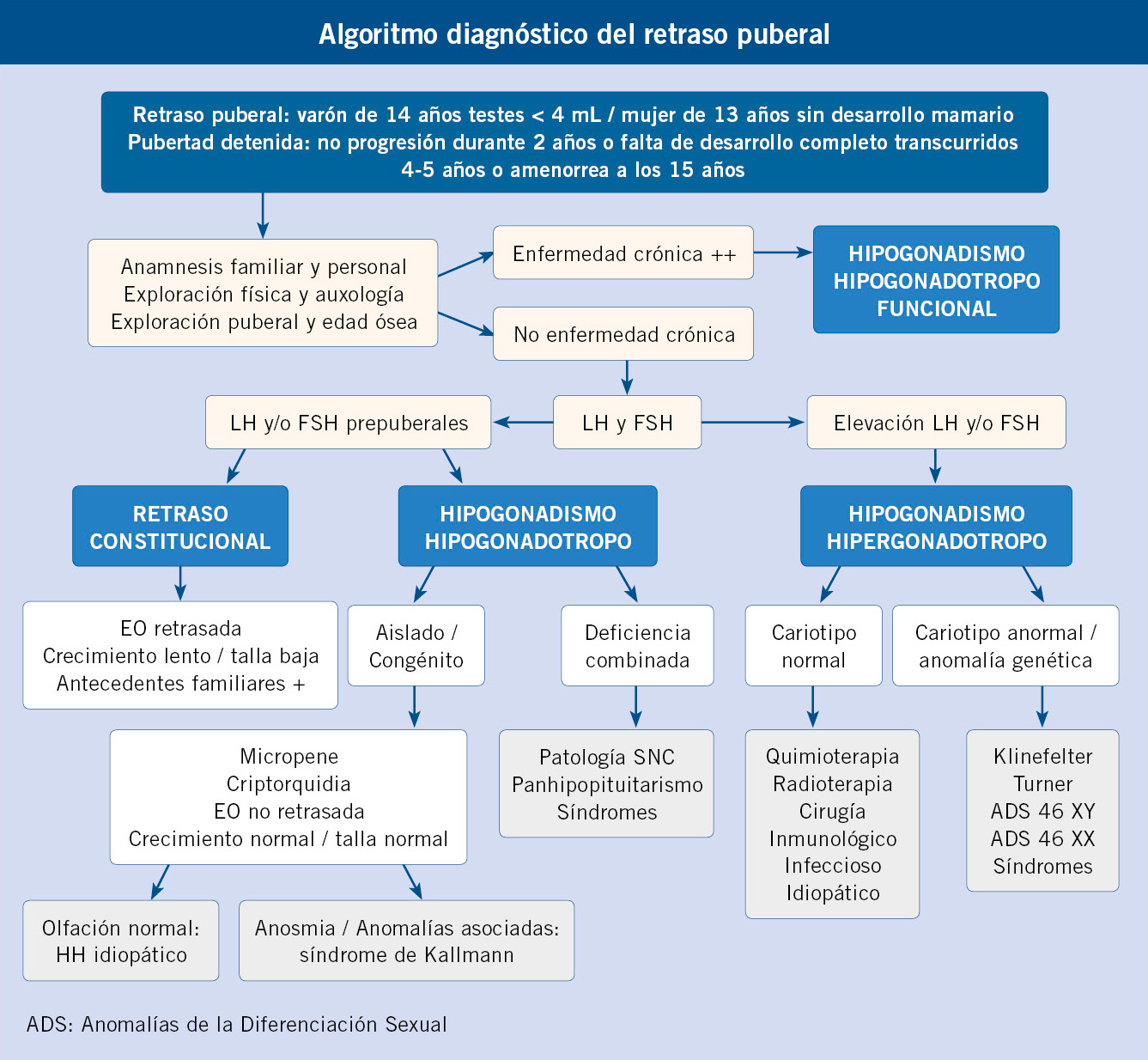
Las causas que pueden provocar un retraso puberal son múltiples, pero se pueden agrupar en tres categorías (Tabla I).

• Retraso constitucional del crecimiento y desarrollo. También se conoce como retraso puberal simple, retraso constitucional de la pubertad o retraso constitucional de la adolescencia. Es una variante de la normalidad que consiste en un trastorno temporal de la secreción de gonadotropinas y esteroides sexuales por retraso madurativo.
• Hipogonadismo hipogonadotropo. Se incluyen todas las condiciones clínicas que asocian, bien de forma permanente o transitoria, ya sea por causa congénita o adquirida, una secreción deficiente de gonadotropinas y secundariamente una insuficiencia gonadal y déficit de esteroides sexuales. En este grupo, se incluye el retraso puberal secundario a enfermedades crónicas, resultado de un hipogonadismo hipogonadotropo funcional.
• Hipogonadismo hipergonadotropo. Se trata de un hipogonadismo producido por daño gonadal primario, que determina unos niveles disminuidos de esteroides gonadales y, secundariamente, una elevación de las gonadotropinas, debido a la pérdida del retrocontrol negativo.
El retraso constitucional del crecimiento y desarrollo (RCCD) es la causa más frecuente de retraso puberal y se produce en un 3-5% de la población. En diferentes series analizadas, supone aproximadamente el 65-82% de las causas de retraso puberal en los varones y el 30-56% en las mujeres. El hipogonadismo hipogonadotropo funcional por enfermedad crónica es la segunda causa más frecuente y supone aproximadamente el 16-20% de los casos, siendo más frecuente en mujeres y ligado a patología nutricional. La tercera causa sería el hipogonadismo hipogonadotropo permanente (congénito y tumoral) y, finalmente, el hipogonadismo hipergonadotropo, especialmente de origen sindrómico(8).
Retraso constitucional del crecimiento y desarrollo
Es la causa más frecuente de retraso puberal y supone una variante de la normalidad, motivada por un retraso madurativo del eje hipotálamo-hipofiso-gonadal. Es frecuente la presencia de antecedentes familiares y un retraso de crecimiento asociado, que puede ser el motivo de consulta junto con la afectación psicológica.
Es un motivo muy frecuente de consulta, tanto de talla baja como de retraso puberal. Es de etiología desconocida y, aunque pueden existir casos esporádicos, en la gran mayoría existen antecedentes familiares semejantes; hasta el 80% de los varones y el 75% de las mujeres tienen un familiar de primer grado afecto de RCCD(9) y se ha propuesto una herencia autosómica dominante. Los varones consultan con más frecuencia que las mujeres, sobre todo, por la talla baja acompañante; con menos frecuencia, el motivo de consulta es la afectación psicológica por el retraso puberal, especialmente cuando la talla no está muy afectada.
El RCCD se caracteriza por presentar un tempo lento de maduración que da lugar a un retraso del crecimiento con un patrón típico, en el que el retraso de la talla es armónico con el retraso de la edad ósea. Existe un retraso de la maduración sexual por falta de activación del eje HHG que conlleva un déficit temporal de GnRH y, secundariamente, de gonadotropinas y de hormonas sexuales. El patrón de crecimiento de estos niños/as es muy característico y se podría resumir de la siguiente manera(10):
• Peso y talla normales al nacimiento.
• Velocidad de crecimiento normal durante los primeros 12-18 meses de vida, con disminución posterior hasta los 2-4 años, en la que el percentil de talla se sitúa en su carril genético.
• Desde los 2-4 años hasta el período prepuberal, la velocidad de crecimiento adquiere un ritmo normal, pero por debajo de la media.
• El fenómeno de depresión prepuberal de la velocidad de crecimiento se encuentra exacerbado, mostrando un distanciamiento más acusado de la curva normal de talla para su edad cronológica.
• Retraso de la talla importante para la edad cronológica, pero no para la edad ósea, la cual presenta un retraso de 2-3 años.
• El estirón puberal es tardío, pero acorde a la edad ósea, y se caracteriza por presentar un menor intervalo de tiempo desde el comienzo de la pubertad hasta iniciar el estirón, y por ser un pico de velocidad de crecimiento inferior y menos intenso cuanto mayor sea el retraso(11).
Los signos clínicos de pubertad empiezan a una edad ósea apropiada, aproximadamente los 11-12 años en las niñas y los 13-14 años en los niños, y no se retrasan más allá de los 16 años y los 18 años de edad cronológica, respectivamente. Todos los niños/as alcanzan de forma espontánea, aunque más tarde, una maduración puberal completa y, la mayoría, una talla adulta normal y adecuada para la talla genética, si bien, en un 15% de los pacientes, la talla adulta se sitúa por debajo de la talla esperada familiar. El impacto que el RCCD tiene en la salud adulta no es totalmente conocido, existiendo algunos trabajos que indican una afectación negativa sobre: talla adulta, densidad mineral ósea, funcionamiento psicosocial, nivel educacional y un mayor riesgo de enfermedad cardiovascular, y metabólica. Por otro lado, ejercería un papel protector para el desarrollo de cáncer de mama y de endometrio, y cáncer de próstata(12).
Hipogonadismo hipogonadotropo
El hipogonadismo hipogonadotropo (HH) se caracteriza por niveles disminuidos o ausentes de LH y/o FSH. El HH puede ser congénito o adquirido, permanente o transitorio, de severidad variable y aislado o asociado a otros defectos hormonales. Las enfermedades crónicas, en función de su severidad y duración, pueden producir un retraso puberal y suponen la segunda causa más frecuente de retraso puberal.
El HH es una condición producida por una insuficiencia hipotálamo-hipofisaria, que da lugar a una secreción insuficiente de gonadotropinas. Su etiología es diversa y puede ser de origen hipotalámico o hipofisario.
Hipogonadismo hipogonadotropo congénito
Deficiencia aislada de gonadotropinas
Se trata de un grupo heterogéneo de trastornos definidos por una disminución de los esteroides gonadales, asociado a una secreción normal o baja de gonadotropinas hipofisarias. Se caracterizan por ausencia parcial o completa de secreción pulsátil endógena de GnRH, ausencia de anormalidades anatómicas del área hipotálamo-hipofisaria y secreción normal del resto de hormonas hipofisarias. El HH congénito se clasifica en dos grandes grupos:
1. HH que cursa con anosmia o hiposmia, conocido como el síndrome de Kallmann (SK).
2. HH con olfación normal y se le denomina HH aislado o idiopático (HHI).
Fisiopatológicamente, el primero se relaciona con una diferenciación y desarrollo anormal de las neuronas GnRH y, el segundo, con trastornos de la secreción y/o acción del GnRH. Hasta la fecha, se han encontrado más de 30 genes diferentes implicados en el SK y/o HHI; sin embargo, en más del 50% de los casos, no se ha identificado un gen responsable(13).
Los hallazgos clínicos del HH varían en función de la edad de presentación, severidad (completa o parcial) y duración. Puede ser sospechado en la edad neonatal, si el paciente presenta micropene y/o criptorquidia. La frecuencia de criptorquidia en pacientes con HH congénito varía entre el 30-50% y, de micropene, entre un 20-40%, que es muy superior a la frecuencia en la población general de 1-3% y 0,015%, respectivamente(11). En la edad prepuberal, no suele haber signos clínicos evocadores. Las manifestaciones clínicas del HH se presentan típicamente en la edad puberal, en forma de retraso o ausencia de aparición de los caracteres sexuales secundarios. En la adolescencia, los varones suelen presentar: hábito eunucoide (relación segmento superior/segmento inferior menor a 1 y una envergadura de los brazos 6 cm mayor que la talla), voz aguda, escaso vello, testes prepuberales y pene infantil. Las formas parciales pueden presentar un inicio puberal hasta un estadio II-III de Tanner pero, posteriormente, se detiene su progresión. La composición corporal muestra menor masa muscular y una distribución del tejido graso tipo ginoide; puede aparecer ginecomastia y la edad ósea está retrasada. Estos pacientes no experimentan el brote puberal y mantienen una velocidad de crecimiento prepuberal. Sin embargo, la talla adulta de estos pacientes no se suele ver afectada. En las mujeres, la presentación clínica habitual es de amenorrea primaria (90%) y menos del 10% tienen alguna forma de sangrado antes de instaurarse la amenorrea. La pubarquia es muy variable, oscilando desde ausencia hasta una aparición normal de vello pubiano(14).
Además de la forma congénita de inicio en la infancia, se ha descrito una forma de HHI de inicio en la edad adulta. En este grupo de pacientes, la pubertad se presenta normalmente pero, posteriormente, existe regresión de la función reproductora, caracterizada por disminución de la libido, impotencia, pérdida de masa ósea y fracturas, oligoazoospermia con testes normales y amenorrea secundaria, e infertilidad. Aunque el HH se había considerado como una condición patológica de por vida, por su etiología genética, existe un 20% de pacientes que exhiben una recuperación del eje HHG tras el tratamiento. No existen factores clínicos predictivos de esta recuperación, si bien, es más frecuente en varones que en mujeres. Estas formas de HH reversibles requieren un seguimiento, ya que se han demostrado recaídas en la evolución posterior(13,15).
Síndrome de Kallmann
El SK es la causa más frecuente de deficiencia aislada de gonadotropinas, aunque en algunas series, el HHI supera al SK. El SK es un cuadro clínico heterogéneo, tanto a nivel clínico como genético, más prevalente en varones que en mujeres, con una incidencia de 1/10.000 y 1/50.000, respectivamente(16). Los casos esporádicos son más frecuentes que las formas hereditarias. Los pacientes con SK tienen manifestaciones clínicas asociadas como defectos craneofaciales (paladar hendido, paladar ojival, hiperterlorismo); agenesia dental (hipodontia); sordera neurosensorial; anomalías digitales (clinodactilia, sindactilia, campodactilia); agenesia renal unilateral; defectos septales cardiacos y defectos neurológicos asociados como: alteraciones oculomotoras (ptosis y movimientos anormales), sinquinesias bimanuales o movimientos en espejo de las manos, y ataxia cerebelosa. Otras anomalías descritas han sido: agenesia de los conductos deferentes y alteraciones esqueléticas (mano/pie hendido o en “pinza de langosta”). Cuando hay grandes deleciones y afectan a genes contiguos, se han descrito otras manifestaciones como ictiosis, retraso mental o talla baja severa(17). En el SK, se han descrito tres modelos de herencia, lo que sugiere la participación de diferentes genes: recesivo ligado al cromosoma X, autosómico dominante y autosómico recesivo.
SK ligado al gen ANOS1. El gen ANOS1 (previamente conocido como KAL1) está localizado en la región pseudoautosómica del cromosoma X (Xp22.3) y codifica la proteína anosmina-1. Entre los miembros de una misma familia y entre familias diferentes, se ha descrito una penetrancia incompleta del hipogonadismo y de la anosmia; sin embargo, los pacientes con mutaciones del gen ANOS1 muestran característicamente una afectación severa de la función reproductora. Las mutaciones en el gen ANOS1 se han encontrado en el 8-11% de las formas esporádicas y en el 14-50% de las formas recesivas ligadas al X.
SK ligado a otros genes. Otros genes implicados en el SK en un modelo de herencia autosómico dominante son: el gen FGR1 (receptor 1 del factor de crecimiento de fibroblastos), responsable de aproximadamente el 10-17% de los SK; FGF8, FGF17 (factor de crecimiento de los fibroblastos 8 y 17); CHD7 (cromodominio helicasa 7 de unión al ADN); HS6ST1 (heparán-sulfato 6-O-sulfotransferasa-1); SOX10 (SRY-box-10); SEMA3A; SEMA7A (semaforina 3A y 7A); WDR11 (proteína 11 contiene repetición dominio WD) e IL17RD (receptor D interleukina 7). Genes implicados en un modelo de herencia autosómico recesivo son: PROKR2 (receptor 2 de la prokineticina), PROK2 (prokineticina-2), FEZ1 (fasciculación y elongación de la proteína zeta 1) y NELF (factor LHRH nasal embriológico)(13).
Hipogonadismo hipogonadotropo idiopático
El déficit aislado de GnRH sin anosmia, conocido como HH aislado o idiopático, suele ser esporádico, aunque puede transmitirse con carácter autosómico recesivo. Desde el punto de vista neuroendocrino, ambos grupos (HHI y SK) son indistinguibles. Entre los genes asociados al HHI se encuentran: GNRHR (receptor del factor liberador de gonadotropinas), GNRH1 (factor liberador de gonadotropinas), KISS1R (receptor 1 de kispeptina), KISS1 (kispeptina), TACR3 (neurokinina B), TAC3 (receptor de neurokinina B), FGFR1, PROK2, PROKR2, FGF8, FGF17, NELF, CHD7, HS6T1 y WDR11. Se estima que aproximadamente el 40-50% de las formas autosómico recesivas y el 10-16% de las formas esporádicas de HHI, se deben a mutaciones en el gen GNRHR. La hipoplasia suprarrenal congénita ligada al X se asocia a hipogonadismo hipogonadotropo y está causada por una mutación en el gen NROB1 (receptor nuclear subfamilia 0 grupo B1, también conocido como DAX1), que se localiza en la región Xp21.3-21.2. También se han descrito otros HHI asociados a déficit del gen de la leptina (LEP), su receptor (LEPR) y del gen de la prohormona convertasa (PCSK1), cuadros que asocian obesidad mórbida de inicio precoz(17,18). El examen detallado de las familias con HHI revela que son genes con baja penetrancia y expresividad variable, lo que ha llevado a la hipótesis del efecto sinérgico aditivo de diferentes genes (oligogenicidad), para explicar fenotipos más severos. Hasta en un 7-15% de los casos de HHI, se ha descrito una compleja transmisión hereditaria con la participación de dos o más genes patogénicos (herencia digénica/oligogénica), entre los que se encuentran fundamentalmente: PROK2, PROKR2 y FGFR1(19).
Hipogonadismo por déficit aislado de LH
Se debe a una mutación del gen LHB (subunidad β de LH). Los varones afectos tienen hábito eunucoide, testículos normales y espermatogénesis reconocible, aunque incompleta, que se restablece con el tratamiento con gonadotropinas (hCG: gonadotropina coriónica humana). Rara vez son fértiles, ya que la espermatogénesis no es activa. La biopsia testicular muestra diversos grados de maduración de la espermatogénesis y escasas células de Leydig. No se ha descrito en mujeres.
Hipogonadismo por déficit aislado de FSH
En mujeres, se presenta como amenorrea e infertilidad, con niveles elevados de LH, indetectables de FSH y disminución de estradiol y progesterona. En varones, se presenta como hipogonadismo con niveles bajos de testosterona y azoospermia. Se debe a una mutación en el gen FSHB (subunidad β de FSH).
Deficiencia combinada de gonadotropinas: panhipopituitarismo
Mutaciones en los factores de transcripción hipofisarios, pueden producir potencialmente hipogonadismo hipogonadotropo aislado o asociado a otros déficits hormonales adenohipofisarios. Aunque en su mayoría son esporádicos, pueden existir casos familiares genéticos. Se han descrito alteraciones en el gen PROP1 (Prophet of Pit 1), LHX3, LHX4 (Lim Homeobox 3 y 4), SOX2 (SRY box 2) y HESX1 (Homeobox ES 1), entre otros. Se han descrito mutaciones en el gen SOX2 y HESX1, en pacientes con displasia septo óptica. La displasia septo óptica está causada por una anomalía del prosencéfalo, consistente en hipoplasia del nervio óptico y ausencia del septum pellucidum. Se asocia a disfunción hipotálamo-hipofisaria y defectos hormonales variables, entre los que puede incluirse el déficit de gonadotropinas. Defectos de línea media como: ausencia del septum pellucidum, labio leporino, paladar hendido o anomalías en el desarrollo del prosencefalo, pueden asociar disfunción hipotálamo-hipofisaria e HH(10,11).
Hipogonadismo asociado a síndromes polimalformativos
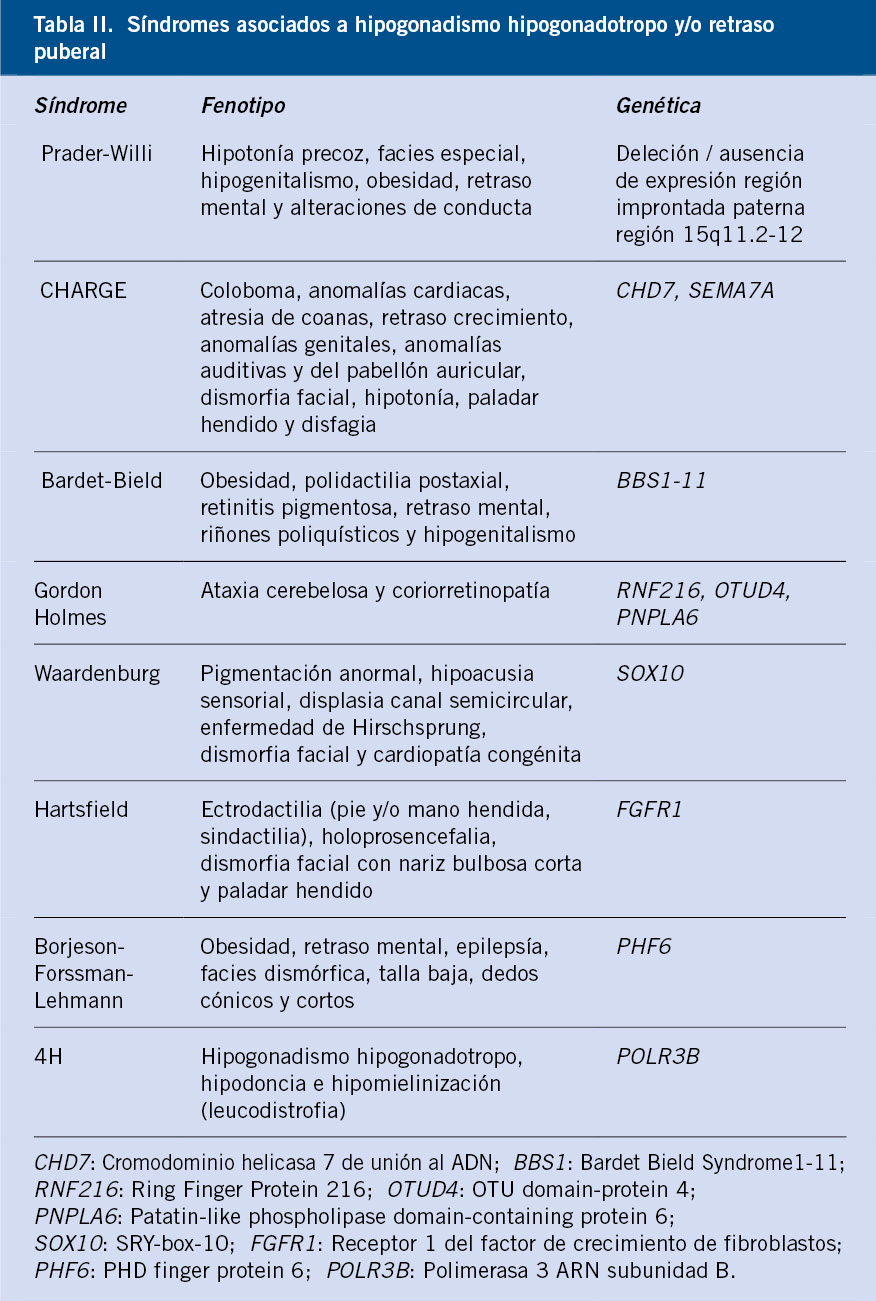
Se han descrito un importante número de cuadros sindrómicos que, junto a otras manifestaciones clínicas, presentan retraso puberal y/o HH (Tabla II).
Hipogonadismo hipogonadotropo adquirido
Orgánico o secundario a lesiones hipotálamo-hipofisarias
El hipogonadismo se puede producir por trastornos o lesiones del SNC que dan lugar a una disfunción hipotálamo-hipofisaria por compresión, infiltración o destrucción, en dependencia de la lesión. Las características más importantes son el comienzo tardío de la sintomatología, que incluye fallo del crecimiento, asociación con defectos hormonales de la hipófisis anterior y posterior, y presencia elevada de trastornos visuales. Los tumores extraselares interfieren con la síntesis de GnRH y, en consecuencia, con la estimulación y secreción de gonadotropinas hipofisarias; los más frecuentes son los craneofaringiomas, seguidos de los germinomas, astrocitomas y gliomas. En el caso de los adenomas hipofisarios, sobre todo, si son macroadenomas, se produce un efecto masa que interfiere con la función hipofisaria. Los prolactinomas pueden debutar como pubertad retrasada o detenida. La histiocitosis de células de Langerhans es una causa poco frecuente de HH, que se produce por infiltración de la región hipotálamo-hipofisaria. Otras enfermedades infiltrativas que pueden causar HH son hemocromatosis y sarcoidosis. La radioterapia craneal (tumores/leucemias) puede ocasionar un descenso gradual de la función hipotálamo-hipofisaria e hipogonadismo. El efecto de la radioterapia es acumulativo y el hipopituitarismo aparece siguiendo un patrón previsible: el déficit de GH aparece en primer lugar, seguido por la afectación de las gonadotropinas, ACTH y TSH. Se aconseja una evaluación anual del eje gonadotropo en pacientes que han recibido dosis >= a 30 Gy y/o con antecedentes de cirugía en el área hipofisaria. La prevalencia de déficit de gonadotropinas en niños supervivientes de cáncer es del 10,8%(20). El uso de radioterapia con protones ha disminuido la prevalencia de hipopituitarismo.
Funcional o secundario a enfermedades crónicas
Todas las enfermedades crónicas pueden ocasionar un retraso del crecimiento y de la pubertad en función de la severidad y de la duración de la noxa, y suponen un motivo muy frecuente de consulta (Tabla III).
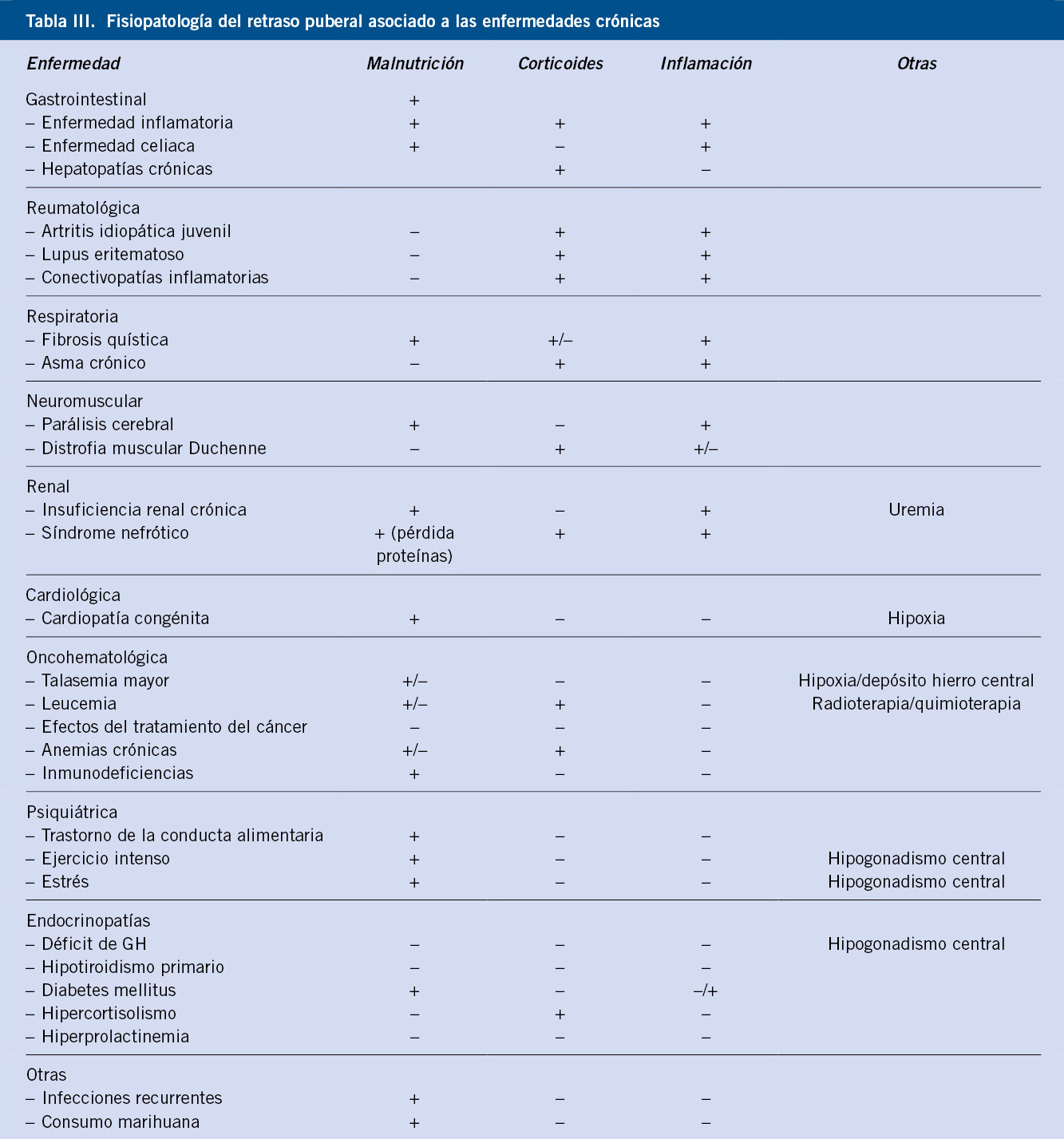
Los mecanismos fisiopatológicos son múltiples e implican modificaciones del eje somatotropo (GH-IGF1) y gonadal a través tres factores: estado de inflamación crónica (citoquinas proinflamatorias como TNF-α , interleukina-1 y -6), déficit nutricional (hipoleptinemia) y tratamiento crónico con glucocorticoides(21). Ambos fenómenos, hipocrecimiento y retraso puberal, se consideran mecanismos adaptativos a una situación de desnutrición, absoluta o relativa, que revierten al tratar la causa. Estos pacientes tienen, además, riesgo de sufrir una menor ganancia de masa ósea. La presentación clínica simula el RCCD con hipocrecimiento, edad ósea retrasada y niveles bajos de gonadotropinas y esteroides. La anorexia nerviosa y los trastornos de la conducta alimentaria pueden cursar con amenorrea primaria o secundaria, en ocasiones, incluso antes de la pérdida de peso; en otras ocasiones, presentan una pubertad detenida con posterior progresión de los caracteres sexuales secundarios al mejorar la enfermedad de base. El ejercicio físico intenso, particularmente en actividades que requieren bajo peso, como el ballet y gimnasia rítmica, puede alterar la pubertad, especialmente en el sexo femenino, en forma de retraso y enlentecimiento de la pubertad, amenorrea primaria y una mayor incidencia de irregularidades menstruales tras la menarquia. El estrés mantenido interfiere en el ciclo menstrual y puede producir anovulación crónica y amenorrea hipotalámica funcional(11,21).
Es especialmente importante descartar causas anatómicas en las mujeres que presentan desarrollo puberal normal y amenorrea primaria como himen imperforado, septum transverso vaginal o síndrome de Rokitansky (espectro de anomalías en el desarrollo de los conductos de Muller, caracterizado por aplasia congénita de útero y de los 2/3 superiores de vagina que puede ir o no asociado a otras malformaciones). En estas pacientes, el eje HHG es normal y el defecto es una anomalía anatómica.
Hipogonadismo hipergonadotropo
El hipogonadismo hipergonadotropo se caracteriza por un fallo gonadal primario y niveles elevados de LH y/o FSH. Puede ser de causa congénita, como el síndrome de Turner y el síndrome de Klinefelter, o adquirida en donde el tratamiento del cáncer por radioterapia y/o quimioterapia, adquiere especial importancia.
El hipogonadismo hipergonadotropo se debe a una insuficiencia gonadal primaria, bien por causas congénitas o adquiridas, con una actividad normal del eje hipotálamo-hipofisario, en el que la disminución de esteroides gonadales produce una pérdida del retrocontrol negativo de las gonadotropinas, por lo que se incrementan sus niveles plasmáticos.
Hipogonadismo hipergonadotropo congénito
Sexo masculino
Síndrome de Klinefelter
El síndrome de Klinefelter es la causa más frecuente de hipogonadismo masculino, presente en 1/1.000 y 1/500 recién nacidos vivos varones. La anomalía cromosómica más frecuente es el cariotipo 47,XXY (80%). Se han descrito formas variantes como cariotipo normal; 48,XXXY; mosaicismos (47,XXY/46,XY; 47,XXY/46,XX; 47,XXY/46,XY/45,X); y formas con varios gonosomas X o Y. La talla suele estar en percentiles normales o altos, habitualmente por encima de lo esperado por su familia, y, en la edad adulta, alcanzan una talla superior a la talla genética. La expresión clínica varía en función de la edad. En la edad prepuberal puede pasar desapercibido y se suele presentar de tres formas:
1. Asintomático o con alteraciones en el fenotipo tipo dismorfia facial discreta (epicanto, mentón pequeño, hipertelorismo).
2. Alteraciones neuropsicológicas, ya que, aunque el coeficiente intelectual suele ser normal, o estar en el límite inferior, es característico el retraso en el área del lenguaje, detectable ya en edades tempranas.
3. Alteraciones de los genitales externos en forma de hipogenitalismo y/o criptorquidia, pero ello es muy poco frecuente.
En la edad de la adolescencia, se observa generalmente desarrollo puberal espontáneo, bien a una edad normal o retrasada, pero en cualquier caso, la pubertad no es progresiva, ya que los testículos permanecen pequeños, entre 3 y 6 ml, siendo de consistencia dura a la palpación. El grado de virilización suele ser satisfactorio con pene de tamaño normal, ya que la función de la célula de Leydig se afecta tardíamente. Es frecuente la ginecomastia uni o bilateral (60%). La mayoría de los pacientes tiene infertilidad por azoospermia, aunque se han descrito casos de espermatogénesis incompleta y de paternidad. Presentan con más frecuencia que la población general cáncer de mama, enfermedades autoinmunes, síndrome metabólico, osteoporosis y trastornos venosos(6).
Síndromes de regresión testicular
Incluyen una serie de trastornos caracterizados porque, en un momento determinado del desarrollo fetal y por un mecanismo desconocido (oclusión vascular o torsión), tiene lugar la regresión de la gónada. Si el trastorno se produce después de la fase crítica de diferenciación sexual, entre la semana 12 y 14, da lugar a la anorquia congénita (1/20.000 varones). Cuando se produce después de la semana 14, da lugar al síndrome de testículos rudimentarios que se caracteriza por testes pequeños, criptorquidia y micropene, generalmente hipospádico, por insuficiencia intersticial y tubular en grado variable.
Otros
Las mutaciones inactivadoras del gen del receptor de LH (resistencia a la LH), en función de su severidad, causan un espectro variable de manifestaciones, desde una feminización completa a una insuficiente virilización con micropene y retraso puberal; este cuadro se asocia con hipoplasia de las células de Leydig. Sin embargo, hasta en un 30-50% de casos con fenotipo compatible con hipoplasia de células de Leydig, no se han encontrado mutaciones en el gen del receptor de LH, indicando que otros genes están implicados en la resistencia a las gonadotropinas. La deficiencia del receptor de FSH (resistencia a FSH) puede producir en el varón: oligozoospermia y subfertilidad. También destacan los defectos enzimáticos de la biosíntesis de testosterona o de la acción de la testosterona, bien por deficiencia de la 5α reductasa o por ausencia del receptor de andrógenos (síndrome de insensibilidad a los andrógenos) que, en las formas parciales, cursa con una insuficiente virilización(10).
Sexo femenino
Síndrome de Turner
Se define por la combinación de una clínica determinada (talla baja, disgenesia gonadal, fenotipo característico) y ausencia total o parcial de un cromosoma X, bien como línea regular o como mosaicismo. Su incidencia es de 1/2.500 RN vivas. Un 50-60% de las pacientes presentan una fórmula 45,X y, en el resto, se han descrito otras anomalías citogenéticas (deleción, isocromosoma y cromosoma en anillo). Con síntomas menos evidentes aparecen los mosaicismos, siendo el más frecuente 45,X/46,XX. El retraso de crecimiento es muy constante y la mayoría se sitúan por debajo del percentil 3 antes de la edad puberal y alcanzan una talla adulta 20 cm inferior a la media. Presentan fenotipo característico (hábito recio, cara triangular y cuello alado) y anomalías radiológicas como: paladar ojival, cúbito valgo o acortamiento del 4º metacarpiano. La disgenesia gonadal determina una ausencia de pubertad e infertilidad, si bien, hasta un 20% puede iniciar un desarrollo puberal espontáneo, pero no es progresiva. Presentan mayor prevalencia de alteraciones cardiovasculares (válvula aórtica bicúspide, coartación de aorta e hipertensión), malformaciones renales, otitis de repetición y pueden tener otras endocrinopatías asociadas como: tiroiditis de Hashimoto, obesidad e intolerancia a los hidratos de carbono y diabetes tipo 2(22).
Disgenesia gonadal pura
La padecen las pacientes que tienen fenotipo femenino normal e infantilismo sexual, sin alteraciones cromosómicas estructurales y su cariotipo puede ser XX o XY. En la forma XX, los genitales externos e internos son femeninos, semejantes al síndrome de Turner, con: cintillas gonadales, vagina, útero y trompas. No tienen fenotipo turneriano. La talla es normal o alta, con hábito eunucoide. No tienen desarrollo puberal y presentan amenorrea primaria. También hay formas incompletas, aunque terminan presentando amenorrea secundaria. En esta forma es rara la transformación maligna de las gónadas, por lo que no está indicada la gonadectomía. El diagnóstico se confirma por laparoscopia. La forma XY fue descrita por Swyer en 1955; las pacientes tienen cintillas gonadales y genitales externos femeninos. La mayoría presenta pubertad retrasada y niveles anormalmente elevados de gonadotropinas séricas. El tratamiento incluye la extirpación quirúrgica de las cintillas gonadales, debido al riesgo de malignización, siendo el gonadoblastoma el tumor más frecuente(10).
Otros
La deficiencia del receptor de LH produce en la mujer oligomenorrea, amenorrea secundaria y anovulación. La deficiencia del receptor de FSH causa un fenotipo moderado con desarrollo puberal normal y amenorrea primaria o secundaria. El déficit de síntesis de estrógenos, bien por deficiencia de 17α hidroxilasa/17-20 desmolasa (gen CYP17A1) o por déficit de aromatasa (gen CYP19A1), puede producir hipogonadismo hipergonadotropo. La ausencia completa del receptor de andrógenos cursa en sujetos XY, con un fenotipo de mujer y amenorrea primaria (síndrome de insensibilidad completa a los andrógenos).
Síndromes como Noonan, distrofia miotónica de Steintert, Smith-Lemli-Opitz o Alström, pueden asociar fallo gonadal primario en ambos sexos.
Hipogonadismo hipergonadotropo adquirido
En el varón, una causa frecuente es la orquitis y, de estas, la más típica es la secundaria a la parotiditis. Puede producirse atrofia testicular por castración traumática o quirúrgica, y las lesiones han de ser bilaterales para producir hipogonadismo. El tratamiento quimioterápico puede ocasionar daño gonadal directo en función de la dosis y dar lugar, a largo plazo, a: oligospermia o azoospermia, disminución del tamaño testicular y elevación de la FSH. Al igual que en la mujer, los agentes alquilantes son los más lesivos. La radioterapia puede dañar la función testicular. En el varón, y a diferencia de la mujer, el daño gonadal puede afectar de manera diferente a la función endocrina (la esteroidogenésis por la célula de Leydig es más resistente) y a la fertilidad (el túbulo seminífero y la célula de Sertoli son más sensibles). Dosis de 2-4 Gy se han asociado con daño transitorio de la espermatogénesis y dosis de 12 Gy pueden producir una disfunción de la célula de Leydig(20). En ocasiones, puede ocurrir un hipogonadismo mixto (primario y central), cuando se combinan los efectos de la quimioterapia y la radioterapia corporal total, sobre todo, si se ha administrado un refuerzo de radioterapia craneal. El testículo es más radiosensible a edades infantiles, a diferencia de lo que ocurre en la mujer, ya que el ovario es más sensible a edades puberales o postpuberales.
En la mujer, puede producirse también hipogonadismo hipergonadotropo por una castración traumática o quirúrgica. La ooforitis autoinmunitaria produce lesión ovárica por infiltración de linfocitos, dando lugar a detención de la pubertad y amenorrea primaria; puede presentarse de forma aislada, pero la mayoría de las veces, se asocia a otras endocrinopatías autoinmunitarias. El tratamiento con quimioterapia produce menos daño en el ovario que en el testículo. Los agentes alquilantes son especialmente dañinos para la reserva folicular (ciclofosfamida, busulfan, ifosfamida y procarbazina) de una manera dosis-dependiente. La afectación del ovario es mayor si se dan en estadio puberal avanzado y a mayor edad; si se da en la prepubertad, el daño ovárico puede recuperarse. El daño es más prevalente si se asocia radioterapia en forma de irradiación corporal total, previa a un trasplante de células hematopoyéticas, o de radioterapia craneoespinal para tumores del SNC. El daño ovárico ocasionado por la radioterapia depende de la dosis. Es frecuente la insuficiencia ovárica completa con dosis superiores a 5-10 Gy directas y se manifiesta clínicamente por detención de la pubertad, amenorrea, infertilidad y menopausia precoz(20). Otras causas de hipogonadismo primario son la insuficiencia ovárica prematura idiopática y la galactosemia, único error congénito del metabolismo conocido, que tiene un efecto directo sobre la gónada; actúa sobre el ovario, pero no sobre el testículo(10,11).
Diagnóstico
La evaluación diagnóstica incluye una anamnesis familiar y personal detallada, y exploración clínica completa (desarrollo genital, crecimiento, búsqueda de anomalías asociada y edad ósea). Las pruebas complementarias deben estar orientadas en función de la sospecha clínica y el diagnóstico diferencial entre RCCD y el HH idiopático requiere una valoración especializada.
Los signos clínicos de alerta del retraso puberal que obligan a emprender un estudio son: la falta de desarrollo mamario en una chica 13 años de edad y un volumen testicular inferior a 4 ml en un chico de 14 años de edad. También requiere evaluación, la falta de progresión de la pubertad, incluso si la edad de inicio ha sido normal. El diagnóstico se basa en: una evaluación clínica, pruebas complementarias generales y especificas del eje HHG y en estudios orientados a realizar un diagnóstico etiológico (v. Algoritmo al final del artículo).
Evaluación clínica
En la historia clínica, la anamnesis familiar ha de investigar la presencia de pubertad retrasada en familiares de primer y segundo grado (edad de afeitado, edad de la menarquia, talla familiar) y búsqueda de otros signos orientativos (infertilidad/subfertilidad, hiposmia/anosmia, consanguinidad). La anamnesis personal debe hacer hincapié en: datos perinatales (embarazo, parto, auxología perinatal); primera infancia (micropene, criptorquidia); historia de enfermedades previas; tratamientos crónicos; patología tumoral (cirugía, radioterapia, quimioterapia); trastornos de la conducta alimentaria; estado nutricional; psicológico; ejercicio intenso y/o estrés; y evaluación de la olfación (hiposmia/anosmia).
El examen físico incluye: exploración física general, genital y auxológica, además del estudio de la maduración ósea. Es necesario una reconstrucción de la gráfica de crecimiento (talla, peso, IMC, segmentos corporales); ya que, el retraso de crecimiento suele ir asociado al retraso puberal, especialmente en: RCCD, enfermedades crónicas y síndrome de Turner. En los pacientes con HH congénito, la talla y la velocidad de crecimiento suelen ser normales en la infancia y tienen ausencia de brote puberal. La adrenarquia está retrasada en el RCCD y suele ocurrir a una edad normal en el HH. Se deben buscar signos específicos de: enfermedades crónicas (enfermedad inflamatoria, desnutrición, enfermedades sistémicas, etc.); rasgos fenotípicos específicos (síndromes de Klinefelter, Turner, CHARGE); evaluación de la presencia de obesidad y retraso cognitivo (síndrome Prader Willi, Bardet Bield); y anomalías propias del SK (hiposmia/anosmia, agenesia renal/malformación renal unilateral, paladar hendido, hipoacusia sensorial uni o bilateral). Se requiere una exploración detallada del desarrollo puberal (volumen testicular, desarrollo mamario, inicio, progresión) y, en mujeres con amenorrea primaria, descartar causas anatómicas.
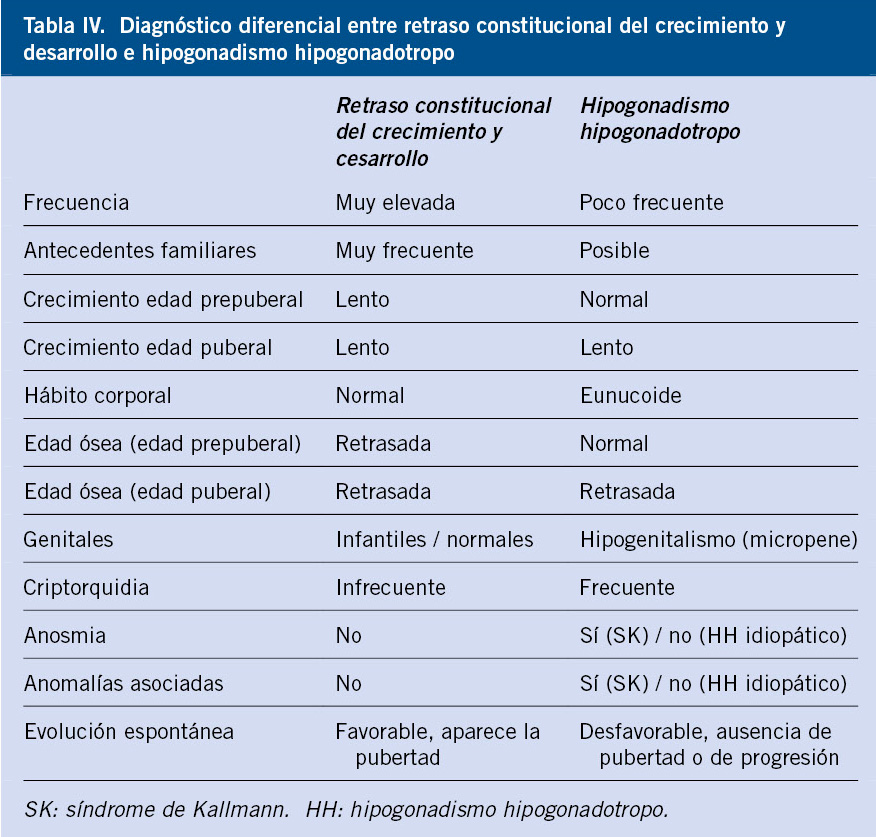
La edad ósea está retrasada de manera importante en el RCCD y es acorde al desarrollo puberal. En las enfermedades crónicas y déficits hormonales (GH y/o TSH), también está retasada y refleja el tiempo de evolución. En el HH suele estar normal o ligeramente retrasada y, a diferencia del RCCD, no se observa desarrollo puberal espontáneo cuando la edad ósea es de 13 años en la mujer y de 14 años en el varón(11). En la tabla IV se presenta el diagnóstico diferencial clínico entre RCCD e HH.
Pruebas complementarias generales
Si la historia y exploración hacen pensar en una enfermedad crónica, habrá que realizar pruebas orientadas a la sospecha clínica. Inicialmente, puede ser necesario realizar ciertos exámenes básicos como: bioquímica general, marcadores de enfermedad celiaca, hemograma, hierro, ferritina, VSG y, cuando se sospeche déficit hormonal, realizar estudio de hormonas tiroideas, prolactina y de IGF-I.
Estudio del eje hipotálamo-hipofiso-gonadal
El nivel sérico de gonadotropinas define el hipogonadismo hipogonadotropo e hipergonadotropo al estar disminuidas y aumentadas, respectivamente. El valor de su determinación depende de la edad y de la intensidad del trastorno. En la edad prepuberal, la determinación basal de las gonadotropinas no es informativa, ya que no discrimina entre la normalidad y patología. En la adolescencia, las determinaciones basales de gonadotropinas tienen un valor limitado, ya que no son capaces de discriminar entre estadio prepuberal y estadios iniciales de la pubertad. En el HH se observan valores muy bajos o indetectables, pero dichos valores pueden ser normales en las formas parciales. Unos niveles de basales de LH > 0,65 UI/L excluyen un HH grave, no así una forma parcial(23); otros autores han propuesto unos niveles de FSH < 1,2 UI/L, como punto de corte para diferenciar los HH en los varones(24).
En el lactante existe un período inicial tras el nacimiento en el que el eje HHG está activado, periodo conocido como minipubertad, que dura 6 meses en el varón y 24 meses en la mujer. En ese periodo, las gonadotropinas basales sí son informativas y unos niveles disminuidos en los primeros 3-6 meses de vida orientan hacia un HH congénito. En el hipogonadismo hipergonadotropo (disgenesia gonadal, anorquia, síndrome de regresión testicular, síndrome de Klinefelter, Turner), los niveles de gonadotropinas, especialmente la FSH, pueden estar elevados en la minipubertad (<2-3 años) y/o a partir de los 10 años de edad en las niñas.
Los niveles de testosterona y estradiol son de poca utilidad en la edad prepuberal y en las fases iniciales de la pubertad, ya que sus niveles séricos se encuentran por debajo del nivel de detección en la mayoría de los inmunoanálisis. Cuando se usan inmunoensayos sensibles con umbral de detección de 10 pg/ml, las mujeres con HH tienen niveles de estradiol inferiores o en el límite bajo de la normalidad. En el varón, cuando el testículo alcanza un volumen testicular de 8-10 ml, el nivel de testosterona aumenta por encima del intervalo prepuberal (1,7 nmol/l o 0,5 ng/ml) y, en ese estadio, su determinación puede ser útil para descartar un hipogonadismo. Habitualmente, los pacientes con HH congénito tienen niveles de testosterona inferiores a 0,86 ng/ml o 3 nmol/l(14).
La principal dificultad es el diagnóstico diferencial entre el RCCD y el HH, sobre todo en su forma idiopática, aislada y parcial, y cuando la edad ósea no está en rango puberal(23). La realización del test de GnRH permite evaluar la integridad del eje y valorar su respuesta, que es dosis dependiente y variable en función de la edad. En condiciones normales con una edad ósea superior a 11 años, se demuestra un incremento significativo de la respuesta de LH (multiplica por 3-6 veces su valor con respecto al nivel basal) y de FSH (multiplica por 1,5-2 veces su valor basal). Los pacientes con RCCD presentan una respuesta más intensa que los pacientes con HH. En el HH, la respuesta al test de GnRH es nula; pero, en función del nivel de afectación (hipotalámico o hipofisario) y de la severidad del mismo, se pueden observar respuestas variables; en un 30% de casos, se pueden solapar sus valores con los encontrados en el RCCD. Se ha propuesto una respuesta de LH < 5,8 UI/l y FSH < 4,6 UI/l como los puntos de corte de mejor sensibilidad y especificidad(24,25). La variabilidad en la respuesta observada, hace que en la práctica esta prueba no permita diferenciar estas dos situaciones. Se han utilizado modificaciones del test de GnRH convencional administrando GnRH en infusión iv durante 4 horas o mediante la administración de bolos repetidos. También, se han utilizado diferentes análogos de GnRH como estímulo liberador de gonadotropinas como: naftarelina, triptorelina o leuprolide, pero la falta de validación de los resultados obtenidos en series pequeñas, ha limitado mucho su aplicación. El test de hCG, restringido al sexo masculino, mide la respuesta de testosterona por estimulación de las células de Leydig; los pacientes con RCCD muestran una respuesta positiva y los HH ausente o débil (< 2,3 ng/mL o 7,97 nmol/l). El test de GnRH cuando se combina con un test de hCG (medición de testosterona a los 3 y 19 días), ofrece una mayor significación diagnóstica con una sensibilidad y especificidad cercana al 100%, describiéndose en el HH, una respuesta significativamente inferior al RCCD(26). La secreción pulsátil de gonadotropinas durante el sueño puede ser de utilidad. Los niños con retraso puberal muestran una secreción pulsátil de LH cuando alcanzan una edad ósea de 12-13 años, no así en los HHI; si bien, con métodos ultrasensibles, se han detectado pulsos de LH de baja amplitud en los HHI. En el varón, las determinaciones de inhibina B y de hormona antimulleriana son útiles para determinar la función de las células de Sertoli durante la infancia y la prepubertad, al estar disminuidas en los hipogonadismos. La determinación de la hormona antimulleriana ha mostrado tener menor sensibilidad y especificidad que la inhibina B. Unos niveles de inhibina B inferiores a 35 pg/ml en varones con un volumen testicular inferior o igual a 3 ml o menor de 65 pg/ml si el volumen es de 3-6 ml orientan hacia un HH(27); si bien, se ha descrito que hasta un 40% de los HH (formas parciales o adquiridas) tienen valores superiores y un 7% de los RCCD tienen valores inferiores(8). No todos los estudios son concordantes con estos resultados. La asociación de inhibina B < a 110 pg/mL y LH basal < 0,3 UI/L aumenta la especificidad al 98%(28). Para otros autores, la combinación de testes <1,1 mL, inhibina B disminuida y LH máxima en el test de GnRH < 4,3 UI/L, se ha propuesto como la mejor manera para diferenciar el HH del RCCD en el varón adolescente(11). Los niveles de inhibina B se correlacionan con el volumen testicular y la función tubular, y unos niveles disminuidos son un factor predictivo de infertilidad; en la mujer, unos niveles disminuidos de hormona antimulleriana, se correlacionan con: menor volumen ovárico, menor reserva de folículos antrales y niveles disminuidos de FSH(14). Recientemente, se ha descrito cómo la infusión de kispeptina produce un pico de LH en el RCCD, pero no en el HHC(8); si bien, formas parciales podrían responder a la infusión continua. La determinación de INSL3 (factor similar a la insulina 3), basal o tras estímulo con hCG, es un marcador en estudio de la función de la célula de Leydig(13), que permitiría diferenciar los RCCD de los HH congénitos. En definitiva, no existe ninguna prueba que permita distinguir con certeza un RCCD de un HH. La prueba de oro es la observación clínica con la constatación de un desarrollo puberal completo a los 18 años (RCCD), frente a un desarrollo ausente (HH severo) o incompleto (HH parcial). Sin embargo, la opción de “esperar y ver” no es posible, ya que los pacientes exigen un diagnóstico precoz. El diagnóstico de RCCD es, en definitiva, un diagnóstico de exclusión(29).
El RCCD cuando se acompaña de talla baja e hipocrecimiento, en ausencia de enfermedad crónica, obliga a realizar un diagnóstico diferencial con el déficit de GH. Ello es especialmente difícil y se fundamenta, sobre todo, en el estudio del patrón de crecimiento, que debe ser interpretado acorde a su ritmo madurativo y a la edad ósea. Para diferenciar estas dos situaciones, se ha propuesto clásicamente realizar un estudio del eje GH-IGF tras primación con esteroides sexuales, pero al no existir criterios de normalidad para interpretar los resultados, su utilización ha caído en desuso.
Estudio etiológico
En ocasiones, el cariotipo puede ayudar al diagnóstico (síndrome de Turner, Klinefelter), pero puede ser necesario realizar estudios genéticos más avanzados (estudio de genes candidatos, panel de genes, exoma, MLPA o cGH array). Es difícil establecer una correlación genotipo-fenotipo en el SK, pero hay ciertas anomalías que se asocian con más prevalencia a ciertos genotipos como: sinquinesia (ANOS1), agenesia dental (FGF8 y FGFR1), anormalidades digitales (FGF8 y FGFR1) e hipoacusia (CHD7)(30). La RM craneal permite el estudio de patología orgánica intracraneal, anomalías del área hipotálamo hipofisaria o de la línea media y estudio de la vía olfatoria (agenesia o hipoplasia de los bulbos o nervios olfatorios típica del SK). La ecografía abdominal puede ser necesaria para descartar: anomalías vaginouterinas (agenesia o malformación uterina, septum vaginal); estudio de tamaño y morfología de los ovarios (disgenesias gonadales); descartar malformaciones asociadas (renales) o localización y tamaño de testes criptorquídicos. En ocasiones, puede ser necesaria realizar una laparoscopia (disgenesias gonadales, síndrome de Swyer y síndrome de Rokitansky).
Tratamiento
El RCCD no suele requerir tratamiento, salvo que exista una afectación psicológica importante, y bastará con tranquilizar a la familia y al paciente. El tratamiento del hipogonadismo depende de la etiología y tiene como objetivos fundamentales inducir el desarrollo puberal, establecer una pauta de mantenimiento en la edad adulta y preservar la fertilidad.
Retraso constitucional del crecimiento y el desarrollo
El RCCD es una variante de la normalidad y, por lo tanto, en la gran mayoría de casos, únicamente será necesario tranquilizar al adolescente y a la familia, explicarle la benignidad y transitoriedad del proceso, y bastará con hacer un seguimiento clínico para comprobar un desarrollo puberal normal. Aquellos casos en los que exista afectación psicológica y repercusiones emocionales (menor autoestima, fracaso escolar) y sociales (aislamiento), serán candidatos a un tratamiento hormonal(31).
En el varón se administran dosis bajas de ésteres de testosterona en forma de preparados depot (50 mg/mes intramuscular en forma de cipionato), a partir de los 14 años de edad cronológica, o 12-12,5 años de edad ósea, durante un periodo de 3-6 meses. La testosterona estimula el crecimiento, induce la aparición de caracteres sexuales secundarios y como a dosis bajas no se inhiben las gonadotropinas, se favorece el desarrollo puberal. No se considera necesario iniciar el tratamiento por debajo de esa edad, por el riesgo de acelerar la edad ósea y afectar a la talla adulta. El primer ciclo de tratamiento debe ir seguido de un periodo de observación clínica (6 meses) y, si es necesario, se puede repetir la administración en forma de un segundo ciclo; si tras ello no se produce un inicio del desarrollo puberal, es muy posible que se trate de un HH. La oxandrolona ha quedado en desuso y la utilización de los inhibidores de aromatasa de nueva generación (letrozole) permanece en investigación, habiéndose publicado estudios prometedores(32).
En la mujer es menos frecuente su indicación y se aconsejarían dosis bajas de estrógenos a partir de los 13 años de edad cronológica u 11 años de edad ósea. Se utiliza 17β estradiol (vía oral 5 microgramos/kg/d o, preferiblemente, transdérmico 3,1-6,2 microgramos/día que equivale a 1/8-1/4 de parches de 25 microgramos), durante 3-6 meses, para ir aumentando en ciclos sucesivos, si es necesario.
Hipogonadismos
La etiología marca la pauta de actuación. El retraso puberal de las enfermedades crónicas exige el tratamiento de la enfermedad de base y, si es necesario inducir el desarrollo puberal, se utilizan pautas semejantes al RCCD. En el HH congénito del varón, el tratamiento del micropene se ha basado clásicamente en el uso de ésteres de testosterona intramuscular a unas dosis de 25 mg/mes durante 3 meses en el primer año de vida; otras opciones, son el uso tópico de cremas de testosterona al 5% o de dihidrotestosterona. La criptorquidia requiere el uso de la cirugía antes de los 12-18 meses de vida y el tratamiento con hCG o GnRH ha mostrado muy poca utilidad. Existen estudios que han utilizado la administración sc de gonadotropinas (rLH y rFSH) en el primer año de edad para simular la minipubertad fisiológica del lactante. Se ha demostrado un efecto beneficioso sobre el crecimiento del pene y la función testicular, valorada por el incremento del volumen testicular, ya que se estimula la proliferación de las células de Sertoli y de los túbulos de seminíferos, y aumento de los niveles de inhibina B, testosterona y hormona antimulleriana. Todavía no se conoce si esta pauta puede mejorar la respuesta posterior al tratamiento para inducir el desarrollo puberal y la fertilidad pero en principio sería más beneficiosa que la administración de testosterona(14,33)y sería la primera opción a considerar.
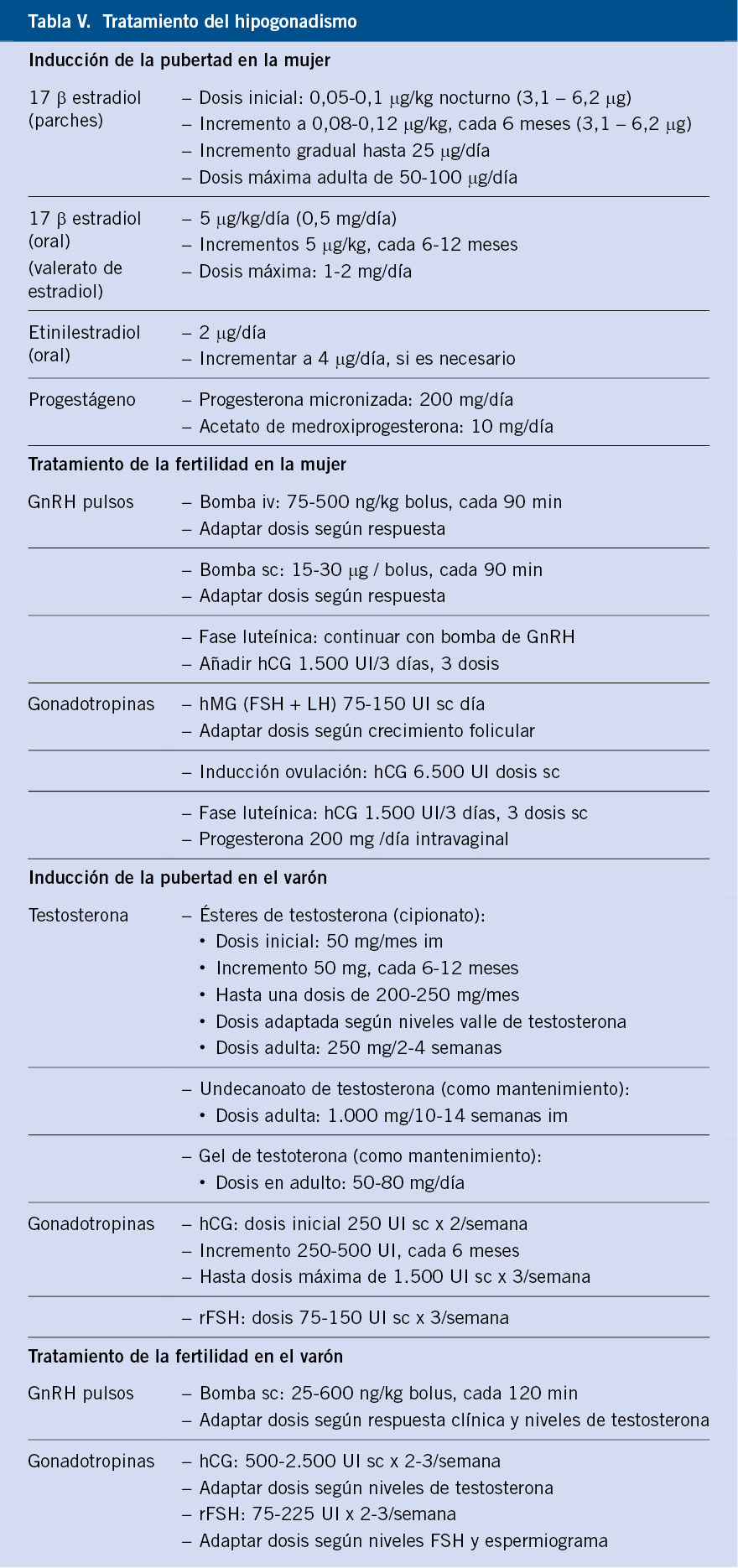
Los objetivos del tratamiento del hipogonadismo en el adolescente y edad adulta son: inducir el desarrollo de los caracteres sexuales secundarios, acelerar el crecimiento, inducir el estirón puberal y optimizar la talla adulta, asegurar una libido y una potencia sexual normal en el varón, preservar la fertilidad cuando sea posible y evitar las complicaciones cardiovasculares, metabólicas, psicosociales y osteoporosis (Tabla V).
Inducción de los caracteres sexuales secundarios
En el varón, la testosterona es un tratamiento eficaz para la inducción y el mantenimiento de los caracteres sexuales secundarios, y para la estimulación del crecimiento. Existen preparados por vía oral, parenteral y transdérmica. Se inicia a una edad ósea de 12-12,5 años. La mayoría de autores utilizan cipionato de testosterona a una dosis inicial de 50 mg/mes (35 mg/m2/mes) durante 6 meses, seguido por 100 mg/mes (70 mg/m2/mes) durante un año, hasta llegar a 200-250 mg/mes (140-150 mg/m2/mes) en los primeros dos años de tratamiento, y así alcanzar unos niveles de testosterona entre 15-20 nmol/l (4,3-5,7 ng/ml); la dosis adulta es de 200-250 mg/15-21 días. En adolescentes con diagnóstico tardío, se puede iniciar el tratamiento a dosis mayores (100 mg/mes) para conseguir una virilización más rápida. Una alternativa a la vía parenteral, es la administración oral de undecanoato de testosterona a la edad de 13 años y en dosis de 20-40 mg/día durante 6-12 meses, incrementando la dosis a 80 mg/día a los 14 años de edad; tiene el inconveniente de que tiene una vida media muy corta, produce niveles de testosterona erráticos y en España no está comercializada. En la edad adulta y como mantenimiento, se prefiere el uso de 1.000 mg/12 semanas de undecanoato de testosterona im. Hay preparados de testosterona de uso tópico (gel o parches) que son más útiles como mantenimiento que como inducción de la pubertad. La testosterona no produce aumento del volumen testicular ni estimula la espermatogénesis, ya que para ello se requiere un incremento de la testosterona intratesticular. Por ello, si en el curso del tratamiento con testosterona se observa un aumento del volumen testicular, se debe pensar en una forma reversible de HH; en ese caso, se debe suspender el tratamiento y realizar seguimiento, ya que estas formas pueden recidivar más tarde.
En los HH, se pueden utilizar las gonadotropinas para inducir el desarrollo puberal. Existen diversos protocolos, bien de hCG sola (dosis inicial de 250 UI sc x 2/semana y aumento progresivo, según respuesta clínica y analítica) o combinada con rFSH (75-150 UI sc x 3/semana), que se añadiría cuando se alcanzan unos niveles de testosterona de 5,2 nmol/l (1,5 ng/mL). En pacientes con formas severas de HH de inicio prepuberal, se ha demostrado beneficioso la primación previa con rFSH antes del tratamiento combinado de hGC + rFSH(34) monitorizado por el volumen testicular y la inhibina B. La administración de gonadotropinas tiene, como inconveniente, la necesidad de una buena adherencia, ya que requiere de inyecciones frecuentes durante un periodo prolongado(33,35).
En la mujer, los estrógenos están disponibles en preparados naturales (17β estradiol y estrógenos equinos) y sintéticos (etinil estradiol). En la mujer, los objetivos del tratamiento son alcanzar un adecuado desarrollo mamario que asegure un bienestar psicológico, estimular el crecimiento uterino, optimizar el crecimiento y asegurar una buena ganancia de masa ósea. La pauta de tratamiento debe iniciarse con dosis bajas, incrementándose progresivamente, con el propósito de imitar la evolución puberal fisiológica. No se ha definido la edad idónea de comienzo, pero se recomienda hacia los 11 años de edad ósea. La pauta de elección es la vía transdérmica con 17β estradiol. Se prefiere la vía transdérmica, ya que es más fisiológica y natural, tiene menor hepatotoxicidad al evitar el paso inicial por el hígado, mejor perfil cardiovascular y menor riesgo trombótico, menor interferencia con la secreción de IGF-I y mejor feminización que los estrógenos equinos o sintéticos. Se inicia con dosis bajas (aproximadamente 1/8 o 1/4 de la dosis adulta) para irla aumentando gradualmente cada 6-12 meses. La dosis inicial es de 0,05-0,1 μg/kg/día (equivale aproximadamente a 3 μg día o 1/8 de parche de 25 μg); en los primeros 4-6 meses, se administra solo en periodo nocturno. Posteriormente, se mantendría todo el día y se administraría 2 veces por semana; en el segundo año, se aumentaría a 0,2 μg/kg/día (6 μg/día o 1/4 de parche de 25 μg). La dosis se iría aumentando progresivamente (incrementos de 0,05-0,1 μg/kg cada 6 meses) hasta llegar gradualmente a la dosis de 25 μg/día. La dosis adulta de sustitución es de 50-100 μg/día. La monitorización del tratamiento se realiza con la clínica (desarrollo mamario y velocidad de crecimiento), niveles de estradiol, volumen uterino, tensión arterial, edad ósea y densidad mineral ósea. Tras 2-3 años de tratamiento sustitutivo, o antes si aparece sangrado menstrual, es necesario añadir un progestágeno cíclico (en forma progesterona natural micronizada 200 mg/día o acetato de medroxiprogesterona 10 mg/día, del día 10 al día 21 de cada ciclo) para protección uterina y establecer ciclos menstruales regulares. Una vez completado el desarrollo puberal, es necesario establecer terapia hormonal sustitutiva de mantenimiento (asociación estrógenos-progestágenos en forma cíclica o continua)(36,37).
Tratamiento de la fertilidad
Las gonadotropinas o la administración pulsátil de GnRH se pueden utilizar para inducir la fertilidad en el HH, pero no en el hipogonadismo primario.
En el varón, la espermatogénesis depende de la concentración intratesticular de testosterona sintetizada en la célula de Leydig tras la acción de la LH, y no del nivel de testosterona circulante, y de la acción de la FSH sobre la célula de Sertoli y la maduración de las espermatogonias; por ello, la fertilidad depende del uso de GnRH o de la administración exógena de gonadotropinas (hCG sola o combinada con FSH). El tratamiento con hCG puede ser útil para completar el desarrollo puberal e inducir la fertilidad; si tras 6-12 meses de monoterapia no se constata una respuesta positiva, se podría añadir rFSH. Se han publicado diferentes pautas y, en la actualidad, se prefiere la vía sc antes que la im. La administración de GnRH es una opción lógica en casos de lesiones de origen hipotalámico y ausencia de daño hipofisario y, debido a su secreción episódica, se requiere una administración pulsátil en forma de minibomba de infusión vía sc o iv; de esta manera, se incrementa la síntesis de gonadotropinas y la maduración testicular. Los pacientes que peor responden son aquellos con signos clínicos de ausencia de “minipubertad” (criptorquidia, micropene, testes pequeños, niveles bajos de inhibina B). Por el contrario, en las formas parciales o adquiridas, con inicio del desarrollo testicular, volumen superior a 4 mL y posterior detenimiento, responden mucho mejor. En el adolescente sin desarrollo puberal, el uso de gonadotropinas (hCG sola o combinada con FSH) puede inducir la pubertad y estimular la fertilidad, y se ha demostrado que la terapia combinada hCG+FSH tiene mejor resultado sobre la fertilidad que la monoterapia de hCG(38). En los casos severos se ha visto que el tratamiento previo con FSH, antes de los pulsos de GnRH o de las gonadotropinas, y un inicio precoz del tratamiento, son dos factores que se relacionan con una mejor fertilidad(31). En general, se puede decir que el tratamiento con gonadotropinas mejora la espermatogénesis(34), pero raras veces la normaliza; si bien, un recuento bajo de espermatozoides no presupone infertilidad. En el síndrome de Klinefelter, la extracción y criopreservación de espermatozoides previa a la degeneración de los túbulos seminíferos y al uso de testosterona, es una opción futura y en investigación para el tratamiento de la fertilidad.
En la mujer con hipogonadismo hipogonadotropo, el tratamiento con bolos de GnRH o gonadotropinas puede permitir inducir la ovulación y alcanzar un embarazo(31); el pronóstico y éxito del tratamiento es bueno, ya que el HH no se asocia con una menor reserva ovárica. En la mujer, la aplicación de GnRH pulsátil, si se dispone, es el tratamiento ideal de la infertilidad producida por insuficiencia hipotalámica, y su administración prolongada normaliza generalmente las secreciones hipofisaria y gonadal, incluyendo ovulación y embarazos en una tasa próxima a la observada en mujeres normales. En las mujeres con fallo gonadal primario, las técnicas de reproducción asistida ofrecen una solución a la infertilidad; la criopreservación de oocitos antes del tratamiento gonadotóxico es una opción a considerar en mujeres con enfermedades oncológicas.
Función del pediatra de Atención Primaria
El pediatra de Atención Primaria es quién primero se va a enfrentar con el posible caso de un adolescente con retraso puberal que habitualmente consultará, bien por afectación psicológica o por retraso de crecimiento, y talla baja. Debe ser capaz de identificar a un varón que, a los 14 años, tenga un volumen testicular inferior a 4 ml o a una mujer que, a los 13 años, no haya iniciado el desarrollo mamario. Una vez detectado el caso debería realizar un diagnóstico diferencial que diferencie tres situaciones posibles. La posibilidad más frecuente es que se trate de un RCCD y, para ello, se apoyará en anamnesis familiar, exploración física, valoración auxológica y realización de edad ósea; en segundo lugar, descartar una posible enfermedad crónica de base y, para ello, realizará una anamnesis personal completa, especialmente dirigida al ámbito nutricional; y en tercer lugar, pensar en la posibilidad de un hipogonadismo. El conocimiento del RCCD, como una variante de la normalidad del desarrollo puberal, es labor del pediatra de Atención Primaria quién tranquilizará a la familia y al adolescente, haciéndoles ver la benignidad y la transitoriedad de dicha condición. En la valoración del desarrollo puberal, tan importante es detectar un retraso en su inicio como una detención en su progresión y/ una falta de desarrollo puberal completo (testes de > de 15 ml en varón o menarquia en la mujer). La detención de una pubertad iniciada es preocupante y sugestivo de patología adquirida y, por ello, debe ser motivo de derivación. Es importante saber diferenciar una variante de la normalidad de trastorno patológico del desarrollo puberal y, en este segundo caso, derivar a una Unidad de Endocrinología Pediátrica precozmente, para realizar un diagnóstico y tratamiento precoz.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1. Soriano-Guillén L. Pubertad normal y variantes. Pediatr Integral. 2015; 19(6): 380-8.
2.** Ojeda S, Lomniczi A. Puberty in 2013: unravelling the mystery of puberty. Nat Rev Endocrinol. 2014; 10: 67-9.
3. Ferrández Longás A, Baguer L, Labarta JI, Labena C, Mayayo E, Puga B, et al. Longitudinal study of normal spanish children from birth to adulthood: anthropometric, puberty, radiological and intellectual data. Ped Endocrinol Rev. 2005; 2: 425-62.
4. Ferrández A, Carrascosa A, Audí L, Baguer L, Rueda C, Bosch-Castañe J, et al. Longitudinal pubertal growth according to age at pubertal growth spurt onset: data from a spanish study including 458 children (223 boys and 235 girls). J Pediatr Endocrinol Metab. 2009; 22: 715-26.
5.** Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the World, secular trends, and changes alter migration. Endocr Rev. 2003; 24: 668-93.
6.** Pozo Román J, Muñoz Calvo MT. Pubertad precoz y retraso puberal. Pediatr Integral. 2015; 19(6): 389-410.
7.** Dye AM, Nelson GB, Diaz-Thomas A. Delayed puberty. Pediatr Ann. 2018; 47: e16-e22.
8.** Raivio T, Miettinen PJ. Constitutional delay of growth versus congenital hypogonadotropic hypogonadism: genetics, management and updates. Best Pract Res Clin Endocrinol Metab. 2019. doi.org/10.1016/j.beem.2019.101316.
9. Wehkalampi K, Widén E, Laine T, Palotie A, Dunkel L. Pattern of inheritance of constitucional delay of growth and puberty in families of adolescente girls and boys referred to specialist pediatric care. J Clin Endocrinol Metab. 2008; 93: 723-28.
10.** Mayayo E, Labarta JI, Sinués B, Ferrández A. Pubertad retrasada. Hipogonadismos. En: Tratado de Endocrinología Pediátrica. Pombo M (4ª ed.) Madrid: McGraw-Hill Interamericana; 2009. p. 524-50.
11.*** Howard SR, Dunkel L. Delayed puberty: phenotypic diversity, molecular genetic mechanisms, and recent discoveries. Endocr Rev. 2019; 40: 1285-317.
12. Zhu Y, Chan YM. Adult consequences of self-limited delayed puberty. Pediatrics. 2017; 139: e20163177.
13.** Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. European consensus statement on congenital hypogonadotropic hypogonadism: pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015; 11: 547-64.
14.*** Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev. 2019; 40: 669-710.
15. Ribeiro SR, Vieira TC, Abucham J. Reversible Kallmann syndrome: report of the first case with a KAL1 mutation and literature review. Eur J Endocrinol. 2007; 156: 285-90.
16. Dodé C, Hardelin JP. Kallmann syndrome. Eur J Hum Genet. 2009; 17: 139-46.
17. Maione L, Dwyer AD, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, et al. Genetic conuseling of congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. 2018; 178: R55-R80.
18. Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol. 2018; 178: 377-88.
19. Francou B, Paul C, Amazit L, Cartes A, Bouvattier C, Albarel F, et al. Prevalence of KISS1 Receptor mutations in a series of 603 patients with normosmic congenital hypogonadotrophic hypogonadism and characterization of novel mutations: a single-centre study. Hum Reprod. 2016; 31: 1363-74.
20.*** Wei C, Crowne E. The impact of childhood cancer and its treatment on puberty and subsequent hypothalamic pituitary and gonadal function, in both boys and girls. Best Pract Res Clin Endocrinol Metab. 2019. doi.org/10.106/j.beem.2019.101291.
21.*** Kao KT, Zacharin M, Wong SC. Pubertal anomalies in adolescents with chronic disease. Best Pract Res Clin Endocrinol Metab. 2019. doi.org/10.106/j.beem.2019.04.009.
22. Gravholt C, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017; 177: G1-G70.
23.*** Harrington J, Palmert MR. Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available tests. Harrington J, Palmert MR. J Clin Endocrinol Metab. 2012; 97: 3056-67.
24. Grinspon RP, Ropelato G, Gottlieb S, Keselman A, Martínez A, et al. Basal follicle-stimulating hormone and peak gonadotropin levels after gonadotropin-releasing hormone infusion show high diagnostic accuracy in boys with suspicion of hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2010; 95: 2811-8.
25.** Albitbol L, Zborovski S, Palmert MR. Evaluation of delayed puberty: what diagnostic tests should be performed in the seemingly otherwise well adolescent? Arch Dis Child. 2016; 101: 767-71.
26. Segal TY, Mehta A, Anazodo A, Hindmarsch PC, Dattani MT. Role of GnRH and hCG stimulation test in differentiating patients with hypogonadotropic hypogonadism from those with constitucional delay of growth and puberty. J Clin Endocrinol Metab. 2009; 94: 780-5.
27. Coutant R, Biette-Demeneix E, Bouvattier C. Baseline inhibin B and anti-Mullerian hormone measurements for diagnosis of hypogonadotropic hypogonadism in boys with delayed puberty. J Clin Endocrinol Metab. 2010; 95: 5225-32.
28. Binder G, Schweizer R, Blumenstock G, Braun R. Inhibin B plus LH vs GnRH agonist test for distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism in boys. Clin Endocrinol. 2015; 82: 100-5.
29.** Wei C, Crowne EC. Recent advances in the understanding and management of delayed puberty. Arch Dis Child. 2016; 101: 481-8.
30. Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, et al. Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab. 2013; 98: E943-E953.
31.*** Howard SR, Dunkel L. Management of hypogonadism from birth to adolescence. Best Pract Res Clin Endocrinol Metab. 2018; 32: 355-72.
32. Varimo T, Huopio H, Kariola L, Tenhola S, Voutilainen R, Toppari J, et al. Letrozole versus testosterone for promotion of endogenous puberty in boys with constitutional delay of growth and puberty: a randomised controlled trial phase 3 trial. Lancet Child Adolesc Health. 2019. doi.org/10.1016/S2352-4642(18)30377-8.
33.** Zacharin M. Pubertal induction in hypogonadism: current approaches including use of gonadotropins. Best Pract Res Clin Endocrinol Metab. 2015; 29: 367-83.
34.** Raivio R, Wikstrom AM, Dunkel L. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol. 2007; 156: 105-11.
35.** Dunkel L, Quinton R. Induction of puberty. Eur J Endocrinol. 2014; 170: R229-R239.
36.*** Matthews D, Bath L, Högler W, Mason A, Smyth A, Skae M. Hormone supplementation for pubertal induction in girls. Arch Dis Child. 2017; 0: 1-6. doi: 10.1136/archdischild-2016-311372.
37.** Klein KO, Rosenfield RL, Santen RJ, Gawlik AM, Backeljauw PF, Gravholt CH, et al. Estrogen replacement in Turner syndrome: literature review and practical considerations. J Clin Endocrinol Metab. 2018; 103: 1790-803.
38.** Rohayem J, Hauffa BP, Zacharin M, Kliesch S, Zitzmann M. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism ?. A multicentre prospective study of hGC/rFSH treatment outcomes during adolescence. Clin Endocrinol. 2017; 86: 75-87.
Bibliografía recomendada
– Howard SR, Dunkel L. Delayed puberty: phenotypic diversity, molecular genetic mechanisms, and recent discoveries. Endocr Rev. 2019; 40: 1285-317.
Este trabajo es una excelente revisión de los mecanismos fisiopatológicos del retraso constitucional de la pubertad y realiza una actualización de las diferentes causas y síndromes que producen un retraso puberal. Estos autores son líderes en el conocimiento de esta patología y es un artículo muy completo, que combina el abordaje clínico con los aspectos de fisiopatológicos.
– Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev. 2019; 40: 669-710.
Excelente revisión del hipogonadismo hipogonadotropo congénito, señalando los aspectos clínicos y genéticos más relevantes. Realiza igualmente una profunda revisión de los aspectos diagnósticos y terapéuticos. Artículo de obligada lectura y referencia.
– Kao KT, Zacharin M, Wong SC. Pubertal anomalies in adolescents with chronic disease. Best Pract Res Clin Endocrinol Metab. 2019. doi.org/10.106/j.beem.2019.04.009.
Revisión exhaustiva del retraso puberal secundario a enfermedades crónicas de elevado interés, tanto a nivel clínico como fisiopatológico. Muy necesaria su lectura para el pediatra de Atención Primaria.
– Harrington J, Palmert MR. Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available tests. Harrington J, Palmert MR. J Clin Endocrinol Metab. 2012; 97: 3056-67.
Excelente revisión de la literatura sobre el diagnóstico diferencial entre el retraso constitucional del crecimiento y desarrollo, y el hipogonadismo hipopogonadotropo. Es un artículo muy didáctico que presenta las diferentes pruebas utilizadas para realizar este diagnóstico diferencial, sus limitaciones y su aplicabilidad. Es un artículo muy citado por todos los autores lo que da idea de su interés.
| Caso clínico |
|
Motivo de consulta Varón remitido por su pediatra a los 14,5 años por presentar ausencia de progresión del desarrollo puberal y detención del crecimiento. Inició la pubertad a los 12,5 años (testes 6 ml y talla 156 cm), lo envía por no progresión del volumen testicular. Antecedentes personales Embarazo y parto normales. Peso: 3.970 g. Longitud: 53 cm. Perímetro cefálico: 34 cm. No anoxia. Adenoidectomizado a los 2,5 años. Traumatismo craneoencefálico con pérdida de conciencia y TAC normal a los 8 años. Asintomático. Antecedentes familiares Padre: 186 cm (percentil 90); pubertad normal. Madre: 173,5 cm (percentil 97); menarquia a los 12,5 años. Talla genética: 186,25 cm (percentil 90). G.A.V. 2.0.2. Hermano afecto de síndrome de Klinefelter. Exploración física Edad cronológica: 14,5 años. Talla: 157,5 cm (percentil 10-25). Peso: 57 kg (percentil 50). Testes de 6 ml, pene II, pubarquia II (Tanner II). Refiere estar asintomático, escolaridad normal, episodio aislado de cefalea hace 2 semanas. En la exploración física, el paciente tiene buen estado general, auscultación cardiopulmonar normal, tensión arterial en percentiles normales y, en la exploración neurológica, no hay nada destacable.
|