 |
| Temas de FC |
G.Á. Martos-Morenoa,b,c,
J. Argentea,b,c,d
aServicios de Pediatría y Endocrinología. Hospital Infantil Universitario Niño Jesús. Instituto de Investigación La Princesa. Madrid.bDepartamento de Pediatría. Universidad Autónoma de Madrid.cCIBER Fisiopatología Obesidad y Nutrición (CIBEROBN). Instituto de Salud Carlos III.dIMDEA Research Institute on Food and Health Sciences
| Resumen
La obesidad constituye uno de los motivos de consulta más frecuentes en la práctica clínica pediátrica en nuestro medio, debido a su elevada prevalencia. |
| Abstract
Obesity is currently one of the most common consultations in pediatrics due to its high prevalence rate in our environment. |
Palabras clave: Obesidad infantil; Genética; Sindrómica; Comorbilidades; Tratamiento de la obesidad.
Key words: Childhood obesity; Genetic; Syndromic; Comorbidities; Obesity management.
Pediatr Integral 2020; XXIV (4): 220 – 230
Obesidades en la infancia
Introducción y concepto de obesidad infantil
La obesidad en el niño engloba enfermedades etiológicamente muy heterogéneas y constituye una enfermedad ya durante la infancia, no solo un factor de riesgo para la vida adulta.
El concepto de “obesidad infantil” hace referencia a la acumulación excesiva de tejido adiposo que determina afectación física y/o psicológica del niño, conduciendo a un incremento en el riesgo, tanto presente como futuro, de padecer patologías asociadas, así como de mortalidad precoz(1,2).
La cuantificación del contenido graso corporal del niño, necesaria para la definición de obesidad, puede ser realizada mediante la medición directa y precisa, empleando técnicas específicas (bioimpedanciometría, densitometría de absorción dual de rayos X [DEXA] o hidrodensitometría). No obstante, su limitada disponibilidad, duración y coste económico han hecho que, en la práctica clínica, se universalice la estimación indirecta del contenido graso corporal mediante el empleo del índice de masa corporal (IMC = peso [kg] / (talla [m])2), pese a las limitaciones conocidas de esta estimación frente a los métodos de medición directa(1,2).
Las diferencias en la composición corporal determinadas por edad, sexo y grado de maduración puberal en el niño y adolescente, hacen necesario el empleo de un valor estandarizado de IMC en función de la edad y el sexo del niño respecto a unas referencias poblacionales. Este hecho genera intensa controversia referente al establecimiento de los “puntos de corte” y de las referencias poblacionales a emplear. De hecho, pese a los intentos de unificación de distintas instituciones como la OMS (Organización Mundial de la Salud) o la IOTF (International Obesity Task Force)(2), aún a día de hoy, no goza de consenso internacional. En nuestro medio, la Guía de Práctica Clínica para la Prevención y Tratamiento de la Obesidad Infanto-juvenil (actualmente en revisión) postula, como criterios para definir el sobrepeso y la obesidad, los valores de los percentiles 90 y 97, respectivamente, específicos por edad y sexo de la distribución del IMC referido a los datos y curvas de Hernández y cols., de 1988(3).
De acuerdo con lo anteriormente expuesto y, teniendo en cuenta que el establecimiento de comorbilidades asociadas a la obesidad ocurre, con frecuencia, en etapas posteriores de la vida, tampoco existe consenso respecto a la definición del concepto de obesidad mórbida en la infancia y adolescencia. Aunque en la actualidad no existe un consenso unánime para definir la obesidad mórbida en la infancia y la adolescencia, algunos autores y sociedades científicas sugieren que cualquier niño o adolescente con un IMC > a +3,5 DE debería ser incluido en este grupo, ya que este valor es equivalente a la edad de 18 años, con la definición de obesidad de clase III en poblaciones adultas (IMC ≥ 40 kg/m2)(22). Otros autores estiman que las desviaciones del IMC para una determinada edad y sexo expresadas en porcentajes, constituyen una forma práctica de conocer el sobrepeso y de valorar el grado de obesidad. Desviaciones comprendidas entre el 120 y el 140% del percentil 95 del IMC definen la obesidad severa, si están comprendidas entre el 140 y el 160% la obesidad mórbida y si son superiores al 160% la obesidad extrema(23).
Finalmente, tampoco existe consenso sobre la definición de obesidad de inicio precoz, sugiriéndose edades orientativas (inicio por debajo de los 5 años o de los 2-3 años para los más conservadores) para establecer dicho límite(1).
En general, el término “obesidad infantil” hace referencia a un rasgo fenotípico (presencia de excesiva acumulación de tejido adiposo corporal) que, en algunos casos, ya está presente desde edades tempranas de la vida; sin embargo, esta condición puede ser el resultado de un conjunto de patologías heterogéneo y que, por lo tanto, precisan estrategias diagnósticas y terapéuticas orientadas a cada una de sus potenciales etiologías(1,2).
Por lo anteriormente expuesto, en los últimos años se han desarrollado múltiples líneas de investigación que nos han permitido profundizar en el conocimiento de los mecanismos reguladores del balance energético, especialmente de las bases genéticas sobre las que se produce la acumulación patológica de tejido adiposo, así como sobre el análisis de la función (y disfunción como consecuencia de la obesidad) endocrinológica de este(4).
Relevancia epidemiológica de las obesidades en la infancia en nuestro medio
Tras una tendencia ascendente en los 30 años previos, en los últimos 10 años se ha producido una estabilización de la prevalencia de obesidad y sobrepeso infantil.
La prevalencia de la obesidad infantil en nuestro medio es difícil de precisar debido, fundamentalmente: a la escasez de registros epidemiológicos nacionales seriados, a las diferencias metodológicas entre los estudios disponibles y al elevado número de factores (socioeconómicos, culturales y étnicos, entre otros) que pueden sesgar los datos obtenidos en distintas muestras poblacionales. Recientemente se ha publicado (abril 2020 en Rev Esp Card) un trabajo sobre prevalencia de sobrepeso, obesidad y obesidad abdominal en población española entre 3 y 24 años. Estudio ENPE de Javier Aranceta-Bartrina.
El punto de partida fue establecido por los datos aportados por el estudio PAIDOS’84, que reflejaba una prevalencia de obesidad en España del 4,9% en niños de ambos sexos y, posteriormente, el estudio enKid, desarrollado de forma multicéntrica entre los años 1998 y 2000 en 3.534 individuos con edades comprendidas entre los 2 y los 24 años, que proporcionaba una prevalencia global de obesidad infantil del 13,9% (12 y 15,6% en niñas y niños, respectivamente), así como del 12,4% en lo referente a sobrepeso(5).
Entre los datos más recientes, cabe destacar el estudio ALADINO (ALimentación, Actividad física, Desarrollo INfantil y Obesidad), desarrollado en el periodo 2010-2011, sobre una muestra de 7.659 niños y niñas de 6 a 9,9 años. Este estudio estimaba la existencia de un 44,5% de la población con exceso de peso, un 26,2% con sobrepeso y un 18,3% con obesidad(6). Asimismo, son reseñables los datos que muestran la influencia del nivel socioeconómico en la prevalencia de esta enfermedad en nuestro medio, con un incremento de la misma, sobre todo en familias con nivel educativo más bajo(7).
Los datos seriados nacionales más recientes son los comunicados en la Encuesta Nacional de Salud 2017 (publicados por el Instituto Nacional de Estadística en julio de 2018)(8), que refleja una prevalencia de obesidad (9,13 y 9,33%), y de sobrepeso (16,30 y 16,42%) en niñas y niños de 2 a 17 años, respectivamente, similar a la comunicada en los años 2006 y 2012, mostrándose la tendencia a la estabilización de la prevalencia de obesidad infantil en nuestro medio en la última década, de forma similar a lo observado en otros países occidentales(2).
Clasificación etiológica / fisiopatológica de las obesidades pediátricas
En el año 2020, es necesario investigar la etiología subyacente a la obesidad en niños, particularmente cuando esta es muy grave y de inicio precoz.
Entre las causas etiológicas de la obesidad en la edad pediátrica destaca la existencia de alteraciones: genéticas, genómicas, epigenéticas, endocrinológicas o sindrómicas subyacentes que, si bien, constituyen un porcentaje limitado del total de casos de obesidad infantil, este crece de forma continuada al tiempo que lo hacen nuestros conocimientos fisiopatológicos de esta enfermedad(1,2). Sin embargo, en el momento actual, no es posible establecer un diagnóstico etiológico preciso en la mayor parte de los pacientes que desarrollan obesidad en la edad pediátrica, habiéndose empleado tradicionalmente términos como “común” o “exógena”, para definir a esta entidad; si bien, esto solo constituye un reflejo de nuestra limitación para la adecuada caracterización de dicha etiología.
Obesidad “exógena”, “común”, ¿o poligénica?, ¿o idiopática?
Este grupo englobaría a todos aquellos pacientes en los que no es posible establecer de forma unívoca una etiología demostrable de su exceso de peso. En estos pacientes, de forma habitual, la coexistencia de una nutrición hipercalórica e inadecuadamente estructurada y de unos niveles reducidos de actividad física, determinan la acumulación del exceso de energía en forma de tejido adiposo. Sin embargo, no todos los sujetos expuestos al mismo ambiente nutricional y a similares limitaciones de actividad física desarrollan obesidad o lo hacen en similar grado. Esto es debido a que estos factores “exógenos” actúan sobre una base “endógena”, la información genética propia de cada individuo, lo cual explicaría, al menos, en parte, la gran heredabilidad familiar de la obesidad, que se estima en un 50-75% de los casos, incluso probablemente más en los casos graves de inicio precoz, con un creciente número de genes y variantes postulados, entre los que destacan algunos polimorfismos en genes como FTO(1).
Por este motivo, este tipo más común de obesidad debería denominarse “obesidad poligénica”, pues es esta base genética la que determina la susceptibilidad del paciente ante los estímulos ambientales(1). Además, las modificaciones epigenéticas; es decir, aquellas ejercidas por factores ambientales sobre el genoma, sobre todo en fases tempranas del desarrollo, parecen desempeñar una función relevante en el riesgo individual para el desarrollo de obesidad(9).
Por consiguiente, el desarrollo de la obesidad en la mayoría de los niños afectos tiene una etiología multifactorial, que asienta sobre una base poligénica. Esta, tiene per se un efecto limitado sobre el fenotipo y únicamente su combinación con otras variantes predisponentes y, sobre todo, la concurrencia de factores ambientales favorecedores de obesidad, determinarán finalmente el desarrollo de la misma. Por tanto, al no poderse establecer una etiología identificable con los recursos diagnósticos existentes en la actualidad (o aplicables según el entorno asistencial), se podría decir que la etiología de la obesidad en estos pacientes es “idiopática” (lo cual estaría sometido a constante revisión en virtud de los avances en el conocimiento de esta patología y en los procedimientos diagnósticos).
Obesidad de etiología genómica / epigenética / monogénica
Se ha constatado la existencia de variaciones en el número de copias (CNVs, duplicaciones o deleciones) de regiones cromosómicas específicas en pacientes con obesidad grave de inicio precoz, algunas de ellas con fenotipos superponibles a los de entidades sindrómicas conocidas, y con problemas cognitivos asociados, que pueden plantear el diagnóstico diferencial con las mismas. En estas duplicaciones o deleciones, uno o varios genes pueden verse implicados, constituyendo un buen ejemplo de estas causas genómicas de obesidad las deleciones en la región 16p11.2 que incluyen al gen SH2B1(10).
Asimismo, los mecanismos epigenéticos, consistentes en alteraciones en el patrón de metilación de determinados loci, subyacen al desarrollo de algunas entidades sindrómicas que incluyen la obesidad como uno de sus rasgos fenotípicos, como los síndromes de Prader-Willi, Beckwith-Wiedemann o el pseudohipoparatiroidismo; si bien, estos pacientes pueden mostrar escasos o nulos estigmas malformativos que sugieran una entidad sindrómica(11).
La obesidad de etiología monogénica se define como, aquélla que es consecuencia de la presencia de variantes de secuencia patogénicas en un único gen. Excede las pretensiones de este artículo su análisis exhaustivo, que puede ser consultado en artículos de revisión previamente publicados(1,2,12). En cambio, con un objetivo docente, las formas monogénicas de obesidad conocidas hasta la fecha se podrían sistematizar resumidamente en tres grupos.
Mutaciones en genes del sistema adipocito-hipotalámico (eje leptina-melanocortina)
La red específica de neuronas productoras de proopiomelanocortina (POMC) se localiza primordialmente en el núcleo arcuato del hipotálamo, integra la información aferente sobre la energía almacenada periféricamente en el tejido adiposo que ofrece la leptina producida en aquél y señalizan mediante los productos derivados de la POMC tras su fraccionamiento por acción de la proconvertasa 1 (PCSK1), principalmente mediante la fracción alfa de la hormona estimulante de melanocitos (α -MSH). La α -MSH actúa sobre otros núcleos hipotalámicos (fundamentalmente el núcleo paraventricular) por medio de los receptores de melanocortina (MCR), cuya isoforma número 4 (MC4R) es el principal transductor de los impulsos anorexigénicos(1,2,13). Los pacientes con mutaciones en los componentes de esta vía presentan obesidad grave de inicio precoz.
• Leptina (LEP): estos pacientes presentan un peso normal al nacer, incrementándose de forma sustancial durante los 3 primeros meses de vida, así como retraso o ausencia de desarrollo puberal, como consecuencia del hipogonadismo hipogonadotropo que frecuentemente presentan.
• Receptor de leptina (LEPR): la deficiencia del receptor de leptina determina una obesidad muy intensa de inicio temprano, con peso normal al nacimiento, pero con una rápida ganancia antes de los seis meses de edad, acompañada de una intensa resistencia a la acción de la insulina. Estos pacientes pueden presentar hipogonadismo hipogonadotropo, así como otras deficiencias hormonales adenohipofisarias.
• Proopiomelanocortina (POMC): la deficiencia completa de POMC ocasiona insuficiencia suprarrenal en el período neonatal, por falta de síntesis de la hormona adrenocorticotrópica (ACTH) en las células antehipofisarias. Por consiguiente, estos pacientes requieren tratamiento con corticosteroides para prevenir crisis de insuficiencia suprarrenal.
Las mutaciones en POMC se describieron inicialmente asociadas a hipopigmentación del cabello, interpretándose este rasgo como consecuencia de la eventual ausencia de MC1R en el melanocito; sin embargo, este rasgo no está presente en todos los pacientes. Suelen mostrar un peso normal al nacimiento, con ganancia ponderal rápida en los primeros seis meses de vida.
Recientemente, se han descrito los primeros casos de pacientes con obesidad grave portadores de variantes en el gen del coactivador del receptor de esteroides número 1 (SRC1), que modula la actividad de POMC inducida por leptina (14).
• Convertasa de proproteínas tipo subtilisina kexina 1 (PCSK1): la inactivación del gen PCSK1 determina obesidad extrema de inicio en etapas muy tempranas de la infancia, que pueden acompañarse de alteraciones en el metabolismo de: hidratos de carbono, hipogonadismo, hipocortisolismo, diabetes insípida, síntomas gastrointestinales (diarrea en la infancia) y concentraciones plasmáticas elevadas de POMC y proinsulina, así como hipoinsulinemia (pues la PCSK1 cataliza el tránsito de proinsulina a insulina).
• Receptor 4 de melanocortina (MC4R): las mutaciones en el gen MC4R cursan con gran obesidad e hiperfagia y, frecuentemente, con hipercrecimiento. Constituyen la causa más frecuente de obesidad humana de etiología monogénica.
En comparación con las mutaciones autosómicas recesivas raras en los genes de LEP, LEPR, POMC y PCSK1, la prevalencia de obesidad causada por mutaciones en heterocigosis en MC4R se estima en torno al 2-5%(1,2,12). La mayoría de las mutaciones de MC4R son heterocigotas, heredadas de forma dominante; si bien, se han descrito casos aislados de homocigosidad o heterocigosidad compuesta con patrón de herencia autosómico recesivo.
Aunque los datos son menos concluyentes que los relativos a MC4R, las mutaciones en el gen del receptor 3 de melanocortina (MC3R) también se relacionan con el desarrollo de obesidad.
Patología en los genes asociados con el desarrollo del hipotálamo
En los últimos años se han descrito, en relación con el desarrollo de obesidad en el ser humano, anomalías en varios genes relevantes en el proceso de desarrollo del hipotálamo, que constituye el centro fundamental para el control de la conducta alimentaria en el sistema nervioso central: SIM1, BDNF, NTRK2 y SH2B1. Sin embargo, los mecanismos precisos por los que sus mutaciones se asocian al desarrollo de obesidad aún están insuficientemente caracterizados. De entre ellos, además de SH2B1 (afectado en las referidas deleciones en 16p11.2), destacan las descripciones de mutaciones puntuales en SIM1 asociadas con el desarrollo de obesidad y rasgos fenotípicos sugerentes de síndrome Prader-Willi (por lo que se sugirió el término fenotipo “Prader-Willi like”). No obstante, en otras familias, la presencia de mutaciones en SIM1 se ha visto asociada exclusivamente a un incremento del riesgo de desarrollo de obesidad, en ausencia de estigmas malformativos ni alteraciones del desarrollo(15).
Obesidad asociada a síndromes polimalformativos
Son muchos los síndromes que se transmiten con un patrón de herencia mendeliano y que cursan con obesidad, como uno de sus rasgos fenotípicos (Tabla I).
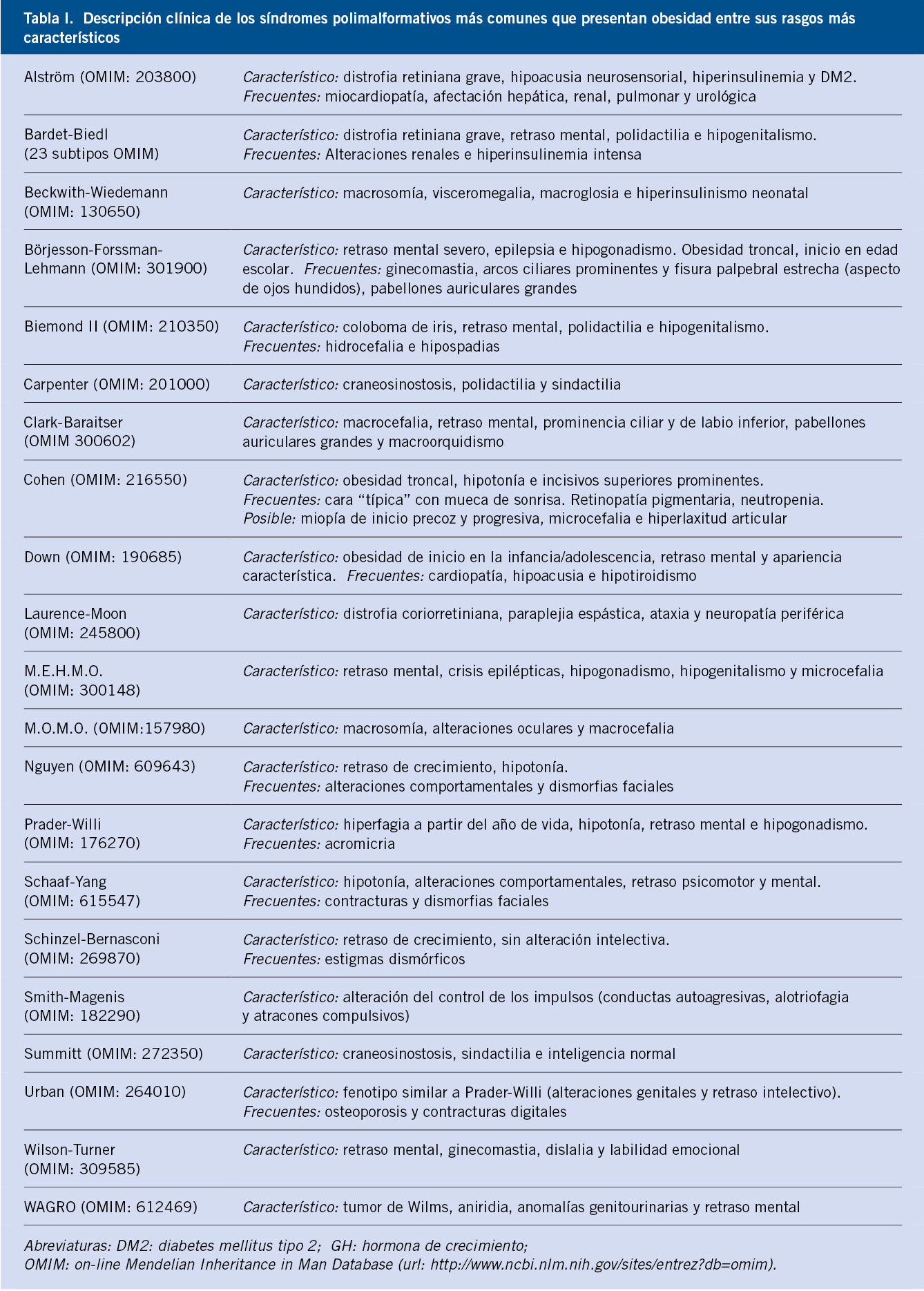
El análisis detallado de todos ellos excede las pretensiones de esta revisión, pero sus características fenotípicas deben ser consideradas para la realización de una anamnesis y exploración física adecuadas. Dentro de su infrecuencia, presentan una mayor prevalencia los síndromes de: Prader-Willi, Bardet-Biedl, Alström y Carpenter, en los que la obesidad constituye uno de los rasgos más destacados.
Debido a las limitaciones intelectuales y físicas que presentan la mayor parte de pacientes afectos de estos síndromes, así como a los tratamientos psicofarmacológicos que con frecuencia reciben por sus alteraciones comportamentales, el desarrollo de obesidad en estos pacientes está influido, en gran medida, por su limitación para la actividad física y las alteraciones en sus patrones de ingesta alimentaria. Sin embargo, algunos pacientes, como los afectos de síndrome de: Prader-Willi, Bardet-Biedl, Almström o Schaaf-Yang, acompañan alteraciones de la señalización de la vía melanocortínica hipotalámica determinantes de su hiperfagia y, al menos, en parte, de su obesidad.
Obesidad secundaria
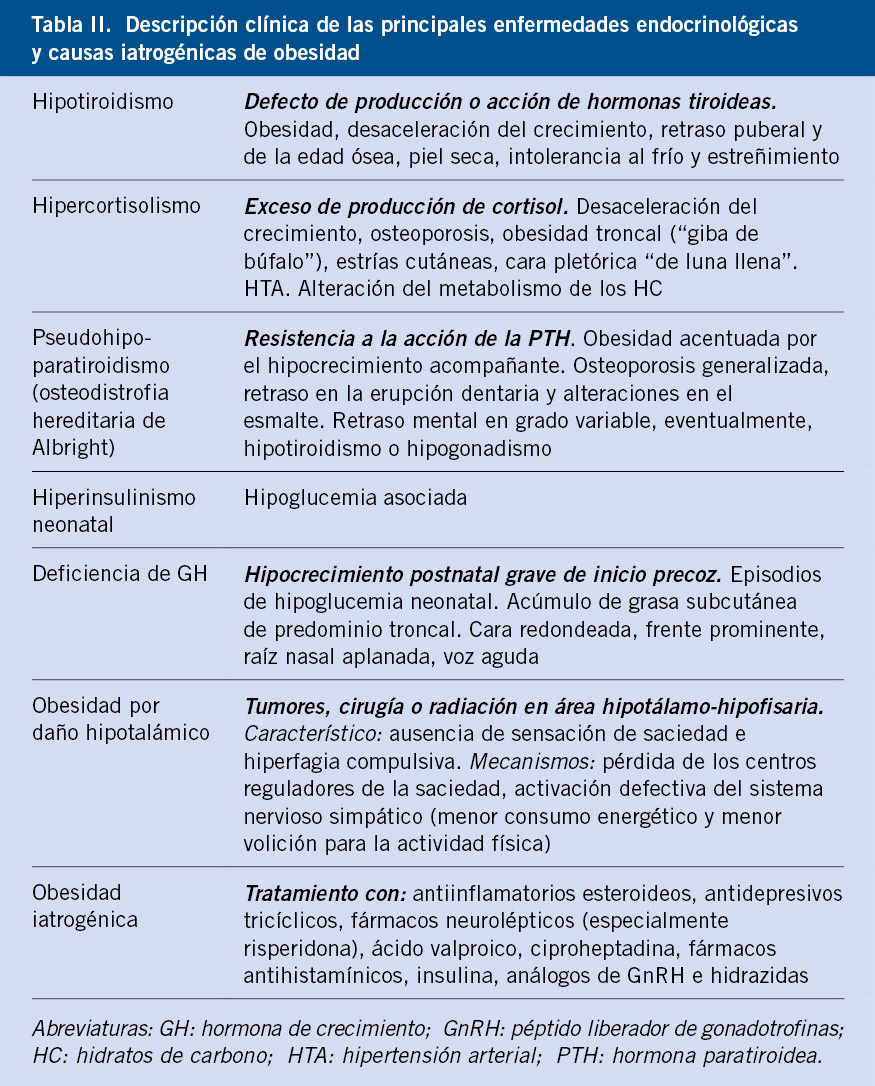
Independientemente del sustrato genético individual y del balance entre ingesta y gasto energético, la presencia de obesidad en el niño puede ser consecuencia de distintas enfermedades, entre las que destacan las patologías endocrinológicas (hipotiroidismo, hipercortisolismo, deficiencia de GH o pseudohipoparatiroidismo), los procesos patológicos o procedimientos terapéuticos que afectan al área hipotálamo-hipofisaria, patologías neurológicas y oncológicas y los tratamientos farmacológicos empleados en las mismas, especialmente con principios psicoactivos (Tabla II)(1,2).
Manifestaciones clínicas y comorbilidades
Como consecuencia del exceso de tejido adiposo, es posible objetivar una serie de alteraciones en los diferentes órganos y sistemas, hacia cuyos signos y síntomas es preciso orientar la anamnesis, la exploración física y las eventuales exploraciones complementarias necesarias. Un análisis exhaustivo de las mismas excede nuestras pretensiones; si bien, las comorbilidades metabólicas merecen especial atención, debido a su eventual papel en el riesgo cardiovascular en la vida adulta. En la tabla III, se enumeran algunas de las comorbilidades más frecuentemente observadas, organizadas por órganos y aparatos, remitiendo al lector, para más información, a revisiones previas donde se detallan las mismas(1,2).
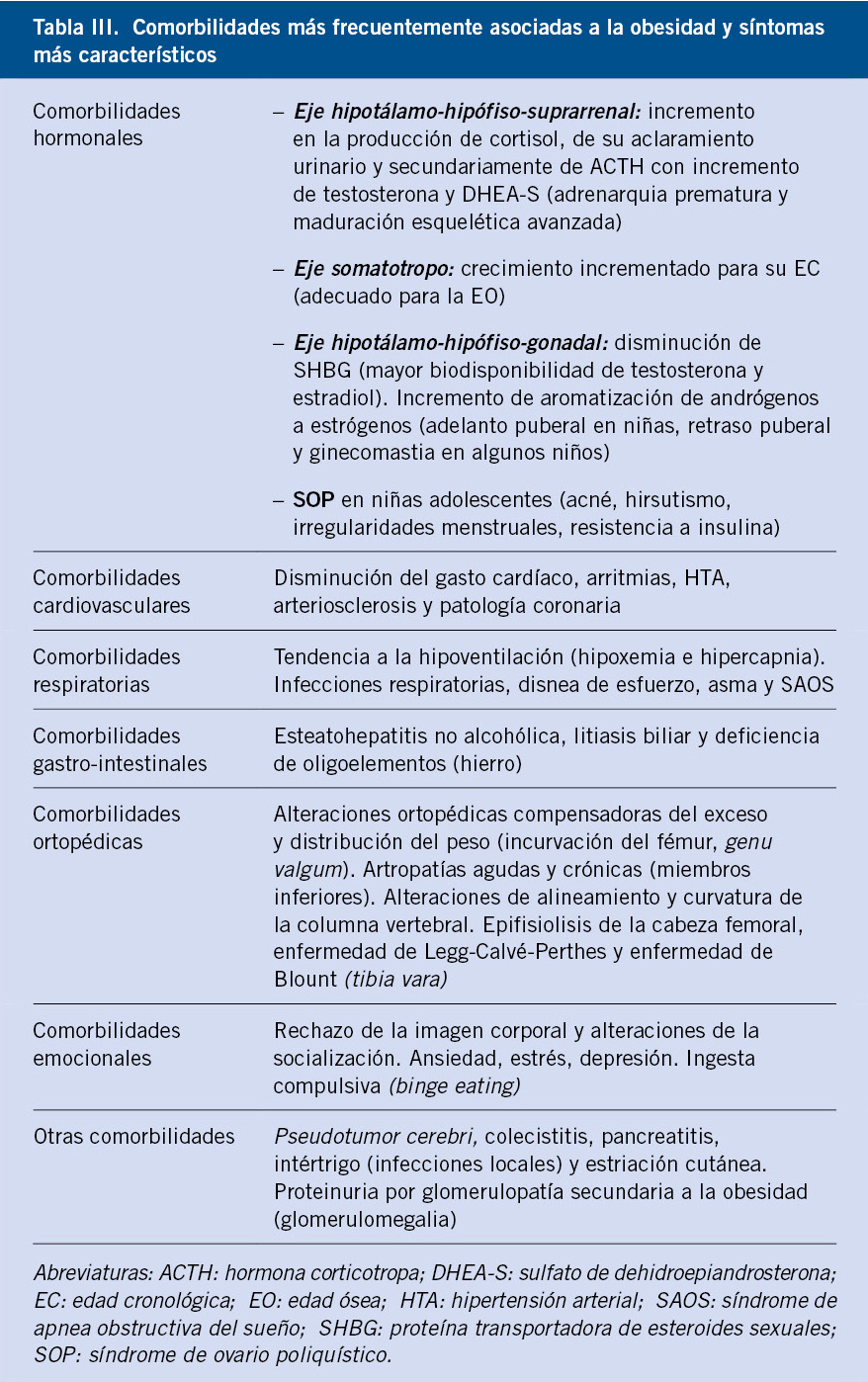
La complicación metabólica más precoz y prevalente en la obesidad infantil en nuestro medio, es la resistencia a la captación de glucosa inducida por insulina o resistencia insulínica (RI), junto con la hipertrigliceridemia y la disminución de los niveles de HDL colesterol.
Es extraordinariamente infrecuente la diabetes mellitus tipo 2 (DM2), aún en presencia de obesidad grave(16). Entre las condiciones consideradas como “estados prediabéticos” por la Asociación Americana de Diabetes(17), la presencia aislada de una alteración de glucemia en ayunas (AGA, 100-110 mg/dl) o de una HbA1c entre 5,7 y 6,4% no suelen mostrar asociación significativa con otras comorbilidades metabólicas, mientras que sí lo hace la existencia de intolerancia a hidratos de carbono (glucemia entre 140 y 199 mg/dl a los 120´ en el test de tolerancia oral a la glucosa)(16).
Para establecer el diagnóstico de Síndrome Metabólico, la International Diabetes Federation (IDF) propone evaluar la presencia de obesidad troncal (perímetro de cintura) junto con las alteraciones glucémicas (AGA o DM2), lipídicas (triglicéridos y fracción HDL de colesterol) e hipertensión arterial, exclusivamente a partir de los 10 años de edad(17); sin embargo, el retraso en la aparición de las alteraciones glucémicas y la alta prevalencia e inicio precoz de la RI en niños obesos en nuestro medio, sugiere la necesidad de revisar estos criterios para su adaptación a la edad pediátrica(16).
Evaluación y diagnóstico
La anamnesis y el examen físico se orientarán a los antecedentes, signos y síntomas que mejor permitan caracterizar la obesidad y comorbilidades del paciente.
Además de la estimación indirecta (IMC) o cuantificación (métodos directos) del contenido graso corporal, es preciso evaluar su distribución. Para ello, la medición de los perímetros de cintura y cadera es el método más accesible en la práctica clínica (existiendo referencias poblacionales para su estandarización), mientras que la tomografía axial computarizada o la resonancia magnética son más precisos.
Asimismo, es menester examinar la presencia de antecedentes clínicos o rasgos físicos sugerentes de una causa subyacente a la obesidad potencialmente identificable; si bien, excepción hecha de las entidades sindrómicas, la mayoría de los pacientes con causas genéticas de obesidad no muestran un fenotipo específico, diferencial e identificable.
Así, la anamnesis incidirá en aquellos antecedentes, tanto familiares como personales, que nos puedan orientar respecto a la etiología de la obesidad que presenta el paciente, haciendo especial hincapié en los hábitos nutricionales y de actividad física del niño y de la unidad familiar, particularmente en el patrón alimentario, con especial atención a la presencia de hiperfagia y a la caracterización de la sensación de saciedad en el niño (Tabla IV)(1,17).

Junto a esta anamnesis detallada, se debe realizar una exploración pediátrica general, pero específicamente dirigida a la detección de cualquier signo que pueda orientar hacia la causa de la obesidad o a la existencia de comorbilidades asociadas(1,17). En ella, hay que considerar especialmente:
• Aspecto y actitud general: distribución y zonas preferenciales de deposición del tejido adiposo (obesidad abdominal y giba de búfalo en hipercortisolismo / acumulación en muslos y caderas en varones con hipoandrogenismo), tono muscular estático y coordinación dinámica, signos de retraso psicomotor o intelectivo.
• Estigmas malformativos displásicos: en las obesidades asociadas a displasias esqueléticas (acortamiento de cuarto y quinto metacarpianos [pseudohipoparatiroidismo]; cubitus valgus e hipocrecimiento disarmónico con acortamiento de miembros [síndrome de Turner]).
• Piel y mucosas: ictericia, piel seca (hipotiroidismo), hiperpigmentación (exceso de hormona estimulante melanocítica [MSH] en la enfermedad de Cushing), hipopigmentación (deficiencia de proopiomelanocortina [POMC]), acantosis nigricans (hiperpigmentación y engrosamiento cutáneo en cuello, axilas y/o ingles, asociada a RI). Presencia de estrías y coloración de las mismas (rojo-vinosas en hipercortisolismo). Acné y/o hirsutismo (síndrome de ovario poliquístico).
• Rasgos dismórficos en cráneo y cara: craneosinostosis. Morfología ocular y/o anomalías en la visión (retinopatía, aniridia o miopía magna asociadas a síndromes polimalformativos) o en el campo visual (procesos expansivos hipofisarios). Líneas de implantación de cabello y de los pabellones auriculares (y morfología de estos); características e implantación de dientes, paladar ojival o hendido (síndromes polimalformativos). Hipoplasia medio-facial, frente prominente, aplanamiento de la raíz nasal, cara “de muñeca” (deficiencia de GH). Plétora facial o cara de “luna llena” (hipercortisolismo).
• Cuello: inspección y palpación de la glándula tiroidea (bocio posible en hipotiroidismo).
• Tórax: defectos morfológicos (pectus excavatum o carinatum); auscultación cardíaca y pulmonar (cardiopatías asociadas a síndromes polimalformativos).
• Abdomen: defectos de la pared abdominal (hernias); presencia de hepatomegalia (esteatohepatitis no alcohólica).
• Genitales externos y valoración del estadio puberal: (adelanto o retraso puberal). Presencia y caracterización de ginecomastia en varones. Posible presencia de adipomastia sin telarquia en niñas. Presencia de adrenarquia prematura. Alteraciones morfológicas en genitales externos (hipogenitalismo, escroto hendido, criptorquidia).
• Sistema músculo-esquelético: presencia de alteraciones ortopédicas compensatorias (genu valgum, rectificación de las curvaturas y del alineamiento de la columna vertebral), alteraciones de la marcha o dolor a la movilización de la cadera (epifisiolisis de la cabeza femoral, enfermedad de Legg-Calvé-Perthes), tibia vara, aplanamiento del arco plantar.
Exámenes complementarios
Los exámenes complementarios se dirigirán a la identificación de comorbilidades, principalmente metabólicas y, cuando sea posible, al diagnóstico etiológico de la obesidad.
Es mandatorio registrar la tensión arterial en el niño afecto de obesidad, debido al posible desarrollo de hipertensión arterial (HTA, especialmente en hipercortisolismo). Las determinaciones de tensión arterial deben ser percentiladas en referencia al sexo, edad y talla del individuo, y realizarse con la metodología y en las condiciones adecuadas, debido a la posibilidad de sobreestimación de la misma(16).
La Academia Americana de Pediatría y la Academia Americana del Corazón, recomiendan estudiar en todos los niños con obesidad los niveles de transaminasas (AST y ALT), glucemia basal y perfil lipídico(17). Sin embargo, se ha podido comprobar cómo la aparición de RI, asociada a alteraciones más intensas del metabolismo de los hidratos de carbono y de los lípidos, es un proceso progresivo, pudiendo existir una hiperinsulinemia franca y otras comorbilidades metabólicas, en ausencia de alteración de la glucemia en ayunas(16). Por este motivo, sería aconsejable incluir la determinación de insulinemia basal en la evaluación de los niños y adolescentes obesos, lo que, a su vez, permitirá el cálculo del índice HOMA (glucosa [mmol/l] x insulina [μU/ml] / 22,5), indicador de resistencia a insulina; si bien, no todas las guías diagnóstico-terapéuticas avalan esta recomendación(17).
El resto de exploraciones complementarias a realizar estarán determinadas por los datos relevantes de la anamnesis y los hallazgos de la exploración física.
Así, se debe considerar la necesidad de realización de un test de tolerancia oral a la glucosa (TTOG) en aquellos casos en los que el paciente pertenezca a un grupo étnico de riesgo (hispano, afroamericano) y/o existan alteraciones de la glucemia o insulinemia basales, dislipidemia, HTA, antecedentes familiares de DM2, o condiciones asociadas a la RI, tales como acantosis nigricans o síntomas del síndrome de ovario poliquístico (SOP).
La realización de una radiografía de mano y muñeca izquierdas permite establecer la “edad ósea” (EO) para evaluar el ritmo madurativo del paciente en relación con su talla y edad cronológica (EC). Es particularmente informativa en el período prepuberal. En la obesidad infantil es habitual una EO acelerada respecto a la EC (pero habitualmente adecuada a la talla del niño)(16); si bien, puede verse influida por la presencia de determinadas etiologías subyacentes a la obesidad (retrasada junto a talla baja en hipercortisolismo).
Pese a la escasa prevalencia de estas entidades, los estudios hormonales deben ir dirigidos a descartar la existencia de hipotiroidismo (tiroxina [T4] libre y hormona estimuladora del tiroides [TSH]) o de hipercortisolismo (excreción urinaria de cortisol en 24 horas), ante la presencia de síntomas y signos sugerentes. Ante la sospecha de deficiencia o insensibilidad a GH, se debe incluir la determinación de los niveles de IGF-I e IGFBP-3. La determinación de los niveles séricos de adipoquinas no tiene una utilidad diagnóstica específica en el momento actual, excepción hecha de los casos infrecuentes de deficiencia de leptina.
La ecografía es la prueba de elección ante la sospecha de la existencia de esteatohepatitis no alcohólica o síndrome de ovario poliquístico.
La presencia de signos o síntomas sugerentes de comorbilidades específicas determinarán la necesidad de realizar una evaluación psicológica o de ampliar la evaluación médica especializada (digestiva, cardiológica, ortopédica, nefrológica u oftalmológica) o los exámenes complementarios (p. ej., estudio polisomnográfico en SAOS).
En un porcentaje no despreciable de pacientes con obesidad, puede ser de utilidad la realización de una prueba diagnóstica genética. El cuadro clínico que presenta el paciente, así como la historia médica y familiar, dirigirá la elección del test genético más adecuado. Si hay una clara sospecha de alguna entidad específica, la elección de la prueba genética debería ir dirigida a la etiología de la misma. Por ejemplo: un MLPA específico de metilación para entidades causadas por defectos de impronta o la secuenciación génica en los casos de sospecha de etiología monogénica, bien por método de Sanger (si existe un único gen implicado), bien mediante NGS (next generation sequencing; panel de genes o exoma) en una patología con alta heterogeneidad genética. Asimismo, ante entidades polimalformativas / sindrómicas o cuando no existe una sospecha clínica clara, puede procederse a realizar un estudio de secuenciación del exoma, ya que permite tanto el estudio de mutaciones puntuales como de deleciones y duplicaciones (CNVs), así como el estudio de genes adicionales si la sospecha clínica inicial no se confirmara.
En el caso de la obesidad no sindrómica, cuando se presenta de forma precoz y es muy grave, también el empleo de la NGS (exoma +/- MS-MLPA) puede ser la alternativa diagnóstica más coste-eficaz, toda vez que lo más frecuente es que no exista una correlación fenotipo-genotipo que permita identificar clínicamente a los pacientes afectos de obesidad por mutaciones monogénicas o CNVs.
Tratamiento
La combinación del tratamiento comportamental, nutricional y la actividad física, constituyen la base del tratamiento de la obesidad infanto-juvenil.
El abordaje terapéutico del niño afecto de obesidad debe acometerse en el momento del diagnóstico, sin demorarse hasta edades futuras hipotéticamente más adecuadas para el mismo.
Actualmente, la Agencia Europea del Medicamento no avala el empleo de ningún fármaco para el tratamiento de la obesidad en pacientes menores de 18 años(3) (a diferencia de la FDA en EE.UU. que acepta, a partir de los 12 años, el empleo del orlistat o de la liraglutida si la obesidad se acompaña de DM2), encontrándose en fase de ensayo clínico el empleo de agentes agonistas melanocortínicos en pacientes con afectación de la vía leptina-POMC(19). Por otra parte, la indicación de tratamiento quirúrgico en nuestro medio queda restringida a la adolescencia, una vez conseguida la maduración física y ante circunstancias excepcionales. Así, el tratamiento de la obesidad infantil se basa en tres elementos, que son: la reorganización de los hábitos alimentarios, la actividad física y el tratamiento conductual(3).
Existen múltiples guías de práctica clínica disponibles(18,19), incluyendo la del Ministerio de Sanidad español(3). En todas ellas, se recomienda la acción combinada sobre los tres elementos mencionados al unísono, como modo de conseguir una intervención eficaz.
El objetivo del tratamiento conductual es ayudar al niño a adquirir nuevas habilidades que le permitan alcanzar unos objetivos previamente consensuados. En este abordaje cabe distinguir tres componentes fundamentales: las técnicas de modificación de conducta (basadas en el condicionamiento clásico), la terapia dirigida al estrés (identificación y modificación de los pensamientos negativos automáticos) y el análisis de la recompensa y el refuerzo(1).
Junto a ello, en el niño y el adolescente afecto de obesidad se propone el empleo de una alimentación mixta, variada, cuantitativamente limitada por medio de una restricción calórica moderada. Todo ello unido a un incremento del gasto energético derivado de la limitación del sedentarismo y la inactividad, que favorezca el dinamismo en la actividad cotidiana y un ejercicio físico adaptado a las capacidades del niño, con un incremento progresivo de su intensidad(3,18).
Función del pediatra de Atención Primaria: prevención y tratamiento
El médico pediatra en Atención Primaria debe desempeñar un papel relevante, tanto en la prevención como en el tratamiento de la obesidad en el niño y el adolescente. En el aspecto de prevención, dispone una posición privilegiada en el sistema de salud para promover los hábitos alimentarios y de actividad física saludables (prevención primaria), así como para identificar pacientes con etiologías subyacentes potencialmente diagnosticables y tratables o en riesgo de desarrollar comorbilidades asociadas a la obesidad (prevención secundaria)(20). Asimismo, en relación con el tratamiento de esta enfermedad, su coordinación con otros niveles asistenciales es esencial, pudiendo ofrecerse una acción complementaria entre la asistencia primaria y especializada. Con particular relevancia, el médico pediatra junto con el personal de enfermería de Atención Primaria pueden facilitar, de forma coordinada con las visitas en atención hospitalaria, la continuidad y frecuencia de las revisiones que estos pacientes precisan, particularmente en las fases iniciales del tratamiento y en los periodos más intensos de este y que no resulta factible ofertar desde el entorno hospitalario.
Agradecimientos
CIBER Fisiopatología de la Obesidad y Nutrición (CIBERobn), Instituto de Salud Carlos III, Fondo de Investigación Sanitaria (FIS. PI09/91060; FIS 10/00747; FIS 13/01295 y FIS 16/00485).
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.*** Martos-Moreno GÁ, Argente J. Paediatric obesities: from childhood to adolescence. An Pediatr (Barc). 2011; 75: e1-63.
2. Klish WJ, Skelton JA. Definition, epidemiology, and etiology of obesity in children and adolescents. Up to date (revisión bibliográfica hasta febrero 2020). Disponible en:
https://www.uptodate.com/contents/definition-epidemiology-and-etiology-of-obesity-in-children-and-adolescents.
3. Grupo de trabajo de la guía sobre la prevención y el tratamiento de la obesidad infantojuvenil. Centro Cochrane Iberoamericano, coordinador. Guía de práctica clínica sobre la prevención y el tratamiento de la obesidad infantojuvenil. Madrid: Plan de Calidad para el Sistema Nacional de Salud del Ministerio de Sanidad y Política Social. Agència d´Avaluació de Tecnologia i Recerca Mèdiques; 2009. Existen estudios poblacionales posteriores en nuestro medio. Guía de práctica clínica: AATRM Nº 2007/25. (Actualización 2013 disponible en: The validity of recommendations from clinical guidelines: a survival analysis. CMAJ. 2014; 186: 1211-9).
4. Martos-Moreno GÁ, Barrios V, Chowen JA, Argente J. Adipokines in childhood obesity. Vitam Horm. 2013; 91: 107-42.
5. Serra Majem L, Ribas Barba L, Aranceta Bartrina J, Pérez Rodrigo C, Saavedra Santana P, Pena Quintana L. Childhood and adolescent obesity in Spain. Results of the enKid study (1998-2000). Med Clin (Barc). 2003; 121: 725-32.
6. Ministerio de Sanidad, Política Social e Igualdad. Estudio de prevalencia de obesidad infantil “Aladino”. (Consultado el 7 de diciembre 2019). Disponible en:
http://www.aecosan.msssi.gob.es/AECOSAN/docs/documentos/nutricion/observatorio/estudio_ALADINO_2011.pdf.
7. Miqueleiz E, Lostao L, Ortega P, Santos JM, Astasio PRE. Trends in the prevalence of childhood overweight and obesity according to socioeconomic status: Spain, 1987-2007. Eur J Clin Nutr. 2014; 68: 209-14.
8. Instituto Nacional de Estadística (INE). Encuesta Nacional de Salud 2017 [Consultado 07 diciembre 2019].
Disponible en:
https://www.mscbs.gob.es/estadEstudios/estadisticas/encuestaNacional/encuestaNac2017/ENSE17_pres_web.pdf.
9. Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VWV, Eriksson JG, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017; 5: 53-64.
10. Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010; 463: 666-70.
11. Martos-Moreno GÁ, Serra-Juhé C, Pérez-Jurado LA, Argente J. Underdiagnosed Beckwith-Wiedemann syndrome among early onset obese children. Arch Dis Child. 2014; 99: 965-7.
12. Hofker M, Wijmenga C. A supersized list of obesity genes. Nat Genet. 2009; 41: 139-40.
13.** Coll AP, Farooqi IS, O’Rahilly S. The hormonal control of food intake. Cell. 2007; 129: 251-62.
14. Yang Y, van der Klaauw AA, Zhu L, Cacciottolo TM, He Y, Stadler LKJ, et al. Steroid receptor coactivator-1 modulates the function of Pomc neurons and energy homeostasis. Nat Commun. 2019; 10: 1718.
15. Bonnefond A, Raimondo A, Stutzmann F, Ghoussaini M, Ramachandrappa S, Bersten DC, et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J Clin Invest. 201; 123: 3037-41.
16.*** Martos-Moreno GÁ, Martínez-Villanueva J, González-Leal R, Chowen JA, Argente J. Sex, Puberty, and Ethnicity Have a Strong Influence on Growth and Metabolic Comorbidities in Children and Adolescents With Obesity: Report on 1300 Patients (The Madrid Cohort). Pediatr Obes. 2019; 14: e12565.
17.*** Klish, WJ. Clinical evaluation of the obese child and adolescent. Up to date (revisión bibliográfica hasta enero 2020). Disponible en: https://www.uptodate.com/contents/clinical-evaluation-of-the-obese-child-and-adolescent?source=related_link.
18. Styne DM, Arslanian SA, Connor EL, Farooqi IS, Murad MH, Silverstein JH, et al. Pediatric Obesity-Assessment, Treatment, and Prevention: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2017; 102: 1-49.
19. Kühnen P, Clément K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. N Engl J Med. 2016; 375: 240-6.
20.*** Skelton JA. Management of childhood obesity in the primary care setting. Up to date (revisión bibliográfica hasta enero 2020). Disponible en: https://www.uptodate.com/contents/management-of-childhood-obesity-in-the-primary-care-setting?source=related_link.
21. Güemes-Hidalgo M, Muñoz-Calvo MT. Obesidad en la infancia y adolescencia. Pediatr Integral. 2015; XIX(6): 412-27.
22. Wright N, Wales J. Assessment and management of severely obese children and adolescents. Arch Dis Child. 2016; 101: 1161-7.
23. Flegal KM, Wei R, Ogden CL, Freedman DS, Johnson CL, Curtin LR. Characterizing extreme values of body mass index-for-age by using the 2000 Centers for Disease Control and Prevention. growth charts. Am J Clin Nutr. 2009; 90: 1314-20.
Bibliografía recomendada
– Martos-Moreno GÁ, Argente J. Paediatric obesities: from childhood to adolescence. An Pediatr (Barc). 2011; 75: e1-63.
Exhaustiva y extensa revisión de la mayoría de los aspectos fisiopatológicos, etiológicos, clínicos y terapéuticos relativos a la obesidad infantil planteados en este artículo, para profundizar en aquellos en que se desee.
– Martos-Moreno GÁ, Martínez-Villanueva J, González-Leal R, Chowen JA, Argente J. Sex, Puberty, and Ethnicity Have a Strong Influence on Growth and Metabolic Comorbidities in Children and Adolescents With Obesity: Report on 1300 Patients (The Madrid Cohort). Pediatr Obes. 2019; 14: e12565.
Estudio de las características clínicas y de las comorbilidades metabólicas de los pacientes afectos de obesidad infanto-juvenil atendidos en atención especializada en nuestro medio, actualmente. Incluye el análisis de la eventual relevancia de los hallazgos analíticos en estos pacientes.
– Klish, WJ. Clinical evaluation of the obese child and adolescent. Up to date (revisión bibliográfica hasta enero 2020). Disponible en: https://www.uptodate.com/contents/clinical-evaluation-of-the-obese-child-and-adolescent?source=related_link.
Actualizada revisión bibliográfica y análisis crítico de los aspectos referentes a la evaluación clínica de la obesidad infantil.
– Skelton JA. Management of childhood obesity in the primary care setting. Up to date (revisión bibliográfica hasta enero 2020). Disponible en: https://www.uptodate.com/contents/management-of-childhood-obesity-in-the-primary-care-setting?source=related_link.
Actualizada revisión bibliográfica y análisis crítico de los aspectos referentes al tratamiento de la obesidad infantil, con mención especial al entorno de Atención Primaria.
| Caso clínico |
|
Motivo de consulta: varón de 9 años y 1 mes de edad, evaluado en el Servicio de Endocrinología un mes después de la resección de una masa intracraneal sólido-quística de 2 x 3 cm con calcificaciones en su interior, de localización supraselar, diagnosticada anatomo-patológicamente como craneofaringioma. En el período postoperatorio, se evidenciaron deficiencias de hormona antidiurética (ADH), hormona corticotropa (ACTH) y hormona estimulante del tiroides (TSH), por lo que recibía tratamiento sustitutivo vía oral con: desmopresina (0,1 mg cada 12 horas), hidrocortisona (10 mg/m2/día) y levotiroxina sódica (50 μg/día). Tras la intervención quirúrgica, los padres objetivaron un incremento del apetito del paciente, alteraciones en el ritmo vigilia-sueño, así como presencia cotidiana de fiebre, en ausencia de procesos infecciosos. Antecedentes familiares: no reseñables. Antecedentes personales: embarazo controlado, sin polihidramnios ni incidencias de interés. Percepción de los primeros movimientos fetales al cuarto mes de gestación. Parto eutócico a las 40 semanas de edad gestacional. No precisó reanimación neonatal. Exploración neonatal: peso: 3.100 g (p20). Longitud: 50 cm (p28). Perímetro cefálico: 35 cm (p38). Apgar: 1´: 7; 5´: 9. Sin hallazgos patológicos. Lactancia materna durante 5 meses, alimentación complementaria completa y reglada, sin alergias ni intolerancias alimentarias ni medicamentosas conocidas. Sin patrón hiperfágico de ingesta. Adecuadamente inmunizado. Desarrollo psicomotor normal (sedestación estable a los 6 meses, deambulación libre a los 12 meses). Escolarizado con adecuado rendimiento y bien socializado. Sin antecedentes patológicos, al margen del craneofaringioma intervenido. Exploración física: edad cronológica: 9 años y 1 mes. Talla: 131,3 cm (p29). Peso: 48,9 kg (p>97). IMC: 28,36 kg/m2 (+ 3,66 SDS). Cintura: 91 cm. Cadera: 85 cm. Tensión arterial: 100/45 mmHg. Frecuencia cardíaca: 90 latidos por minuto. Buen estado general, normocoloreado y bien hidratado. Abundante panículo adiposo de predominio abdominal. Sin estrías ni acantosis nigricans. Red venosa superficial colateral visible en abdomen. Sin bocio ni adenopatías palpables. Auscultación cardiopulmonar: normal. Adipomastia bilateral. Abdomen globuloso, blando y depresible sin masas ni visceromegalias palpables. Genitales externos masculinos normales, estadio de Tanner I (G1P1Aa), testes de 2 ml de Prader en bolsas escrotales. Exploraciones complementarias: radiografía de mano y muñeca izquierdas: edad ósea: 7 años. Hemograma, bioquímica general y perfil lipídico: normales. Prueba de tolerancia oral a la glucosa (ingesta de 75 gramos de glucosa): glucemia basal: 66 mg/dl; a los 120 minutos: 76,9 mg/dl (pico máximo: 120 mg/dl). Insulina basal: 7,2 μUI/ml; a los 120 minutos: 71 μUI/ml (pico máximo 96 μUI/ml). Determinaciones hormonales: T4l: 0,84 ng/ml (0,66-1,4); TSH: 0,039 mUI/ml (con tratamiento sustitutivo); IGF-I: 548 ng/ml (220-630); IGFBP-3: 4,2 μg/l (1,7-5,1). Evolución y tratamiento: seis meses después de su primera consulta se detectó, en una resonancia magnética craneal de control, la persistencia de una lesión quística en región supraselar, con captación periférica de contraste. Dicha lesión incrementó su tamaño durante los tres meses siguientes, por lo que, a la edad de 10 años y 4 meses recibió tratamiento radioterápico local (52 Gy). Posteriormente, se objetivó una disminución en la velocidad de crecimiento; por lo que, tras la comprobación de deficiencia de hormona de crecimiento (GH) por medio de dos pruebas de estimulación, se inició tratamiento sustitutivo con GH biosintética, a una dosis de 0,03 mg/kg/día, con restauración de un crecimiento normal en los meses posteriores. Paralelamente, tras el tratamiento radioterápico, se produjo un incremento desmesurado del apetito del paciente, con abolición de la sensación de saciedad, y episodios hiperfágicos que conllevaban conductas agresivas ante la imposibilidad de conseguir alimento, produciéndose un incremento de peso (48 kilos en los cuatro años siguientes). Las medidas dietéticas instauradas y la recomendación de ejercicio físico resultaron ineficaces en el control ponderal. Igualmente, pese a la instauración de apoyo conductual psicológico y tratamiento farmacológico por parte del servicio de Psiquiatría (topiramato, fármacos ansiolíticos e inhibidores selectivos de la recaptación de serotonina), los impulsos hiperfágicos del paciente no pudieron ser controlados. En su visita, a los 14 años de edad, mostraba un índice de masa corporal de 40,17 kg/m2 (+ 6,63 SDS), junto con una insulinemia en ayunas de 54 μU/ml (normal < 15), aunque el resto de estudios metabólicos (glucemia, perfil lipídico, ácido úrico) y polisomnográficos practicados no revelaron alteraciones patológicas.
|
