 |
| Temas de FC |
J. Pozo Román
Médico adjunto del Servicio de Endocrinología Pediátrica del Hospital Infantil Universitario Niño Jesús de Madrid y Profesor asociado de Pediatría de la Universidad Autónoma de Madrid
| Resumen
El crecimiento es el proceso fisiológico más característico de la edad pediátrica. Aunque la talla adulta y la edad a la que esta se alcanza están, en cada sujeto, determinadas genéticamente, el resultado final puede variar dependiendo de la compleja interacción a lo largo de todo el período de crecimiento de factores genéticos y ambientales. La talla baja supone un motivo de preocupación para los padres y es una causa muy frecuente de consulta en Pediatría y en Endocrinología Pediátrica; si bien, en la gran mayoría de los casos, se tratará de variantes normales de talla baja que, salvo una adecuada información, no requerirán tratamiento. El conocimiento de las características normales del crecimiento y de los factores implicados en su regulación en los distintos períodos de la vida es una condición imprescindible para poder valorar la normalidad o anormalidad en el crecimiento de un niño. En esta revisión, se intenta resumir de forma sencilla, los aspectos básicos que, desde el ámbito de la Atención Primaria, permitan al pediatra: 1) valorar si el crecimiento de un niño es normal o no; 2) conocer las causas que con más frecuencia pueden provocar su alteración; 3) saber cómo orientar el diagnóstico y cuándo remitir al especialista (a partir de la historia clínica, de la edad ósea y, en algunos casos, de un reducido número de pruebas complementarias fácilmente disponibles); y 4) conocer las opciones terapéuticas disponibles. |
| Abstract
Growth is the most characteristic physiological process of the pediatric age. Although adult stature and age at which this is reached are genetically determined in each subject, the final result may vary according to the complex interaction of genetic and environmental factors over the growth period. Short stature causes concern for the parents and is a very frequent cause for consultation in Pediatrics and Pediatric Endocrinology. However, in most of the cases, this is due to normal variants of short stature which, except for adequate information, do not require treatment. Knowledge of the normal characteristics of the growth and of the factors involved in its regulation in the different periods of life is an essential condition to be able to evaluate the normality or abnormality in the growth of a child. In this review, an attempt is made to give a simple summary of the basic features which, from a community setting, allow the pediatrician to: 1) evaluate if the growth of a child is normal or not; 2) know the causes that most often may cause its alteration; 3) know how to orient the diagnosis and when to send the patient to the specialist (based on the clinical history, bone age and, in some cases, on a reduced number of easily available complementary tests); and 4) know the available therapeutic options./em> |
Palabras clave: Talla baja; Crecimiento; Hipocrecimiento; Fracaso de crecimiento
Key words: Short stature; Growth; Deficient growth; Growth failure
Pediatr Integral 2015; XIX (6): 411.e1-411.e23
Crecimiento normal y talla baja
Introducción
La talla baja es un motivo habitual de preocupación para los padres, de consulta al pediatra de Atención Primaria y de derivación a consultas de Endocrinología Infantil; sin embargo, la mayoría de los niños que consultan por este motivo son niños sanos cuyo hipocrecimiento resultaría de la variabilidad normal de la talla y del ritmo madurativo.
El crecimiento es el proceso biológico más característico de la edad pediátrica y se extiende desde la concepción hasta la finalización de la maduración esquelética y sexual. Conlleva un incremento de masa, pero también una progresiva maduración que lleva al sujeto a adquirir la plena capacidad funcional. Talla y ritmo madurativo están determinados por multitud de mecanismos genéticos y epigenéticos que interaccionan a lo largo de todo el proceso de crecimiento con factores ambientales intrínsecos y extrínsecos(1); de forma que, para que el potencial genético de crecimiento de un determinado individuo no se vea limitado, es imprescindible que “todo funcione bien”: nutrición, entorno psicosocial, medio interno, estructuras físicas, mecanismos biológicos de regulación, etc. Por ello, la valoración del crecimiento de un niño constituye un indicador sensible de su estado de salud y bienestar y forma parte de todos los programas de prevención de la salud en los niños (“control del niño sano”).
Desde el punto de vista médico, la talla baja puede ser un síntoma de múltiples patologías; sin embargo, en la mayoría de los casos, es simplemente la expresión de uno de los extremos de la distribución normal de la talla y del ritmo madurativo del ser humano. Pese a ello, socialmente, la talla baja es considerada frecuentemente como una causa de estrés psicosocial para el niño y un factor limitante de su futuro éxito social y profesional, especialmente en los varones. Por otra parte, los medios de comunicación y, en ocasiones, los propios médicos, hemos transmitido a la sociedad el concepto erróneo de que toda talla baja puede y debe ser tratada. La consecuencia es que, la mayoría de los niños que consultan por este motivo son niños normales que no requieren tratamiento y en los que la utilización de tratamientos intempestivos no conlleva claros beneficios y sí potenciales efectos secundarios. De ahí, la importancia de establecer un diagnóstico correcto y de transmitir a los padres una información adecuada y veraz.
Bases fisiopatológicas del crecimiento normal
El conocimiento del patrón normal de crecimiento de un niño y de sus factores reguladores es la base para poder detectar situaciones patológicas. No es posible valorar si el crecimiento de un niño es normal o no, si no se conocen las características normales del proceso, los principales factores que lo regulan y cómo la variabilidad normal o determinadas patologías pueden modificarlo.
Patrón de crecimiento
La curva que representa el crecimiento humano normal muestra una morfología sigmoide. A finales de los años ochenta, Kalberg(2) propuso un modelo matemático de la curva conocido como “modelo ICP” (Fetal-Infancy, Childhood, Puberty), según el cual, la curva de crecimiento normal estaría formada por la suma y solapamiento de tres componentes: 1) fetal-primera infancia; 2) prepuberal o de la segunda infancia; y 3) puberal, (Figs. 1 y 2). El objetivo de este modelo es relacionar sus distintos componentes con los factores biológicos de los que dependen; de forma que, la alteración o el retraso en la aparición de uno de estos componentes permitiera una orientación diagnóstica más rápida de las distintas patologías.
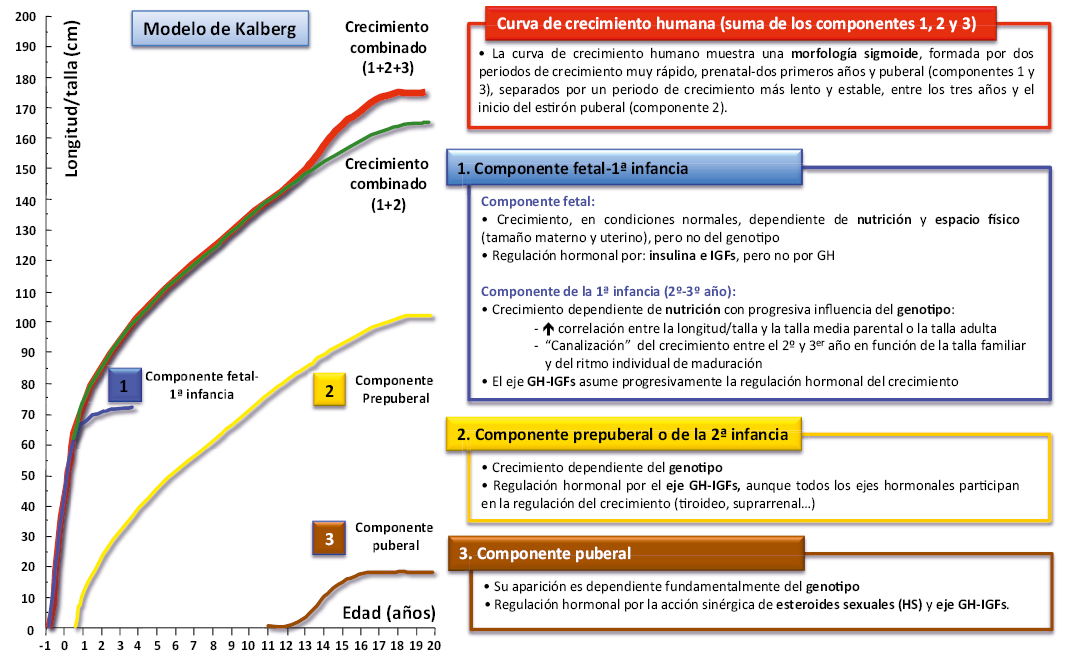
Figura 1. Gráfica de Kalberg (talla/longitud), donde se ponen de manifiesto los tres componentes de la curva normal de crecimiento humano: 1) fetal-primera infancia; 2) prepuberal o segunda infancia; y 3) puberal, así como los principales factores de los que dependen y los mecanismos hormonales que regulan el crecimiento en estas tres etapas de la vida: factores de crecimiento semejantes a la insulina (IGFs), hormona de crecimiento (GH) y hormonas sexuales (HS).
Componente fetal-primera infancia
La deceleración normal en la velocidad de crecimiento que experimentan los niños con talla baja familiar y/o maduración tardía, habitualmente, entre los 1-3 años de vida, es responsable de que numerosos niños sean erróneamente catalogados como “fracasos de crecimiento”.
El componente fetal-1ª infancia se extendería desde la media gestación hasta aproximadamente los 2-3 años. La velocidad de crecimiento (VC: ? talla/año) durante el periodo fetal es muy rápida y continúa siéndolo durante los tres primeros años de vida; si bien, la caída postnatal de la VC es clara y progresiva respecto a la vida intrauterina (Fig. 2).
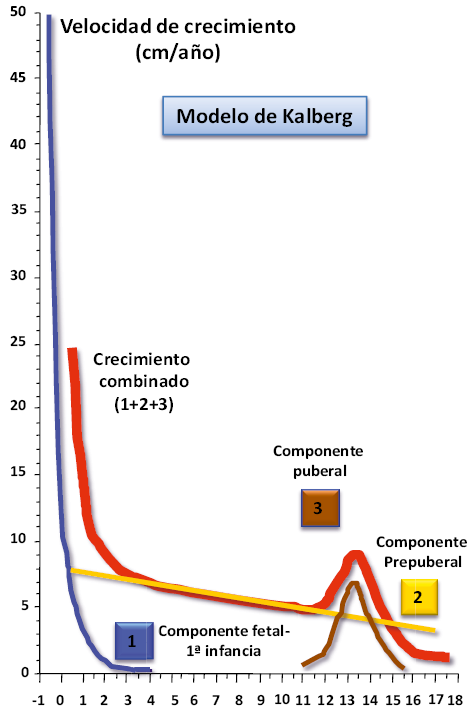
Figura 2. Gráfica de Kalberg (velocidad de crecimiento), donde se ponen de manifiesto los tres componentes de la curva normal de crecimiento humano: 1) fetal-primera infancia; 2) prepuberal o segunda infancia; y 3) puberal.
El crecimiento fetal, en ausencia de anomalías genéticas, no depende prácticamente del genotipo, sino del espacio disponible para crecer (tamaño uterino y materno) y de la nutrición, que depende, a su vez, de la función placentaria (aporte de oxígeno y nutrientes). Después del nacimiento y durante los primeros meses de vida, la nutrición continúa siendo el principal factor determinante del crecimiento, pero la influencia del genotipo se va poniendo progresivamente de manifiesto y, como consecuencia, se producen aceleraciones o deceleraciones en el ritmo de crecimiento de los niños en función de su potencial de crecimiento (talla familiar alta, media o baja) y/o de su ritmo de maduración (rápido, normal o lento), rasgos ambos determinados genéticamente, que les llevarán a establecer su propio canal de crecimiento (fenómeno de “canalización”), habitualmente, entre el 2º y el 3er año de vida. Pasado este periodo de ajuste, la correlación (r) entre la talla del niño y la talla media parental (TMP) o la talla adulta se incrementa; de forma que, que pasa de 0,2-0,3 en el recién nacido (RN) a 0,7-0,8 a los 3 años de edad.
Los mecanismos hormonales que regulan el crecimiento fetal son, en gran medida, desconocidos; no obstante, la insulina y el sistema de los factores de crecimiento semejantes a la insulina (IGFs –insulin-like growth factors–), especialmente los IGFs números 1 y 2 (IGF-1 e IGF-2), parecen tener un papel relevante(3). Al contrario de lo que ocurre en la vida postnatal, la producción fetal de IGFs no es dependiente de hormona de crecimiento (GH); de hecho, la GH no es determinante en el crecimiento fetal. Esta situación se mantiene durante los primeros meses de vida extrauterina, aunque la influencia de la GH en el control del crecimiento se va poniendo de manifiesto de forma gradual y progresiva y está claramente presente, tan pronto como, a los 6 meses de vida.
Componente prepuberal o de la segunda infancia
La deceleración postnatal de la VC continúa durante todo el periodo prepuberal, pero de forma más lenta, debido a la adición del componente prepuberal (Figs. 1 y 2). Este se pone de manifiesto, habitualmente, entre los 6 y 12 meses de edad postnatal y se extiende hasta la finalización del crecimiento. La reducción progresiva del ritmo de crecimiento durante el periodo prepuberal mantiene las VC entre 5-7 cm/año durante la mayor parte del tiempo (Fig. 2) y persiste hasta el inicio del estirón puberal, momento en que se alcanza el nadir (4,5 y 5 cm/año). Este fenómeno, conocido como “depresión prepuberal de la VC”, es especialmente manifiesto en los maduradores tardíos, que llegan a alcanzar VC de solo 2-3 cm/año.
Durante el período prepuberal, el principal regulador del crecimiento es el genotipo. Los niños con talla familiar baja o maduración lenta tienden a crecer con una VC media inferior al percentil 50; mientras que, los niños con talla familiar alta o maduradores rápidos tienden a hacerlo con una VC media por encima del percentil 50. Esta diferencia en la VC, a lo largo de todo el periodo prepuberal, es la principal responsable de la diferencia de talla adulta entre sujetos con talla familiar alta o baja. Por el contrario, las diferencias de VC entre maduradores rápidos y lentos no suelen afectar significativamente la talla adulta; ya que, se compensan, al menos parcialmente, gracias a un mayor o menor número de años de crecimiento prepuberal.
El principal regulador de la VC durante el periodo prepuberal y, en general, en el periodo postnatal, es el eje GH-IGFs (Fig. 3); no obstante, otras muchas hormonas (insulina, hormonas tiroideas, andrógenos suprarrenales, glucocorticoides, leptina, paratohormona, vitamina D…) y factores de crecimiento locales contribuyen también a la regulación del crecimiento, aunque con frecuencia, a través de interacciones con el eje GH-IGFs.
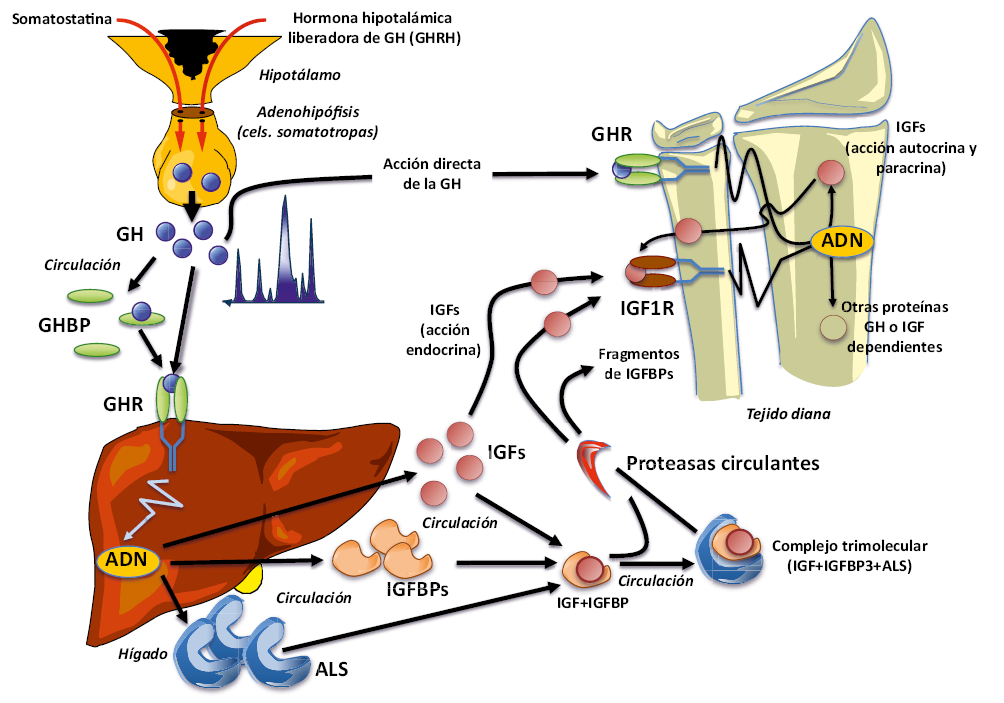
Figura 3. Representación esquemática del sistema GH-IGFs. Se representan las acciones endocrinas, paracrinas y autocrinas de IGF-I, así como la formación del compejo trimolecular de alto peso molecular, formado por IGF-I, IGFBP-3 y ALS.
GHRH: hormona hipotalámica liberadora de hormona de crecimiento (GH); SRIH: hormona inhibidora de la liberación de GH, somatostatina; GHBP: proteína trasportadora de GH; GHR: receptor de GH; IGF-I: factor de crecimiento semejante a la insulina número 1; IGFBP-3: proteína trasportadora de IGFs número 3; ALS: subunidad ácido lábil; IGFIR: receptor de IGF-I.
La GH es sintetizada y secretada de forma pulsátil (4-6 pulsos secretorios/día) y con un predominio nocturno por las células somatotropas de la adenohipófisis(4). Posee acciones directas estimulantes del crecimiento, pero la mayoría de ellas son mediadas por los IGFs y especialmente por el IGF-1. Alrededor del 50% de la GH circula en la sangre unida a una proteína de transporte específica, la GHBP (growth hormone binding protein), que es idéntica a la porción extracelular del receptor de GH (GHR), del que deriva por rotura proteolítica. Cuando la GH se une al RGH, este debe dimerizar para poder transmitir el mensaje al interior de la célula, que no es otro que inducir la expresión de determinados genes, como es el caso del IGF-1, a través de diferentes vías intracelulares, de las cuales, la vía MAPK (mitogen-activated protein kinase), la STAT (signal transducers and activators of transcription) y la PI3-K (phosphatidylinositol 3-OH kinase) son las mejor caracterizadas.
Los IGFs (IGF-1 e IGF-2), previamente conocidos como somatomedinas, son péptidos de estructura similar a la insulina producidos en el hígado y en los tejidos periféricos, especialmente en hueso y músculo, que, de forma endocrina (IGFs circulantes) o paracrina-autocrina (IGFs sintetizados en los tejidos), ejercen acciones mitogénicas y anabolizantes sobre la mayoría de las células(4,5). Aproximadamente, el 99% de los IGFs que circulan en la sangre lo hacen unidos a proteínas de transporte específicas, las IGFBPs (IGFs binding proteins). La más importante desde el punto de vista clínico es la IGFBP-3, que transporta un 75-90% de los IGFs circulantes. El complejo IGF-IGFBP-3 se une en la sangre con otra proteína, la ALS (subunidad ácido-lábil), formando un complejo trimolecular (IGF-IGFB-3-ALS) de alto peso molecular, lo que limita la salida de los IGFs a los tejidos, aumentando su vida media y su concentración sérica, al tiempo que evita sus efectos hipoglucemiantes derivados de su similitud molecular con la insulina. Proteasas circulantes rompen lentamente estos complejos y permiten la liberación de los IGFs y su salida a los tejidos. Tanto la IGF-1 como la IGFBP-3 y la ALS son proteínas producidas en el hígado por estímulo directo de la GH y mantienen niveles séricos más o menos estables a lo largo del día; de forma que, su determinación puede utilizarse como un reflejo indirecto de la secreción de GH. Por el contrario, las características pulsátiles de la secreción normal de GH hacen que su determinación basal carezca de utilidad en el diagnóstico de los hipocrecimientos. De estas tres proteínas dependientes de GH, la más utilizada clínicamente ha sido la determinación de IGF-1. La IGFBP-3 presenta, sobre todo en menores de 3 años, algunas ventajas sobre la IGF-1 como marcador de deficiencia de GH: concentraciones séricas más elevadas (menor solapamiento entre sujetos normales y deficitarios), menor variación con la edad y el desarrollo puberal y menor influencia del estado nutricional sobre sus niveles séricos (IGF-1 e IGFBP-3 disminuyen en situaciones de malnutrición); en general, IGFBP3 es más específico pero menos sensible que IGF-1 para el diagnóstico de deficiencia de GH.
Componente puberal
Cuando la pubertad se inicia a una edad normal, aunque esta sea adelantada o retrasada respecto a la media, la talla final no se modifica, aunque sí el momento en que esta se alcanza.
El componente puberal se suma al prepuberal a una edad variable, que es determinada por el genotipo, responsable del ritmo de maduración individual y del momento en que se inicia la pubertad. Su aparición se manifiesta por una clara inflexión en la curva de crecimiento, el llamado “estirón puberal”. En las niñas, la aparición del botón mamario (estadio II de Tanner) marca el inicio puberal, a una edad media de 10,5-11 años, y suele coincidir con el inicio del “estirón puberal”. La menarquia, por el contrario, es un fenómeno tardío, que se produce alrededor de 2 años después del inicio puberal (estadio IV de Tanner), cuando el estirón está prácticamente finalizado. El crecimiento postmenarquia es muy variable, entre 4 y 11 cm (media de 6-7 cm). En los varones, el inicio de la pubertad lo marca el incremento del volumen testicular (=4 ml), que se produce a una edad media de 11,5-12 años. A diferencia de las niñas, el estirón puberal no coincide con el inicio de la pubertad, sino que se inicia, aproximadamente, un año después (12,5-13 años), en un estadio III de Tanner y unos 2 años más tarde que en las niñas.
El crecimiento cesa en los varones y mujeres a una edad media de 21 y 17 años, respectivamente. La ganancia de talla, desde el inicio del estirón hasta la finalización del crecimiento, supone unos 25-30 cm en los varones y unos 23-27 en las mujeres (15-20% de la talla adulta) y predomina proporcionalmente el aumento de tronco sobre el de extremidades. La magnitud del estirón es similar en sujetos con talla familiar alta o baja. En lo que al ritmo de maduración se refiere, los maduradores rápidos no solo crecen a un mayor ritmo durante el periodo prepuberal, sino que tienen un estirón puberal más potente que los maduradores normales y estos, a su vez, mayor que el de los maduradores tardíos, compensándose así el mayor o menor número de años de crecimiento prepuberal. Por ello, una maduración normal, pero adelantada o retrasada respecto a la media, modifica la edad a la que se alcanza la talla final, pero no la magnitud de esta. Los dos años más de crecimiento prepuberal que tienen los niños (8-10 cm) y la mayor amplitud de su estirón puberal (3-5 cm) determinan los 12,5-13 cm de diferencia entre la talla adulta de ambos sexos, así como la mayor longitud proporcional en los varones de las extremidades respecto al tronco (mayor periodo de crecimiento prepuberal).
La responsabilidad de la aparición del componente puberal de la curva de crecimiento corresponde a los esteroides sexuales, no solo porque poseen acciones anabolizantes directas sobre el cartílago de crecimiento, sino, también, porque incrementan la secreción de GH; de forma que, la actuación sinérgica de esteroides sexuales (especialmente estradiol) y GH sería la responsable del estirón puberal, pero también del cierre de los cartílagos de crecimiento y de la finalización de este.
Fenómenos de “crecimiento de recuperación”, “canalización” y “programación”
La capacidad de recuperación de la talla perdida tras la curación o mejoría de una enfermedad crónica es lo que se denomina “crecimiento de recuperación”(6). Puede definirse como: el incremento en el ritmo de crecimiento por encima de los límites normales, que se produce durante un tiempo limitado después de un periodo de detención del crecimiento y que permite recuperar total o parcialmente el crecimiento perdido. Su existencia presupone que cada niño, en condiciones normales, tendería a mantenerse dentro de un estrecho carril de crecimiento, un fenómeno que se conoce como “canalización del crecimiento”. Según esta hipótesis, cuando un sujeto, por la acción de una determinada noxa, se desvía de su canal de crecimiento, el organismo no solo es capaz de percibirlo, sino que, cuando la noxa desaparece, tiende a recuperarlo. Los mecanismos fisiológicos que median el fenómeno de la “canalización” no son bien conocidos y la recuperación del canal de crecimiento, dependiendo del momento de actuación de la noxa, así como de su gravedad y duración, puede no ser completo; de hecho, se ha postulado la existencia de “periodos críticos” o “de ventana”, durante los cuales, la actuación de una noxa podría alterar definitivamente la talla adulta. Estos periodos de alta vulnerabilidad, corresponderían a los momentos de máximo crecimiento, como es la pubertad y, especialmente, el periodo fetal y primer año de vida, cuando los fenómenos de proliferación celular son más intensos. Según la hipótesis de la “programación”, la repercusión de estas agresiones en periodos críticos sería mucho más amplia que la afectación aislada del crecimiento, ya que podrían inducir cambios permanentes, morfológicos y funcionales (expresión génica, replicación celular, estructura y función de órganos, composición corporal, secreción/acción de hormonas y factores del crecimiento…), que modificarían la susceptibilidad al desarrollo en la vida adulta de determinadas enfermedades degenerativas (hipótesis de Barker o del origen fetal de las enfermedades del adulto(7)), como sería el caso de: obesidad, síndrome metabólico o enfermedades cardiovasculares.
Concepto y epidemiología del hipocrecimiento
Establecer la normalidad en el crecimiento de un niño puede, en algunos casos, ser un problema complejo; ya que, no existe un punto de corte que discrimine de forma nítida entre tallas y VC “normales” y “patológicas”.
La “normalidad” de una talla no puede establecerse solo en relación con la edad, el sexo y la etnia del sujeto (normalidad estadística), sino que, al ser un rasgo genético e influenciado por el ritmo madurativo, debe analizarse también en su contexto familiar y en función del ritmo madurativo individual de cada sujeto(8,9).
La correlación de la talla de un niño con su contexto familiar suele hacerse comparando su “talla diana” (TD) con su expectativa de talla adulta (predicción de talla). La TD, también denominada talla genética, es, simplemente, la talla esperable para los hijos de una pareja determinada, asumiendo un proceso normal de herencia y unos efectos ambientales sobre el crecimiento, similares en ambas generaciones(9). La fórmula más comúnmente empleada para calcularla es la desarrollada por Tanner a partir de la talla media parental (TMP) ajustada al sexo del niño (Tabla I). En cuanto a la predicción de talla adulta (PTA), esta puede hacerse de diferentes maneras e incluir o no en los cálculos la TMP(9); pero todas ellas tienen en cuenta el ritmo madurativo individual, que se valora en la práctica clínica mediante la “edad ósea” (EO). La fiabilidad de los métodos de PTA es relativamente escasa y tanto menor cuanto menor es la edad del niño. En cualquier caso, la PTA no debería hacerse antes de que los ajustes en función del contexto familiar y del ritmo madurativo (canalización) se hayan realizado y el crecimiento sea más estable. El método más sencillo es utilizar la “talla proyectada”, que se calcula extrapolando la talla final que alcanzará el niño a partir del carril de crecimiento para la edad en el que se encuentra y siguiéndolo hasta los 18-20 años. Si la EO está retrasada o adelantada, es preferible extrapolar la talla final a partir de la talla para la EO. La talla final también puede predecirse mediante diferentes métodos matemáticos, todos los cuales tienen en consideración la EO como indicador del grado de maduración alcanzada. El más utilizado en la clínica, por su sencillez y fiabilidad, es el método de Bayley-Pinneau, que tiene en consideración, únicamente, la talla del niño y su EO determinada por el atlas de Greulich-Pyle(9).
Otro aspecto a la hora de valorar si el crecimiento de un niño es normal o no, es tener en consideración que el crecimiento es un proceso dinámico; mientras que, la talla es un parámetro estático y que, por ello, la observación en un momento dado de una talla “normal” no excluye la posibilidad de que se esté produciendo un fracaso de crecimiento. Es necesario analizar, también, si la VC a lo largo del tiempo es normal. La menor fiabilidad en el cálculo de la VC y las múltiples variaciones que, en condiciones normales, esta experimenta hacen que, con frecuencia, sea también difícil determinar su normalidad o anormalidad(8). Los criterios que, en la práctica clínica, utilizamos habitualmente para establecer cuándo un niño debe ser estudiado por un problema de hipocrecimiento(1) quedan reflejados en la tabla I.

La prevalencia de hipocrecimiento en un momento dado es difícil de establecer, no solo por las dificultades metodológicas inherentes a su definición, sino por la aparición, a lo largo de la infancia, de nuevos casos (patología crónica, alteraciones hormonales…) y por la recuperación de otros (pequeños para su edad gestacional que se recuperan, patologías crónicas que se curan o tratan…). Si consideramos aisladamente el concepto estadístico de talla baja, como una talla por debajo de ?2 SDS para su edad, sexo y etnia, el 2,3% de la población entraría en el concepto de hipocrecimiento; no obstante, el número de sujetos que se miden en los estudios poblacionales es insuficiente para determinar con fiabilidad los extremos de la curva de Gauss que representa la distribución de la talla y, dado que, es más factible que el crecimiento se vea gravemente frenado que acelerado, es probable que el porcentaje de tallas bajas sea superior al 2,3%. Si consideramos, además, las otras posibles definiciones de hipocrecimiento (Tabla I), es muy probable que el número de niños con hipocrecimiento sea superior al 3-5%.
Etiopatogenia y clasificación de los hipocrecimientos
Dentro de los hipocrecimientos, clásicamente, se han diferenciado dos grandes grupos etiopatogénicos: los hipocrecimientos normales o variantes normales de talla baja (VNTB) y los hipocrecimientos patológicos. En la actualidad, tienden a clasificarse más como: hipocrecimientos de causa conocida e hipocrecimientos de causa desconocida o idiopáticos (TBI), entre los que se incluirían las VNTB.
Hipocrecimientos de causa conocida
Representan alrededor del 20% de los hipocrecimientos(1) y serían la consecuencia de trastornos patológicos que alterarían la capacidad de crecimiento intrínseca de los tejidos (osteocondrodisplasias, alteraciones del metabolismo óseo, retrasos de crecimiento intrauterino, cromosomopatías y síndromes dismórficos), sus mecanismos reguladores (hipocrecimientos de causa endocrinológica) o el ambiente interno (malnutrición y patología crónica en diferentes órganos y sistemas) y emocional del niño (hipocrecimiento psicosocial).
Desde un punto de vista etiopatogénico y diagnóstico (Tabla II) es útil, diferenciar, dentro de los hipocrecimientos, el momento de su inicio (pre o postnatal) y si se conservan o no las proporciones corporales normales (armónico o disarmónico). Los hipocrecimientos de inicio prenatal suelen ser el resultado de una agresión al ambiente fetal o de un defecto genético; por el contrario, los de inicio postnatal suelen responder, en general, a una agresión iniciada fuera del período de vida intrauterino. En lo que se refiere al mantenimiento o no de la armonía corporal, los hipocrecimientos disarmónicos son siempre patológicos y pueden ser congénitos (osteocondrodisplasias) o, menos frecuentemente, adquiridos (p. ej., los secundarios a radioterapia espinal); por el contrario, los armónicos pueden ser normales o patológicos.

Hipocrecimientos de inicio prenatal
Son conocidos como recién nacidos pequeños para su edad gestacional (RNPEG) y el 80-90% experimentarán un crecimiento de recuperación, parcial o total, durante el primer o segundo año de vida y alcanzarán una talla dentro del rango normal (entre ± 2 SDS), aunque con frecuencia por debajo de su contexto familiar.
Se considera a un RN como RNPEG cuando su peso (PRN) y/o su longitud al nacimiento (LRN) se encuentran, al menos, 2 SDS por debajo de la media para su edad gestacional. En España, las tablas y graficas recomendadas para la clasificación de los RN como RNPEG son las correspondientes al Estudio Español de Crecimiento de 2010, que abarcan desde la 26 a las 42 semanas de edad gestacional.
En los países desarrollados, solo el 4-7% de los RN son RNPEG; sin embargo, es una de las causas más importantes de talla baja, ya que, el antecedente de RNPEG se encuentra en el 20% de los adultos con talla baja(10). Los RNPEG presentan una elevada morbimortalidad perinatal, así como una mayor frecuencia de secuelas a largo plazo, como son, entre otras: hipocrecimiento postnatal, disminución del rendimiento intelectual y psicológico y desarrollo de anomalías hormonales y metabólicas en la infancia y edad adulta (adrenarquia prematura, resistencia a la insulina, hiperandrogenismo ovárico, obesidad, diabetes mellitus tipo 2, dislipemia, hipertensión arterial y enfermedad cardiovascular), que han sido atribuidas a la modificación del “programa” genéticamente establecido como consecuencia de la alteración intrauterina (hipótesis de Baker)(7).
El patrón de crecimiento de los RNPEG suele ser característico. El 80-90% experimentan un crecimiento de recuperación, parcial o total, durante el primer y segundo año de vida, que puede prolongarse algo más en los RNPEG prematuros, y alcanzan una talla dentro del rango normal, entre ± 2 SDS, aunque con frecuencia por debajo de su contexto familiar. El 10-20% restante mantendrán la talla baja (< 2 SDS) después de los 2 años y en, al menos, un 50% de estos, la talla final será baja. La EO suele estar retrasada, pese a lo cual la pubertad se inicia habitualmente a una edad normal o incluso ligeramente adelantada, corrigiéndose rápidamente el retraso en la maduración ósea y alcanzando una talla final baja que, en la mayoría de los casos, es similar, en SDS, a la talla prepuberal. También, los niños prematuros con muy bajo peso al nacimiento (PRN < 1.500 g), aunque este sea adecuado a su EG, pueden mostrar también un patrón de crecimiento similar al de los RNPEG, con ausencia de un crecimiento de recuperación adecuado y talla final baja.
Cuando el RN presenta una disminución combinada del PRN y de la LRN (RNPEG armónico) el riesgo de alcanzar una talla adulta baja es mayor que cuando solo se afecta el PRN (RNPEG disarmónico).
Desde el punto de vista etiopatogénico (Tabla II), una tercera parte de los RNPEG se deberían a factores fetales (cromosomopatías, anomalías congénitas y síndromes dismórficos) y las otras dos terceras partes a factores maternos (malnutrición, infecciones, tóxicos…) y uterino-placentarios (malformaciones uterinas, arteria umbilical única…); no obstante, hasta en un 40% de los casos no se identifica ninguna causa patológica.
Multitud de cuadros sindrómicos (Russell-Silver, Seckel, Cornelia de Lange, Noonan, Prader-Willi…), muchos de ellos de causa genética (mutaciones, deleciones, disomías uniparentales…), al igual que ocurre en la mayoría de las cromosomopatías, presentan, como una de sus principales manifestaciones clínicas, talla baja, con frecuencia, pero no siempre, de inicio prenatal. El hipocrecimiento se asocia frecuentemente, además de a rasgos dismórficos más o menos específicos y marcados, a malformaciones en diferentes órganos y a un grado variable de retraso mental; de ahí, la importancia de buscar sistemáticamente en la exploración de todo niño con talla baja, la presencia de rasgos sindrómicos que orienten hacia este tipo de diagnósticos (Tabla III).

Entre las cromosomopatías, merecen especial mención por su frecuencia: el síndrome de Down (trisomía 21; 1:600 RN vivos) y el síndrome de Turner (45, X0, y sus variantes; 1:2.500-3.000 RN vivas). Las características fenotípicas del síndrome de Down hacen que su diagnóstico se realice, habitualmente, en el periodo neonatal; por el contrario, el diagnóstico del síndrome de Turner suele hacerse más tardíamente. Sus rasgos sindrómicos característicos (implantación posterior del cabello baja y en forma de “M”, pterigium colli, acortamiento de metacarpianos, cúbito valgo, tórax en coraza…) pueden ser poco manifiestos o pasar desapercibidos, aunque el fracaso de crecimiento, sobre todo a partir de los 2-3 años (solo un 16% son RNPEG), es prácticamente constante.
En cualquier niña con talla baja de causa poco clara, aun en ausencia de cualquier rasgo sindrómico, es obligada la realización de un cariotipo.
Osteocondrodisplasias
La desproporción entre los distintos segmentos corporales (hipocrecimiento disarmónico) debe hacer pensar en la existencia de una displasia ósea u osteocondrodisplasia.
Las displasias óseas (DO) representan anomalías primarias del hueso y del cartílago, de base genética y herencia variable que, habitualmente, dan lugar a una talla baja disarmónica. Individualmente, son entidades raras, pero colectivamente integran un grupo numeroso (2-5:10.000 RN). El hipocrecimiento y la desproporción entre miembros y tronco puede ser debida a un acortamiento preferentemente de los miembros (acondroplasia, hipocondroplasia, pseudoacondroplasia, discondrosteosis de Léri-Weill, condrodisplasia metafisaria…), del tronco (mucopolisacaridosis, displasias espondiloepifisarias…) o de ambos (displasia metatrópica…), y puede o no estar presente en el momento del nacimiento.
En los últimos años, se ha producido un considerable avance en el conocimiento de las bases moleculares de las DO. Su última clasificación distingue, en función de sus características clínicas, bioquímicas, radiológicas y moleculares, 456 entidades diferentes, agrupadas en 40 familias, con 226 genes implicados(11). Pero, si dejamos aparte las mutaciones del gen SHOX (short stature homeobox-containing gen) que no siempre dan lugar a hipocrecimientos disarmónicos, mutaciones en solo dos loci son responsables de las formas más frecuentes y, por consiguiente, de la mayoría de las DO: COL2A1 (gen del colágeno tipo 2, en 12q13.1-q13.3), responsable de la mayoría de las displasias espondiloepifisarias; y FGFR3 (gen del receptor tipo 3 del factor de crecimiento de los fibroblastos, en 4p16.3), responsable, entre otras entidades clínicas, de: acondroplasia (la DO más frecuente y conocida; 1: 15.000-40.000 RN vivos), hipocondroplasia, displasia de SADDAN y displasia tanatofórica.
El diagnóstico genérico de DO puede ser relativamente sencillo ante un paciente con marcada disarmonía corporal; sin embargo, un diagnóstico más preciso puede ser difícil, debido a la multitud de cuadros clínicos y a su variable expresividad clínica. Por otra parte, en algunas DO, la disarmonía corporal puede ser poco manifiesta; de forma que, con facilidad, pueden ser incluidas entre los hipocrecimientos armónicos de etiología incierta o idiopática (TBI). Entre las DO que por su frecuencia, expresividad clínica, herencia dominante y patrón de crecimiento pueden confundirse con un hipocrecimiento armónico, y en especial con una VNTB, se encontrarían la hipocondroplasia y la haploinsuficiencia del gen SHOX.
Las DO no siempre presentan disarmonía corporal o esta es claramente evidente; por ello, algunas formas de DO pueden entrar a formar parte del diagnóstico diferencial de la TBI. La haploinsuficiencia del gen SHOX es la causa monogénica más frecuente de talla baja en la especie humana.
Hipocondroplasia. Ha sido descrita como una “forma leve” de acondroplasia; no obstante, aunque ambas enfermedades se heredan de forma autosómica dominante y se deben a mutaciones en el mismo gen (FGFR3), no se han descrito familias en las que coincidan ambas entidades. La mutación responsable más frecuente es Asn540Lys, pero sus bases moleculares son más heterogéneas que en el caso de la acondroplasia y otros genes podrían estar implicados; de hecho, se han descrito fenotipos compatibles con hipocondroplasia asociados a mutaciones en SHOX. Desde el punto de vista clínico, no presentan el aspecto facial característico de la acondroplasia, el hipocrecimiento y la rizomelia son menos marcados y el incurvamiento de las piernas y el genu varum son frecuentes. Como en la acondroplasia, es característica la reducción progresiva de la distancia interpeduncular lumbar, de L1 a L5, y las anomalías en la pelvis. La talla adulta se sitúa alrededor de 132-150 cm, pero el hipocrecimiento puede no ser manifiesto hasta los dos años y confundirse, dados los antecedentes familiares, con una forma extrema de talla baja familiar.
Síndromes por deficiencia del gen SHOX. Este gen se localiza en la región PAR1 (región pseudoautosómica 1), en el extremo distal de Xp e Yp. Las mutaciones o deleciones en homocigosis o heterocigosis compuesta son excepcionales y dan lugar a una forma severa de DO, la llamada displasia mesomélica de Langer, con talla media inferior a -6 SDS y aplasia de cúbito y peroné. Por el contrario, la haploinsuficiencia del gen SHOX, debida a mutaciones o deleciones en heterocigosis en SHOX/PAR1, con herencia pseudoautosómica dominante, representan la causa monogénica más frecuente de talla baja en la especie humana (1:1.000-2.000) y es responsable: 1) de la mayoría de los casos de discondrosteosis de Léri-Weill (60-80%); 2) del 2-5% de las tallas bajas aparentemente armónicas y consideradas a priori como TBI; y 3) de algunas de las manifestaciones clínicas del síndrome de Turner(12).
Las manifestaciones clínicas más características de la haploinsuficiencia del gen SHOX son: hipocrecimiento mesomélico (acortamiento de antebrazos y tibias), cubitus valgo, deformidad de Madelung (Fig. 4), acortamiento de metacarpianos y metatarsianos, paladar ojival, desarrollo anormal de las orejas, micrognatia y cuello corto. Estas manifestaciones clínicas definen la discondrosteosis o síndrome de Léri-Weill y son más marcadas en mujeres y especialmente después de la pubertad. No obstante, la variabilidad fenotípica es muy amplia, incluso entre los miembros de una misma familia (falta de correlación genotipo-fenotipo); de forma que, el fenotipo clínico en la haploinsuficiencia del gen SHOX se muestra como un continuum (Fig. 4), que abarca desde formas muy graves de talla baja desproporcionada a formas muy leves de talla baja o talla normal-baja, armónica o disarmónica, con o sin otras anomalías clínicas y radiológicas(12).
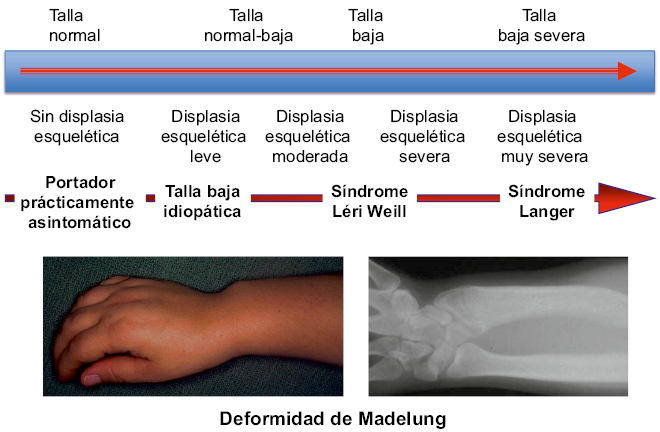
Figura 4. Esquema donde se representa la variable expresividad clínica y fenotípica de la insuficiencia del gen SHOX que viene a ser un continuum que abarca desde formas muy severas de talla baja desproporcionada a formas muy leves de talla baja o normal, armónica o disarmónica, con o sin otras anomalías clínicas y radiológicas. Deformidad de Madelung: acortamiento e incurvación del radio con subluxación dorsal del extremo distal del cúbito, triangularización de los huesos del carpo y fusión prematura de las epífisis.
Por ello, se ha desarrollado un sistema de puntuación, basado en los hallazgos clínicos y antropométricos (Fig. 5), que permitiría optimizar la selección de pacientes en los que debería excluirse una haploinsuficiencia del SHOX y que no presentan las manifestaciones clínicas características del síndrome de Léri-Weill, como la clásica deformidad de Madelung. En lo que se refiere a la talla adulta, la talla media de estos pacientes es de 145 cm en mujeres y de 155 en varones, pero alrededor del 50% de los pacientes muestran una talla normal. Al comparar con los hermanos no afectados, la pérdida de talla supone, de media, unos 14,4 cm (2,4 SDS) en mujeres y unos 5,3 cm (0,8 SDS) en varones.
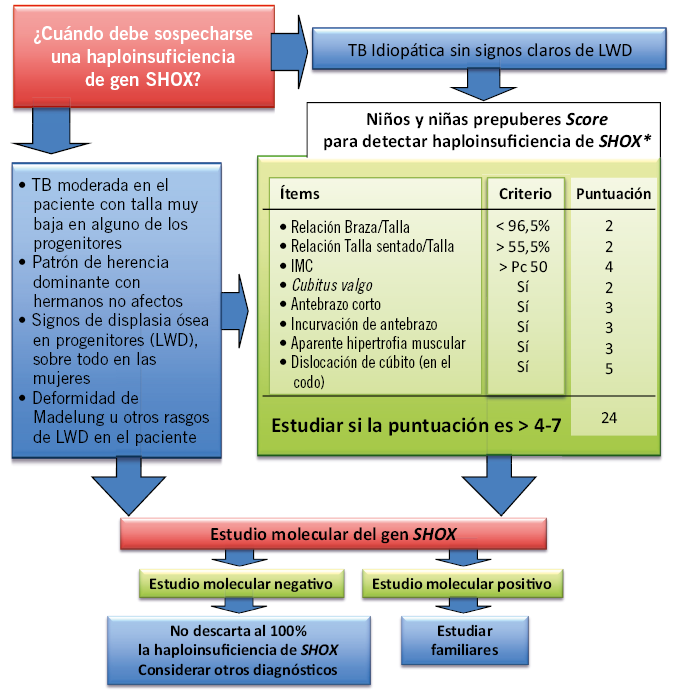
Figura 5. Esquema donde se representan aquellas circunstancias clínicas y antropométricas que deben hacer sospechar una haploinsuficiencia del SHOX y solicitar estudio genético. LWD: discondrosteosis de Léri-Weill; TB: talla baja. *Sistema de puntuación tomado de: Rappold G, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007; 44: 306-13.
Malnutrición
En los países en vías de desarrollo, la malnutrición junto con los procesos infecciosos y parasitarios, especialmente gastrointestinales, actúan de manera sinérgica (binomio infección-malnutrición) en la génesis del fracaso de crecimiento.
En los países subdesarrollados o en vías de desarrollo, el binomio infección-malnutrición (Fig. 6) es con mucho la causa más frecuente de hipocrecimiento. En los países desarrollados, la escasez de alimentos es una situación excepcional y los hipocrecimientos de causa nutricional suelen ser secundarios a: patologías crónicas, dietas inadecuadas en su cantidad (ejercicio excesivo) o composición (vegetarianas estrictas, macrobióticas…) o trastornos más o menos graves de la conducta alimentaria (anorexia nerviosa, síndrome del miedo a la obesidad, fallo de medro…). En cualquier caso, el resultado final de todas estas situaciones es un aporte insuficiente de macro (malnutrición calórico-proteica) o de micronutrientes (minerales y vitaminas).
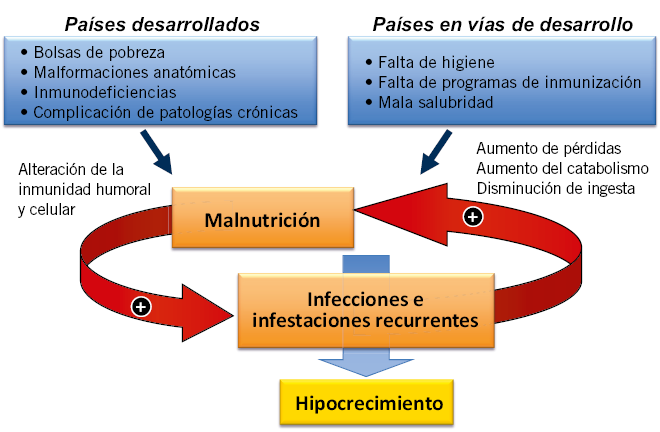
Figura 6. Esquema donde se refleja la interacción entre malnutrición e infección-infestación recurrente en la génesis del hipocrecimiento. La malnutrición, asociada a la pobreza, a enfermedades crónicas y a dietas inadecuadas, determina alteraciones en la inmunidad humoral y celular favoreciendo los procesos infecciosos y parasitarios, especialmente gastrointestinales, lo que a su vez induce una mayor malnutrición (incremento de pérdidas y catabolismo con disminución de aportes), cerrando así el circulo vicioso del binomio malnutrición-infección.
En la malnutrición calórico-proteica, una pérdida o inadecuada ganancia ponderal suele acompañar o preceder a la alteración del crecimiento en uno o dos años; si bien, no siempre es así, y el fracaso de crecimiento, acompañado de retraso de la EO y de la pubertad, puede ser el mecanismo de adaptación a la disminución de nutrientes, sin que se altere la relación peso/talla u otros marcadores clínicos/bioquímicos de malnutrición(13). En este proceso adaptativo, todo el sistema hormonal está implicado, pero son de especial importancia, las alteraciones en el eje GH-IGFs (resistencia a la GH, con niveles séricos normales o elevados de GH y disminuidos de IGF-1 y de IGFBP-3).
En lo referente a los déficits aislados de micronutrientes, se ha demostrado que, además de las deficiencias de calcio y fósforo, algunos oligoelementos, como: iodo, hierro, cobre, cromo y zinc, son capaces de provocar cuadros de retraso de crecimiento en humanos. En los déficits vitamínicos aislados, la alteración del crecimiento no suele ser una manifestación clínica precoz, con excepción hecha de la vitamina D, que al igual que el calcio y el fósforo, está directamente implicada en la mineralización y crecimiento del hueso.
Infecciones e infestaciones recurrentes
Son características de los países subdesarrollados, donde los procesos infecciosos y parasitarios, especialmente gastrointestinales, actúan de manera sinérgica con la malnutrición en la génesis del fracaso de crecimiento. En los países desarrollados, las infecciones recurrentes son poco frecuentes, pero, cuando alteran el crecimiento, suelen reflejar la existencia de malformaciones anatómicas (renales, pulmonares…) o inmunodeficiencias subyacentes (SIDA…) y contribuir al fracaso de crecimiento asociado a otras patologías crónicas. La malnutrición parece ser el principal mecanismo fisiopatológico implicado en estas formas de hipocrecimiento; ya que, la alimentación suplementaria durante los períodos de infección parece evitar o, al menos, disminuir sus repercusiones negativas sobre el crecimiento.
Enfermedades crónicas
Cualquier patología crónica (gastrointestinal, renal, cardíaca, pulmonar, hematooncológica…), si es lo suficientemente grave y prolongada, puede provocar un hipocrecimiento. En general, cuanto más precoz, grave y prolongada sea la enfermedad, menos posibilidades hay de que pueda recuperarse completamente.
El fracaso en el crecimiento y la talla baja consiguiente son hallazgos habituales en los niños que padecen enfermedades crónicas (Tabla II). Se calcula que, alrededor del 10-15% de los hipocrecimientos serían secundarios a enfermedades crónicas; no obstante, es probable que este porcentaje no solo esté infravalorado, sino que se incremente como consecuencia de la mayor supervivencia en muchas enfermedades crónicas. Los factores etiopatogénicos implicados en el fracaso del crecimiento asociado a las enfermedades crónicas son múltiples y varían dependiendo de la enfermedad de base, pero suelen estar en relación con: malnutrición, alteraciones metabólicas, efectos secundarios de la terapia, infecciones sobreañadidas y, posiblemente también, con los trastornos psicológicos que acompañan al padecimiento de una enfermedad crónica. Dentro de ellos, la malnutrición, a la que se puede llegar por múltiples mecanismos (disminución de aportes o incremento de pérdidas o demandas), es uno de los factores más importantes y constantes.
En lo que se refiere a las manifestaciones clínicas, las específicas de la enfermedad de base serán, en la mayoría de los casos, las predominantes en el cuadro clínico y el fracaso del crecimiento será solo un síntoma más. No obstante, en determinadas patologías, como: malnutrición, celiaquía, enfermedad inflamatoria intestinal o acidosis tubular renal, entre otras, el hipocrecimiento puede ser, durante años, el único o el principal síntoma de la enfermedad subyacente.
El patrón de crecimiento suele ser similar en todas ellas. La aparición de la enfermedad va a determinar un enlentecimiento más o menos marcado del ritmo de crecimiento (fracaso de crecimiento) que suele acompañarse de un retraso simultáneo de la EO y del desarrollo puberal. En caso de curación o mejoría significativa, se producirá un “crecimiento de recuperación” que puede permitir recuperar, total o parcialmente, la talla perdida. La medida en que el hipocrecimiento puede ser compensado depende de diferentes factores: edad de inicio, gravedad y duración del fracaso en el crecimiento, etiología y patogénesis de la enfermedad y, también, de factores individuales propios de cada sujeto.
Enfermedades endocrinológicas
El porcentaje de retrasos de crecimiento de origen endocrinológico es muy escaso, alrededor del 5%, y, en concreto, las deficiencias de GH suponen menos de un 1-2% de los hipocrecimientos.
Deficiencia de GH. Su incidencia oscila entre 1:3.500-1:10.000 RN vivos. Puede presentarse de forma aislada o asociado a otras deficiencias hipofisarias (hipopituitarismos) y puede ser congénita (alteraciones genéticas, malformaciones de línea media…) o adquirida (tumores, traumatismos, histiocitosis, infecciones, radioterapia…). En la mayoría de los casos, el déficit es idiopático y solo en aproximadamente un 20% de los casos es posible identificar una causa orgánica responsable. Entre las formas idiopáticas, es frecuente el hallazgo en la RM craneal de determinadas anomalías morfológicas, como son: hipoplasia hipofisaria, tallo hipofisario ausente o muy afilado y neurohipófisis ectópica. Esta asociación, conocida como “síndrome de sección del tallo hipofisario”, aunque de etiopatogenia no aclarada, constituye un dato diagnóstico relevante y conlleva un mayor riesgo de desarrollar en el tiempo otras deficiencias hipofisarias.
Se estima que entre un 5 y un 30% de las formas idiopáticas tendrían una base genética, bien por mutaciones en el gen de GH (GH1; deficiencia o GH biológicamente inactiva) o del receptor de la hormona hipotalámica liberadora de GH (GHRHR), que condicionarían una deficiencia aislada de GH, o bien por mutaciones en genes que codifican para factores de transcripción implicados en el desarrollo de la hipófisis (HESX1, LHX3, LHX4, POUIFI, PROPI…) que condicionarían, habitualmente, deficiencias hipofisarias múltiples(4).
La manifestación clínica más característica de la deficiencia de GH es el fracaso de crecimiento, que se acompaña de una marcada disminución de la VC y de retraso de la EO. La secreción espontánea de GH y/o la respuesta de GH a los diferentes test de estimulación están disminuidas, al igual que los niveles séricos de IGF-1 e IGFBP-3. En las formas congénitas o en las adquiridas de inicio muy precoz, el hipocrecimiento suele acompañarse de un fenotipo característico: cara de “muñeca”, voz aguda, incremento periabdominal de la grasa, manos y pies pequeños y disminución de la masa muscular. El déficit congénito de GH se asocia, frecuentemente, a complicaciones perinatales (sufrimiento fetal, presentación podálica, fórceps, hipoglucemia e hiperbilirrubinemia conjugada), así como a un pene pequeño en los varones (frecuente deficiencia asociada de gonadotropinas). En las formas adquiridas de inicio tardío, el fracaso de crecimiento puede ser la única manifestación clínica.
Insensibilidad a la GH. Se definiría como la ausencia de una apropiada respuesta metabólica y de crecimiento a la GH endógena o a la GH administrada a dosis fisiológica de sustitución. La insensibilidad adquirida a la GH es una situación clínica frecuente; ya que, se asocia a patologías crónicas y especialmente a la malnutrición calórico-proteica. Por el contrario, la formas de insensibilidad congénita a la GH por anomalías en el receptor de GH (RGH), salvo en determinadas poblaciones con alto grado de consanguinidad, son extraordinariamente infrecuentes(4). En la mayoría de los casos, se trata de mutaciones en el gen del RGH (GHR), de herencia autosómica recesiva, que dan lugar a una insensibilidad total a la acción de la GH con hipocrecimiento grave y fenotipo característico (síndrome de Laron), que es similar en muchos aspectos al de la deficiencia completa de GH. Los niveles séricos de GH son normales o elevados y disminuidos los de IGF-1 e IGFBP-3.
Se han descrito, también, formas congénitas de insensibilidad parcial a la GH, debidas a mutaciones en heterocigosis en el GHR, que dan lugar a formas de hipocrecimiento menos graves. Así mismo, se han descrito también situaciones excepcionales de insensibilidad total o parcial a la GH por alteración en los mecanismos post-receptor encargados de la transmisión del mensaje de GH (mutaciones en STAT-5b), así como por deficiencia de ALS (mutaciones en el gen de ALS), deficiencia de IGF-I (mutaciones en el gen de IGF-I) o resistencia a IGF-I (mutaciones en el receptor de IGF-I). La afectación del crecimiento en estos casos puede ser variable, con hipocrecimientos, en ocasiones, dentro del rango bajo de la normalidad y que pueden ser confundidos con VNTB.
Hipotiroidismo. Representa menos de un 1% del total de hipocrecimientos, gracias a la aplicación generalizada del screening neonatal, al mejor control en las áreas de bocio endémico y a la mejoría, en general, en el diagnóstico y tratamiento de los hipotiroidismos adquiridos (tiroiditis linfocitaria crónica, lo más frecuente). El hipocrecimiento, el sobrepeso y el retraso en la EO y en el inicio de la pubertad son hallazgos habituales en las situaciones de hipotiroidismo prolongado.
Exceso de esteroides sexuales. El incremento de esteroides sexuales durante la infancia puede obedecer a numerosas causas y puede resultar de una activación precoz, idiopática u orgánica, del eje hipotálamo-hipófiso-gonadal (pubertad precoz verdadera) o puede producirse independientemente de la secreción de gonadotropinas hipofisarias (pseudopubertad precoz o pubertad precoz periférica). La pubertad y la pseudopubertad precoces son una forma especial de hipocrecimiento; ya que, el exceso de esteroides sexuales durante la fase prepuberal condiciona un hipercrecimiento transitorio, por aceleración anormal de la VC y de la EO, con cierre precoz de los cartílagos de crecimiento y talla final baja.
Hipercortisolismo. El hipocrecimiento en situaciones de hipercortisolismo crónico (síndrome de Cushing) es un fenómeno prácticamente constante y suele ser, junto con la obesidad, su manifestación clínica más precoz. El hipercortisolismo puede ser secundario a: 1) patología hipofisaria (adenoma secretor de ACTH; enfermedad de Cushing); 2) patología suprarrenal (hipercortisolismo ACTH independiente: tumor suprarrenal, hiperplasia suprarrenal macro o micronodular…); o 3) administración exógena y mantenida de glucocorticoides. Salvo en este último caso, el hipercortisolismo es una causa excepcional de hipocrecimiento en la infancia. Los tumores adrenales suelen producir, además de cortisol, cantidades excesivas de andrógenos, lo que puede enmascarar el hipocrecimiento asociado a la hipercortisolemia. Los mecanismos fisiopatológicos que median la alteración del crecimiento en el hipercortisolismo son múltiples, entre otros: la alteración de la secreción-acción de la GH y el trastorno de la osificación y síntesis de colágeno en la placa de crecimiento. La gravedad del hipocrecimiento depende, entre otros factores, de la dosis, duración, vía, tipo de glucocorticoide y sensibilidad individual del paciente; además, puede no recuperarse completamente al cesar su administración y comprometer definitivamente la talla adulta. El efecto negativo sobre el crecimiento es mayor con corticoides de acción prolongada, como la dexametasona, y con la administración diaria frente a la administración a días alternos. Los corticoides inhalados, utilizados con frecuencia en el tratamiento crónico del asma, salvo dosis elevadas y mantenidas o especial sensibilidad del sujeto, suelen tener poca o nula repercusión sobre el crecimiento.
Pseudohipoparatiroidismo (PHP). Representan un grupo heterogéneo de enfermedades, extremadamente infrecuentes caracterizadas por la resistencia a la acción de la paratohormona (PTH) en sus órganos diana (hueso y riñón). Desde el punto de vista bioquímico, se caracterizan por hipocalcemia, hiperfosfatemia y niveles supranormales de PTH, pero sin hiperfosfaturia ni aumento de 1-25 (OH)2 vitamina D. Muchos de estos pacientes presentan, además, un fenotipo peculiar, denominado osteodistrofia hereditaria de Albright (OHA), que se caracteriza por: talla baja, obesidad, cara redonda, calcificaciones en tejidos blandos, anomalías óseas (acortamiento de metacarpianos, metatarsianos y falanges…) y, con frecuencia variable, retraso mental leve-moderado.
El patrón hereditario y fenotípico de los PHP es complejo, ya que, el gen implicado (GNAS; 20q13.32), que codifica para la subunidad alfa estimuladora de la proteína G que interviene en la transmisión del mensaje del receptor de PTH y de otros receptores hormonales (TSH, LH, FSH, GHRH…), está sometido a imprinting paterno; es decir, el alelo procedente del padre no se expresa en condiciones normales en: hipófisis, tiroides, gónadas y túbulo renal. Esta es la causa de que, el fenotipo pueda variar dependiendo de si la causa de la enfermedad es una mutación o una alteración de la impronta genética y de si el gen mutado es de origen materno (OHA con resistencia hormonal) o paterno (OHA sin resistencia hormonal, pseudopseudohipoparatiroidismo). La mayoría de los pacientes presentan mutaciones inactivantes en heterocigosis (herencia autosómica dominante) y pueden presentar resistencia, además de a la PTH, a otras hormonas, especialmente a la TSH.
Hipocrecimiento psicosocial
Se define el hipocrecimiento de causa psicosocial como: “un síndrome de talla baja y/o retraso puberal que se produce en niños y adolescentes en situaciones de hostigamiento psicológico o deprivación afectiva y para el que no se encuentra otra explicación”. Los mecanismos fisiopatológicos que median esta forma de hipocrecimiento son desconocidos, aunque en algunos casos se han implicado alteraciones en el eje de la GH-IGFs. Su diagnóstico es difícil y requiere un alto índice de sospecha.
Hipocrecimientos de causa desconocida (talla baja idiopática)
El concepto de TBI agruparía situaciones normales y patológicas de hipocrecimiento armónico de inicio postnatal, cuyo único denominador común es nuestra incapacidad para alcanzar un diagnóstico etiopatogénico. Se estima que, aproximadamente, el 80% de los niños que consultan por talla baja podrían ser diagnosticados de TBI.
La definición de TBI es el resultado del consenso entre expertos de las sociedades Americana y Europea de Endocrinología Pediátrica, así como de la Growth Hormone Research Society, reunidos en Santa Mónica (California) en octubre de 2007(15). En esta reunión, se definió la TBI como: una condición en la que la talla de un individuo se encuentra más de 2 SDS por debajo de media para su edad, sexo y grupo de población, sin evidencia de anomalías sistémicas, endocrinas, nutricionales o cromosómicas. Esta definición, según establece el propio consenso, incluye a los niños con VNTB y excluye, específicamente, niños con: peso y/o talla baja para su EG (RNPEG), fenotipo dismórfico o disarmónico (DO, síndromes de Turner, de Noonan…), trastorno psiquiátrico o emocional grave u otras causas claramente identificables de talla baja (por ejemplo: enfermedad celíaca, enfermedad inflamatoria intestinal, malnutrición, deficiencia o insensibilidad a la GH, hipotiroidismo, síndrome de Cushing, etc.).
Las VNTB serían los hipocrecimientos armónicos de inicio postnatal que resultarían de la variabilidad normal tanto de la talla como del ritmo madurativo que existe en la especie humana(1). Nuestro desconocimiento de los mecanismos fisiológicos que subyacen al control genético del crecimiento y del ritmo madurativo, así como nuestra incapacidad para predecir con fiabilidad la talla adulta de un niño, han condicionado su inclusión en el grupo de TBI. Dentro de las VNTB se incluyen: la talla baja familiar (TBF), el retraso constitucional del crecimiento y de la pubertad (RCCP) y la frecuente asociación de ambos patrones de crecimiento (Tabla IV).

El término de TBF hace referencia a un grupo de individuos con talla baja, por otro lado sanos, que maduran a un ritmo normal y cuyos familiares más próximos son de talla baja. Por su parte, los pacientes con RCCP serían sujetos sanos que, como consecuencia de un ritmo de maduración más lento que la media de la población (alrededor del 60% tienen antecedentes familiares de maduración tardía), presentan durante la infancia: talla baja, inadecuada para su contexto familiar, retraso de la EO e inicio puberal tardío (2-3 años después que la media), alcanzando la talla adulta a una edad superior a la media de la población. En ambas situaciones, la talla final es acorde con el contexto familiar.
El concepto de TBI es controvertido, artificial y heterogéneo; ya que, incluye situaciones normales y patológicas, cuyo único denominador común es nuestra incapacidad para alcanzar un diagnóstico etiopatogénico. Se estima que, aproximadamente, el 80% de los niños que consultan por talla baja podrían ser diagnosticados de TBI. La inmensa mayoría de estos niños (80-85%) corresponderían a VNTB y un pequeño porcentaje (15-20%) a patologías en las que, por desconocimiento o dificultad diagnóstica, no se llega a alcanzar un diagnóstico, como sería el caso de: hipocrecimientos nutricionales, hipocrecimientos psicosociales, alteraciones infrecuentes o menores en el eje GH-IGFs, así como osteocondrodisplasias, cuadros sindrómicos o alteraciones genéticas (mutaciones, microdeleciones, duplicaciones, disomías uniparentales…) de escasa expresividad clínica, entre otras posibles causas. Por consiguiente, a medida que nuestros conocimientos y métodos de diagnóstico, sobre todo moleculares, mejoren y puedan aplicarse en estos pacientes, muchos de estos niños diagnosticados de TBI deberán ser reclasificados a grupos de patología conocida.
Orientación diagnóstica y pruebas complementarias
La evaluación inicial de todo paciente que consulta por hipocrecimiento incluirá una historia clínica exhaustiva y un examen clínico completo (Tabla V y Algoritmo diagnóstico)(16), al que debe añadirse una valoración auxológica básica(8,9), la determinación de la maduración ósea (EO)(9) y, si es posible, el análisis del patrón de crecimiento a partir de los datos aportados por los padres o acumulados en la historia del niño (Fig. 7). Los datos obtenidos de esta evaluación inicial nos permitirán, al menos, determinar si se trata de un hipocrecimiento armónico o disarmónico y si es de inicio prenatal o postnatal, y así orientar el diagnóstico y las pruebas complementarias.

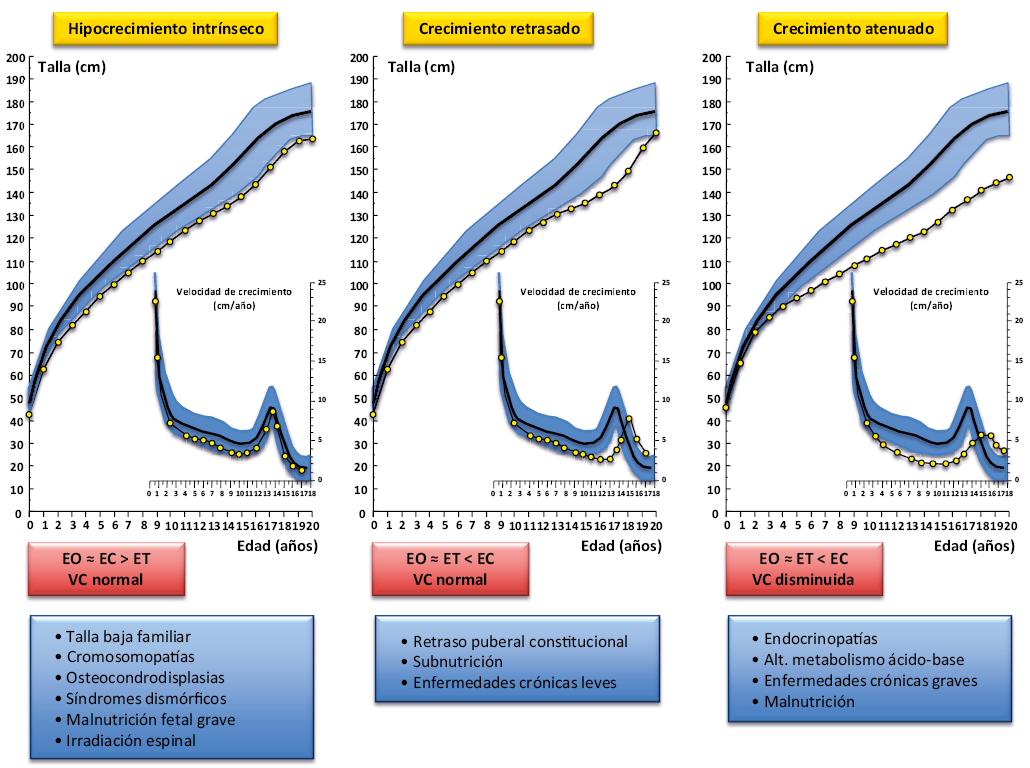
Figura 7. Patrones clínicos básicos de hipocrecimiento. Patrón de hipocrecimiento intrínseco: se observa en aquellas situaciones en las que existe una limitación en el crecimiento inherente a los propios tejidos, especialmente el esquelético, en la mayoría de los casos de base genética o congénita, lo que va a condicionar una talla final baja. Patrón de crecimiento retrasado: se traduce en un retraso puberal con prolongación del periodo de crecimiento y talla final, en la mayoría de los casos, normal. Patrón de crecimiento atenuado: se caracteriza por una velocidad de crecimiento disminuida de forma mantenida en el tiempo, que condiciona un alejamiento progresivo de la talla de los percentiles normales y una talla final baja. EC: edad cronológica; EO: edad ósea; ET: edad talla (edad para la que la talla del sujeto estaría en el percentil 50 de la población para su edad y sexo).
La EO es uno de los parámetros más sencillos e importantes en la valoración del crecimiento de un niño. No solo se utiliza en el cálculo de la PTA, sino que puede orientar a determinadas patologías (Fig. 7). Está habitualmente retrasada en los trastornos de crecimiento secundarios (patologías crónicas, endocrinopatías…) y en muchos de los niños con TBI; más aún, la ausencia de retraso en la EO, prácticamente, excluye una situación de deficiencia de GH. Por el contrario, en la mayoría de los trastornos primarios o intrínsecos del crecimiento (síndromes dismórficos, osteocondrodisplasias…), el retraso de la EO es menor o inexistente. Un cuidadoso análisis de la radiografía de mano puede, además, sugerir la existencia de una DO no diagnósticada (braquidactilias, haploinsuficiencia del SHOX, pseudohipoparatiroidismo…), en cuyo caso, al igual que ocurre cuando el hipocrecimiento es disarmónico, debería realizarse un estudio radiológico del resto del esqueleto.
Si el hipocrecimiento es armónico y de inicio prenatal
Si el hipocrecimiento es armónico y de inicio prenatal, debe observarse la evolución del niño y, si este no recupera su carril de crecimiento en el primer o segundo año de vida, debería remitirse al especialista; habida cuenta de la escasa posibilidad de recuperación espontánea del crecimiento a partir de esa edad y de la posibilidad de tratamiento con GH(10). Si no existe constancia de patología materna o placentaria que justifique el inicio prenatal del hipocrecimiento y especialmente si el niño presenta retraso mental o psicomotor, malformaciones mayores o menores, fenotipo peculiar o algún rasgo sindrómico, debe considerarse la posibilidad de que se trate de una cromosomopatía o de un síndrome dismórfico; por lo que, estaría indicada la realización de un cariotipo y la remisión al especialista.
Existe una multitud de cuadros síndrómicos en los que el hipocrecimiento es una de sus principales características, algunos de los más frecuentes se recogen en la tabla III. Muchos de ellos tienen una base genética, pero la mayoría no muestran alteraciones en el cariotipo convencional y su reconocimiento requiere de una gran experiencia por parte del médico que lo observa. Si se sospecha un síndrome de base genética conocida y la metodología está accesible, es recomendable confirmar molecularmente el diagnóstico clínico, lo que permitirá: establecer un diagnóstico de certeza, orientar a los padres sobre la futura evolución del paciente y realizar un adecuado consejo genético.
En caso de que las características clínicas del paciente no orienten hacia un diagnóstico concreto, se puede considerar, en colaboración con el genetista clínico, la posibilidad de realizar otros estudios genéticos más amplios (revisión en Palacios-Verdú y Pérez-Jurado(17)): estudios de MLPA (Multiplex Ligation-dependent Probe Amplification), que permiten analizar variantes del número de copias (CNV) en 40-50 loci simultáneamente (los loci de los síndromes más frecuentes asociados a hipocrecimiento), o lo que se conoce como “estudios de asociación a lo ancho del genoma” o GWAS (genoma-wide association studies). Dentro de estos estudios, los más utilizados en la detección de estas patologías son: los arrays-SNP (SNP: single nucleotide polymorphism), que permiten detectar cientos o miles de SNPs (variaciones de una sola base en el ADN), que son el tipo de polimorfismo más frecuente; y los arrays-CGH (CGH: hibridación genómica comparada) que permiten detectar variaciones en la dosis o número de copias del genoma (microdeleciones, microduplicaciones o disomías uniparentales). Más recientemente, se están incorporando progresivamente a la práctica clínica los llamados estudios de secuenciación de alto rendimiento (NGS: Next-Generation Sequencing). Este tipo de estudios permite la secuenciación del genoma completo, de capturas selectivas (paneles de genes implicados en determinadas patologías), de regiones concretas o de todas las regiones codificantes y reguladoras conocidas de todo el genoma (exoma). La secuenciación del exoma parece la mejor estrategia inicial o, al menos la de mayor eficiencia, ya que, analizando solo un 1-1,5% del genoma, es posible detectar el 80-90% de las mutaciones causantes de enfermedad, con rendimientos diagnósticos que oscilan según los estudios entre un 10 y un 54%. En cualquier caso, la indicación y, más aún, la interpretación de este tipo de estudios no es fácil y debería hacerse en colaboración con el genetista clínico; ya que, cada tipo de estudio presenta sus propias limitaciones diagnósticas y aunque nos señalan genes que podrían estar implicados en una enfermedad, no establecen una relación causa-efecto, lo que debe comprobarse y no siempre es fácil.
Si el hipocrecimiento es armónico y de inicio postnatal
Si la talla no está gravemente afectada, entre -2 y -3 SDS, existen antecedentes familiares de TBF o de RCDP y no existen datos sugerentes de hipocrecimiento patológico (ver Algoritmo diagnóstico), en la mayoría de los casos se tratará de una VNTB. Estos hallazgos, junto con un patrón de crecimiento característico y una predicción de talla adulta acorde con la talla familiar, permitirán establecer un diagnóstico provisional de VNTB. La constatación, a los 6 y 12 meses, de la existencia de una VC normal, prácticamente confirmará el diagnóstico. Si, por el contrario, la VC es patológica, se deberá reconsiderar el diagnóstico y valorar la realización de estudios complementarios o la remisión del paciente al especialista.
Si la afectación de la talla es severa, por debajo de -3 SDS, o existen datos sugerentes de patología (ver Algoritmo diagnóstico), deberán realizarse pruebas complementarias. En muchos de los casos, la valoración inicial nos permitirá establecer un diagnóstico de sospecha y orientar estas pruebas hacia una patología específica (nutricional, gastrointestinal, respiratoria…). Si no existen datos sugerentes de patología específica, será necesario practicar, junto con un seguimiento estrecho de la evolución clínica y auxológica del paciente, una serie de pruebas complementarias que permitan descartar, aquellas enfermedades en las que el hipocrecimiento pueda ser la manifestación inicial o que, por su escasa expresividad clínica, puedan pasar desapercibidas (Tabla VI). Estas pruebas básicas son las propuestas en la mayoría de los protocolos nacionales e internacionales de talla baja idiopática(16), pero pueden ampliarse o completarse, en función de la historia clínica, de la evolución o de los resultados de las pruebas previas, con otros estudios (Tabla VI).

La deficiencia de GH es muy infrecuente y más aún los cuadros clínicos de resistencia congénita a la GH, por lo que los estudios de valoración del eje GH-IGFs, salvo la determinación basal de IGF-1, no estarían incluidos en la valoración inicial de un niño con talla baja. En cualquier caso, IGF-1 carece de la sensibilidad y especificidad suficientes para establecer o descartar una alteración en el eje de la GH; de hecho, alrededor del 25-50% de las TBI presentan niveles séricos de IGF-1 por debajo de -2 SDS y pacientes deficitarios de GH pueden presentar niveles séricos de IGF-1 dentro del rango bajo de la normalidad(18). Pese a ello, la mayoría de los autores está de acuerdo en que si los niveles séricos de IGF-1 son claramente normales (entre ± 1 SDS para la edad y sexo), la EO no está claramente retrasada y la VC es normal, la probabilidad de que exista un trastorno en el eje GH-IGF-1 es muy escasa y no sería necesario realizar más estudios de valoración. Si por el contrario, los niveles de IGF-1 están por debajo de -2 DE para la edad y sexo o por debajo de la media junto con datos clínicos sugerentes de deficiencia de GH, estos deberían llevarse a cabo(18). La realización e interpretación de los test que valoran el eje GH-IGFs es compleja y tampoco existe un acuerdo sobre qué, cuándo y cómo deberían realizarse estos estudios (test de estimulación farmacológica con/sin primación, qué fármacos o estímulos utilizar, estudios de secreción espontánea, test de generación de IGF-I, niveles séricos de ALS, GHBP…). Por ello, su indicación e interpretación solo deberían realizarse por personal experimentado y siempre en un contexto clínico compatible.
Lo mismo podría decirse de los estudios genéticos orientados al diagnóstico de los hipocrecimientos(19). Deberían estar orientados por el especialista y en colaboración con el genetista clínico hacia genes concretos, en función del diagnóstico y los hallazgos clínicos y hormonales: deficiencia aislada de GH con historia familiar positiva (GH1, GHRH…), deficiencia combinada de hormonas hipofisarias (LHX3, LHX4, PROP1, HESX1…), deficiencia primaria de IGF-1 (GHR, STAT5b, IGF-1, IGF1ALS…) o resistencia a IGF-1 (IGF1R…)… En el caso de pacientes con TBI sin otras alteraciones y no sugerentes de ser VNTB, podría estar indicado el estudio inicial de alguno de los genes más frecuentes asociados a TBI, como sería el caso de SHOX (responsable del 5% de las TBI), FGFR3 o NPR2 (gen del receptor del péptido natriurético de tipo C –CNP–), o la realización de estudios más amplios: GWAS (rendimiento diagnóstico del 5-10% en TBI) o NGS (rendimiento diagnóstico del 10-54%)(19-21).
Si el hipocrecimiento es disarmónico, prenatal o postnatal
Si la talla baja se asocia con unas proporciones corporales anormales, debe remitirse al especialista y considerarse como una DO. Un diagnóstico preciso va a depender en gran medida de la experiencia del especialista y del radiólogo; ya que, el estudio radiológico del esqueleto (mapa óseo) es la prueba diagnóstica más importante. A menudo, y dada la aparición tardía de las deformidades y de algunas de las anomalías radiológicas, es necesario repetir el examen a lo largo de la infancia y valorar cuidadosamente la armonía corporal y la presencia de deformidades en los progenitores. El estudio anatomopatológico del cartílago y hueso puede ser de utilidad en algunos casos, aunque rara vez se lleva a cabo. Cuando existe la sospecha clínica de una osteocondrodisplasia de base genética conocida y la metodología está disponible (acondroplasia, hipocondroplasia, mucopolisacaridosis, haploinsuficiencia de SHOX, entre otras), pueden realizarse estudios bioquímicos o moleculares que confirmen el diagnóstico, lo que permitirá un adecuado consejo genético y un diagnóstico prenatal precoz. Cuando no es así, pueden realizarse estudios genéticos más amplios que incluyan paneles con los genes asociados a las DO más frecuentes o estudios más amplios del genoma completo.
Opciones terapéuticas en la talla baja
Orientación general
La talla baja no es una enfermedad, sino un síntoma. El tratamiento, en la mayoría de los casos (VNTB), consistirá en aportar una adecuada información al niño y a sus padres de las causas y las expectativas de crecimiento y realizar un seguimiento clínico y auxológico. En otros casos, consistirá únicamente en el tratamiento de la enfermedad responsable. Por último, en un pequeño número de pacientes, puede ser necesaria la utilización de tratamientos específicos para estimular o mejorar el crecimiento.
En la práctica clínica, disponemos de un número limitado de terapias capaces de influir beneficiosamente sobre el crecimiento, como son: la GH y el IGF-1 recombinantes, los fármacos moduladores de la pubertad y el alargamiento óseo.
Tratamiento con GH
La GH estimula el crecimiento longitudinal de los huesos largos de manera prácticamente dosis-dependiente y es el tratamiento específico del déficit de GH. Además, desde que la ingeniería genética permitió la biosíntesis de GH en la década de los 80, con su consiguiente disponibilidad solo limitada por su precio y potenciales efectos secundarios, se ha utilizado experimentalmente en la mayoría de las formas de hipocrecimiento (TBI, displasias esqueléticas, síndromes dismórficos, hipercortisolismos, fibrosis quística, PHP…)(22), con resultados, en el mejor de los casos discretos (mejorías medias de 1-1,5 cm/año de tratamiento en la talla adulta). En la actualidad, las indicaciones aceptadas para el tratamiento con GH en España y en Europa, además de la deficiencia de GH en niños y adultos, son: el síndrome de Turner, el hipocrecimiento asociado a insuficiencia renal crónica, la falta de recuperación de la talla en los RNPEG, el síndrome de Prader Willi y la haploinsuficiencia del gen SHOX. La GH se administra por vía subcutánea, diaria, habitualmente antes de acostarse, en una dosis única de 0,025-0,035 mg/kg/día, en los pacientes deficitarios, y de 0,045-0,05 mg/kg/día, en el resto de situaciones no deficitarias. No obstante, las pautas idóneas de tratamiento, en función del diagnóstico, la respuesta terapéutica y el grado de desarrollo puberal, entre otras variables, no han sido plenamente establecidas.
Los riesgos conocidos del tratamiento con GH en los pacientes deficitarios son escasos, los más frecuentes: epifisiolisis de la cabeza femoral y pseudotumor cerebri (hipertensión intracraneal benigna), con una frecuencia aproximada, en ambos casos, de 1:1000 niños tratados. A dosis más altas y mantenidas, puede haber disminución de la sensibilidad a la insulina, desarrollo de rasgos acromegaloides y elevación suprafisiológica de los niveles séricos de IGF-1 de repercusión incierta. En cualquier caso, la experiencia a largo plazo en pacientes no deficitarios y tratados con dosis altas de GH es escasa y sus potenciales riesgos, en gran medida, desconocidos. Esta incertidumbre debería hacer extremar la prudencia sobre su uso indiscriminado en patologías en las que sus hipotéticos beneficios no hayan sido suficientemente probados.
Tratamiento con IGF-1
La utilización de IGF-1 recombinante fue aprobada por la Agencia Europea del Medicamento (AEM) en 2008 para su utilización en los hipocrecimientos por deficiencia primaria grave de IGF-I (anomalías en el receptor de GH, en sus vías de señalización intracelular o mutaciones en el gen de IGF-1, entre otras posibles causas). La dosis recomendada es de 0,04-0,08 mg/kg/día (máximo: 0,12 mg/kg/día), repartida en dos dosis y administrada, cada 12 horas, por vía subcutánea, preferentemente antes de una comida para prevenir sus efectos hipoglucemiantes.
La experiencia clínica en la utilización del IGF-1 recombinante es relativamente escasa y prácticamente limitada al tratamiento de pacientes con insensibilidad congénita a la GH (síndrome de Laron). Los estudios a corto y medio plazo demuestran en ellos la eficacia del tratamiento, aunque sus resultados son menores que los obtenidos con la GH en los pacientes deficitarios. Los efectos secundarios más frecuentes serían: hipoglucemias, aumento del tejido linfático (amígdalas, adenoides, bazo y timo), náuseas, vómitos, cefalea, papiledema (pseudotumor cerebri), prurito y aumento de transaminasas(23).
Recientemente, se ha publicado el primer estudio sobre eficacia y seguridad del tratamiento combinado con GH e IGF-1, llevado a cabo en pacientes con TBI y niveles séricos disminuidos de IGF-1(24). Los resultados del estudio indican una discreta mejoría en el crecimiento respecto a los pacientes tratados solo con GH, que no justificaría, al menos de forma generalizada, la utilización combinada de GH e IGF-1.
Tratamiento con moduladores de la pubertad
Los beneficios de la utilización de fármacos moduladores de la pubertad en el tratamiento de la talla baja no están claramente demostrados(25). Los fármacos más utilizados han sido los análogos de GnRH (aGnRH) y más recientemente los inhibidores de 3ª generación de la aromatasa (IA: anastrozole y letrozole), la enzima que cataliza, en condiciones normales, el paso de andrógenos a estrógenos. Los estudios disponibles indican que, los aGnRH administrados aisladamente en pacientes con talla baja y pubertad normal o adelantada, pese frenar el desarrollo puberal y prolongar el periodo de crecimiento, no son útiles para mejorar la talla adulta, y que asociados con GH conducirían, en el mejor de los casos, a un beneficio modesto en la talla adulta, cuando el tratamiento combinado se mantiene durante, al menos, 3 años. En el caso de los IA, su objetivo sería enlentecer el cierre de las placas de crecimiento (mediado principalmente por los estrógenos) y prolongar el crecimiento lineal. Aunque los estudios iniciales, en varones con RCCP y TBI, eran muy prometedores y sugerían beneficios en la talla adulta de alrededor de 5 cm tras 1-2 años de tratamiento, los resultados disponibles a talla final, aunque todavía insuficientes para poder establecer conclusiones definitivas, no sostienen estos potenciales beneficios y sí posibles efectos secundarios, entre ellos: marcada elevación de los niveles séricos de testosterona, de repercusión incierta, desarrollo de anomalías vertebrales, disminución del colesterol HDL y aumento del hematocrito(26).
Tratamiento quirúrgico de la talla baja
Se basa en la realización de alargamientos óseos, habitualmente en tibias, fémures y húmeros. Su objetivo no es solo obtener una talla más alta, sino también mantener o mejorar la proporcionalidad de los segmentos corporales y, en casos concretos, conseguir, mejorías funcionales. Su indicación más habitual, generalmente a partir de los 9-12 años, son las DO, especialmente aquellas con huesos sólidos y afectación preferente de extremidades, como la acondroplasia. En las tallas bajas no displásicas, la indicación de la elongación ósea es más controvertida y suele realizarse en hipocrecimientos severos (por debajo de -3 SDS), una vez finalizado el crecimiento, cuando otros tratamientos han fracasado en la consecución de una talla “aceptable”. Antes de aconsejar este tipo de terapia, es importante informar al paciente y a sus padres de que las grandes elongaciones (30-35 cm en extremidades inferiores, 10-12 cm en las superiores) conllevan procesos de larga duración, dolorosos, en los que las incidencias y complicaciones (infecciosas, cicatriciales, óseas, musculotendinosas, vasculares y nerviosas) son relativamente frecuentes y, en ocasiones, graves, con resultados no siempre satisfactorios. Por ello, el paciente debe estar muy motivado y solo deberían ser llevadas a cabo por unidades muy experimentadas.
Tratamiento específico de las distintas formas de talla baja
Retrasos de crecimiento intrauterino
El 80-90% de los hipocrecimientos prenatales alcanzan una talla en los 2-4 primeros años de vida dentro de la normalidad y no requieren tratamiento. En el 10-20% que no lo hace, estaría indicado el tratamiento con GH, en Europa, a partir de los 4 años de edad, ya que, no solo mejora la talla final de muchos de estos pacientes, sino que parece ejercer también efectos positivos sobre la composición corporal, la tensión arterial y el metabolismo lipídico(10).
Cuadros sindrómicos y cromosomopatías
El hipocrecimiento asociado a los cuadros sindrómicos y cromosomopatías carece en general de tratamiento específico. En Europa, solo el síndrome de Turner (1,4 mg/m2/día) y el síndrome de Prader-Willi (1 mg/m2/día, sin sobrepasar los 2,7 mg/día) son indicaciones aceptadas para el empleo de la GH. En EE.UU., en 2007, la FDA (Food and Drug Administration) aprobó, también, la utilización de la GH en el síndrome de Noonan. Los resultados disponibles en síndrome de Noonan(27), aunque todavía insuficientes, indican una respuesta positiva, pero variable, entre los diferentes pacientes, con mejorías en la talla final que oscilan entre 0,5-1,4 SDS, sin aparentes repercusiones cardiacas o metabólicas. Tampoco parece que el tratamiento con GH influya sobre el potencial riesgo tumoral que presentan estos pacientes (mayor incidencia de leucemias y de ciertos tumores sólidos), aunque el seguimiento es todavía insuficiente para poder establecer conclusiones definitivas.
Osteocondrodisplasias
El tratamiento de la mayoría de las osteocondrodisplasias es todavía sintomático y ortopédico. Si el hipocrecimiento se produce a expensas del acortamiento de las extremidades inferiores y la calidad del hueso lo permite, el alargamiento óseo puede ser una alternativa. Aunque se ha utilizado la GH de forma experimental en diferentes displasias óseas, con escasos resultados, en Europa, solo en los hipocrecimientos por haploinsuficiencia del SHOX está aceptada la utilización de la GH (0,045-0,050 mg/kg/día). La experiencia a talla final en estos pacientes es todavía escasa, aunque los resultados preliminares sugieren una eficacia a largo plazo similar a la obtenida en las niñas con síndrome de Turner(28). Un ensayo clínico de tratamiento con análogo subcutáneo de CNP (péptido natriurético de tipo C) en pacientes con acondroplasia está en marcha y parece abrir la posibilidad de nuevas vías para el tratamiento de las DO(21).
Hipocrecimientos secundarios a enfermedades crónicas
Cuando el hipocrecimiento es el resultado de una enfermedad crónica (insuficiencia renal, fibrosis quística, malabsorción…), los esfuerzos terapéuticos deben dirigirse a mejorar la nutrición y, si es posible, a curar la enfermedad de base. La sensibilidad creciente respecto a las secuelas que sobre el crecimiento tienen estas enfermedades ha conducido a la modificación de muchos de los protocolos de tratamiento, al objeto de minimizar dichas secuelas e, incluso, a que se instauren terapias específicas encaminadas a mejorar el pronóstico de talla de estos pacientes, como es el caso, en algunas de ellas, de la administración de GH: insuficiencia renal crónica (IRC), enfermedad inflamatoria intestinal, corticoterapia crónica, artritis reumatoide y trasplantes hepático y cardíaco, entre otros. Salvo en el caso de la IRC, donde la indicación de GH ha sido aceptada, la utilización de la GH en el resto de situaciones continúa siendo experimental.
Hipocrecimientos secundarios a patología endocrinológica
La mayoría de los hipocrecimientos secundarios a patología endocrinológica tienen un tratamiento específico: GH en la deficiencia de GH, IGF-1 en la de insensibilidad congénita a la GH, levotiroxina sódica en los hipotiroidismos, hidrocortisona en la hiperplasia suprarrenal congénita, aGnRH en la pubertad precoz central, etc. El tratamiento del hipercortisolismo crónico requiere la supresión de la fuente de cortisol, habitualmente mediante la extirpación del tumor productor de ACTH (adenoma hipofisario) o de la suprarrenal (suprarrenaloma) o suprarrenales responsables de la hipercortisolemia. En aquellos casos en que el hipercortisolismo es iatrogénico, se debe intentar administrar los corticoides a días alternos y reducir, en la medida de lo posible, tanto la duración como la dosis.
Hipocrecimiento psicosocial
El tratamiento del hipocrecimiento de causa psicosocial es, al igual que su diagnóstico, complejo y difícil. En algunos casos, será suficiente con mejorar el aporte nutricional y modificar las actitudes familiares para conseguir un adecuado crecimiento de recuperación, pero, en la mayoría de los casos, solo un adecuado tratamiento psicológico y la separación del niño del ambiente familiar, no siempre posible, permitirá una normalización del crecimiento(14).
Talla baja idiopática
Las VNTB englobadas dentro del concepto de TBI no suelen requerir ningún tipo de tratamiento, salvo el apoyo psicológico y el aporte de una adecuada información a los padres y al paciente sobre la normalidad del cuadro clínico, su previsible patrón de crecimiento y las expectativas de talla adulta. En algunos pacientes con RCCP, el retraso en el crecimiento y, sobre todo, la aparición tardía de los caracteres sexuales secundarios puede determinar una mala adaptación psicosocial y baja autoestima. En estos casos, puede estar indicada la utilización durante un corto periodo de tiempo de esteroides sexuales a dosis bajas.
En el año 2003, la FDA norteamericana y posteriormente otros países han aprobado el uso de la GH (0,035-0,050 mg/kg/día) en pacientes con TBI(15,29). Pocas veces, una indicación de tratamiento ha generado y sigue generando, tanta polémica y tanto papel gastado en consideraciones clínicas, éticas, económicas y de toda índole. En resumen, los dos principales argumentos para esta aprobación fueron: 1) que la talla baja sería responsable de problemas psicosociales tanto en la infancia como en la edad adulta que condicionarían un menor estatus social y una menor calidad de vida; y 2) que desde 1993, se han ido aprobando indicaciones para la GH en situaciones no deficitarias basándose solo en la talla baja y su repercusión psicológica. Por consiguiente, la etiología de la talla baja no debería ser moralmente relevante en la decisión de quién debe o no ser tratado con GH; ya que, el objetivo del tratamiento sería corregir las consecuencias psicosociales negativas de la talla baja.
En España y en el resto de Europa la situación es distinta, y aunque multitud de tratamientos en esta indicación se están realizando off-label, la indicación de GH en la TBI no ha sido todavía aceptada por la AEM. En primer lugar, porque los estudios más recientes indican que la talla baja, aunque puede conllevar alguna situación de estrés emocional, no se asocia a problemas psicopatológicos, ni a menor estatus social, ni a menor calidad de vida. En segundo lugar, porque la relación coste-beneficio es mala: elevado coste económico (6.000-12.000 €/año de tratamiento) con discretos efectos beneficiosos sobre la talla final (1-1,5 cm/año de tratamiento). Por último, porque aunque los efectos secundarios a corto-medio plazo son escasos, existe un cierto grado de incertidumbre sobre sus efectos a largo plazo.
Otras opciones terapéuticas, como los IA o la asociación de GH y aGnRH, son todavía objeto de discusión y deberían ser considerados solo en el marco de estudios controlados(25).
Funciones del pediatra de Atención Primaria (AP)
No existe un consenso establecido sobre cuándo el pediatra de Atención Primaria debe iniciar un estudio por talla baja, qué pruebas deber realizar o qué criterios debe emplear para remitir a un paciente a una consulta especializada. Un intento de aproximación a cuál podría ser su papel en esta patología queda reflejado en el algoritmo diagnóstico. En cualquier caso, sería labor del pediatra de Atención Primaria:
• Monitorizar el crecimiento y desarrollo de todos los niños a lo largo de la infancia y adolescencia.
• Realizar los pasos iniciales en la orientación diagnóstica de estos pacientes: historia clínica, exploración, valoración auxológica y pruebas complementarias básicas, estableciendo el diagnóstico, en muchos casos, y decidiendo, en otros, la necesidad de derivación al especialista.
• Realizar el seguimiento de la mayoría de los niños con VNTB, pero también de otras causas de talla baja que no requieran tratamientos especializados, contando siempre con la potencial colaboración del especialista, si lo considera necesario, en función de la evolución o de las circunstancias individuales de cada caso.
• Por último, y no por ello menos importante, tranquilizar e informar a los padres y al niño, transmitiéndoles una información veraz y realista de las expectativas de crecimiento de sus hijos y de las opciones terapéuticas disponibles en cada caso, evitando crear falsas expectativas respecto a la bondad de tratamientos cuyos efectos sobre la talla final son discretos o escasamente probados y cuyos efectos secundarios a largo plazo son inciertos y, en algunos casos, potencialmente graves.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.*** Pozo J, Argente J. Hipocrecimiento armónico. En: Moro M, Málaga S, Madero L, ed. Cruz Tratado de Pediatría. (11ª edición). Madrid: Editorial Panamericana, S.A.; 2014. p. 1223-33.
2.** Karlberg J. A biologically-oriented mathematical model (ICP) for human growth. Acta Paediatr Scand. 1989; (Suppl 350): 70-94.
3.** Fowden AL, Forhead AJ. Endocrine regulation of feto-placental growth. Horm Res. 2009; 72: 257-65.
4.** Backeljauw PF, Dattani MT, Cohen P, Rosenfeld RG. Disorders of Growth Hormone/Insulin-Like Growth Factor Secretion and Action. En: Sperling MA, ed. Pediatric Endocrinology (fourth edition). Philadelphia: Elsevier Saunders; 2014. p. 292-404.
5.*** Juul A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm IGF Res. 2003; 13: 113-70.
6.** Boersma B, Wit JM. Catch up growth. Endo Rev. 1997; 18: 646-61.
7.** Barker DJP. Maternal nutrition, fetal nutrition, and disease in later life. Nutrition. 1997; 13: 807-13.
8.** Pozo J. Valoración auxológica del crecimiento I. Pediatr Integral. 2011; XV: 590-8.
9.** Pozo J. Valoración auxológica del crecimiento II. Pediatr Integral. 2011; XV: 691-701.
10.*** Clayton PE, Cianfarani S, Czernichow P, Johannsson G, Rapaport R, Rogol A. Consensus Statement: Management of the Child Born Small for Gestational Age through to Adulthood: A Consensus Statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab. 2007; 92: 804-10.
11.** Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet Part A. 2011; 155: 943-68.
12.** Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011; 75: 81-9.
13.** Lifshitz F. Nutrition and growth. J Clin Res Ped Endo. 2009; 1: 157-63.
14.** Pozo J, Argente J. Retraso de crecimiento de causa psicosocial En: Pombo M y cols., ed. Tratado de Endocrinología Pediátrica (4ª edición). Madrid: McGraw-Hill/Interamericana S.A.; 2009; p. 199-203.
15.*** Cohen P, Rogol AD, Deal CL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008; 93: 4210-17.
16.** Oostdijk W, Grote FK, de Muinck Keizer-Schrama SM, Wit JM. Diagnostic approach in children with short stature. Horm Res. 2009; 72: 206-17.
17.*** Palacios-Verdú MG, Pérez-Jurado LA. Nuevas metodologías en el estudio de enfermedades genéticas y sus indicaciones. Pediatr Integral. 2014; XVIII: 515-28.
18.*** Wit JM, Clayton PE, Rogol AD, Savage MO, Saenger PH, Cohen P. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Horm IGF Res. 2008; 18: 89-110.
19.** Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014; 99: 3080-92.
20.** Guo MH, Shen Y, Walvoord EC, et al. Whole exome sequencing to identify genetic causes of short stature. Horm Res Paediatr. 2014; 82: 44-52.
21.** Vasques GA, Arnhold IJ, Jorge AA. Role of the natriuretic peptide system in normal growth and growth disorders. Horm Res Paediatr. 2014; 82: 222-9.
22.** Pozo J, Argente J. La hormona de crecimiento (GH) en Pediatría. Rev Horm Crecim. 2007; X: 108-32.
23.** Bang P, Polak M, Woelfle J, Houchard A. Effectiveness and Safety of rhIGF-1 Therapy in Children: The European Increlex® Growth Forum Database Experience. Horm Res Paediatr. 2015; 83: 345-57.
24.** Backeljauw PF, Miller BS, Dutailly P, et al. Recombinant human growth hormone plus recombinant human insulin-like growth factor-1 coadministration therapy in short children with low insulin-like growth factor-1 and growth hormone sufficiency: results from a randomized, multicenter, open-label, parallel-group, active treatment-controlled trial. Horm Res Paediatr. 2015; 83: 268-79.
25.** Dunkel L. Treatment of Idiopathic Short Stature: Effects of Gonadotropin-Releasing Hormone Analogs, Aromatase Inhibitors and Anabolic Steroids. Horm Res Paediatr. 2011; 76 (suppl 3): 27-9.
26.** Shams K, Cameo T, Fennoy I, et al. Outcome analysis of aromatase inhibitor therapy to increase adult height in males with predicted short adult stature and/or rapid pubertal progress: a retrospective chart review. J Pediatr Endocrinol Metab. 2014; 27: 725-30.
27.** Giacomozzi C, Deodati A, Shaikh MG, Ahmed SF, Cianfarani S. The impact of growth hormone therapy on adult height in Noonan syndrome: a systematic review. Horm Res Paediatr. 2015; 83: 167-76.
28.** Blum WF1, Ross JL, Zimmermann AG, et al. GH treatment to final height produces similar height gains in patients with SHOX deficiency and Turner syndrome: results of a multicenter trial. J Clin Endocrinol Metab. 2013; 98: E1383-92.
29.*** Wit JM, Reiter EO, Ross JL, et al. Idiopathic short stature: Management and growth hormone treatment. Growth Horm IGF Res. 2008; 18: 111-35.
Bibliografía recomendada
- Cruz Tratado de Pediatría. (11ª edición). Madrid: Editorial Panamericana, S.A.; 2014.
La nueva edición (11ª) del “Cruz Tratado de Pediatría”, libro de referencia de la Pediatría española revisa y actualiza, en diferentes capítulos a lo largo de la obra, el crecimiento normal, su valoración y, por supuesto, las alteraciones del crecimiento.
- Wit JM, Clayton PE, Rogol AD, Savage MO, Saenger PH, Cohen P. Idiopathic short stature: Definition, epidemiology, and diagnostic evaluation. Growth Horm IGF Res. 2008; 18: 89-110.
- Wit JM, Reiter EO, Ross JL, et al. Idiopathic short stature: Management and growth hormone treatment. Growth Horm IGF Res. 2008; 18: 111-35.
Estos dos trabajos recogen las reflexiones de expertos de reconocido prestigio internacional, norteamericanos y europeos, reunidos en el año 2007, en Santa Mónica (California), para analizar los diferentes aspectos (conceptuales, diagnósticos y terapéuticos) de la talla baja idiopática (TBI), o lo que es lo mismo, del 80% de los hipocrecimientos. Fueron la base del consenso internacional sobre el diagnóstico y tratamiento de la TBI publicado posteriormente, en el año 2008 (referencia 15 de la Bibliografía). Son dos amplias revisiones que analizan en profundidad todos los aspectos de la TBI y, por ende, de la talla baja en general. Su lectura detenida es muy recomendable para todos aquellos que quieran profundizar en este tema tan controvertido.
- Palacios-Verdú MG, Pérez-Jurado LA. Nuevas metodologías en el estudio de enfermedades genéticas y sus indicaciones. Pediatr Integral. 2014; XVIII: 515-28.
Si algo está cambiando y a pasos muy rápidos en el estudio de los hipocrecimientos, cuadros dismórficos y, en general, en toda la Pediatría, es el desarrollo de nuevas tecnologías capaces de estudiar casi toda la información genética de un individuo. Este trabajo, recientemente publicado en Pediatría Integral describe con sencillez, poco habitual, la batería de recursos tecnológicos disponibles, así como sus indicaciones, limitaciones y utilidad en el estudio de las enfermedades con base genética.
| Caso clínico |
|
Motivo de consulta Varón de 10 años y 2 meses que consulta por talla baja desde siempre. Refiere la madre que ya nació pequeño y que no se ha recuperado. Los datos de su pediatra muestran que ha mantenido un canal de crecimiento paralelo, pero por debajo del P3, alejado del canal de crecimiento que correspondería a su talla diana (170,5 ± 5 cm; P10-25). Antecedentes familiares Madre: hipertensión arterial. G.A.V.: 1/0/1. Menarquia: 13 años. Talla: 153,9 cm. Padre: estenosis mitral por fiebre reumática. Talla: 173 cm. Desconoce la edad de desarrollo puberal. No consanguinidad ni otros antecedentes familiares de interés. Antecedentes personales Embarazo con hipertensión arterial. Parto a las 37 + 3 semanas de gestación (EG), cesárea por fracaso de la inducción. Periodo neonatal: Apgar 7/8. PRN: 2.335 g (-1,59 SDS para EG). LRN: 43 cm (-3,3 SDS para EG). Estuvo en incubadora por hipoglucemia e irritabilidad neuromuscular con ictericia que precisó fototerapia. Pruebas metabólicas normales. Alimentación: sin alergias ni intolerancias alimenticias. Desarrollo psicomotor: normal, aunque refieren mal rendimiento escolar que ha requerido adaptación curricular y apoyos en el colegio. Ha tenido problemas psicológicos en relación, al parecer, con separación de los padres y fallecimiento de familiar próximo, por lo que se encuentra en seguimiento por un psicopedagogo. Enfermedades anteriores: estenosis pulmonar severa (valvuloplastia a los 5 años) y comunicación auricular de tipo ostium secundum (precisó también cierre quirúrgico). Criptorquidia bilateral con respuesta positiva a gonadotropina coriónica. Resto de antecedentes personales: sin interés. Exploración Edad: 10 años y 2 meses. Talla 122,7 (P < 3/-2,63 SDS). Peso 27,1 kg (P10-25). T/A: 112/66 mm Hg. FC: 78 lpm. Buen estado general. Hábito armónico con algún rasgo fenotípico peculiar con: implantación posterior del cabello baja, hendiduras palpebrales antimongoloides, mentón prominente y nevus múltiples. Sin adenias significativas. Cuello: normal, no se palpa bocio. Tórax normal con pectus excavatum leve. Auscultación cardiopulmonar: normal. Abdomen: normal. Genitales externos masculinos normales, en estadio puberal I de Tanner (G1, P1, Aa), con testes en bolsas de 2 mL de volumen. Resto de la exploración normal. Pruebas complementarias iniciales • Hemograma, ferritina, VSG, lipidograma y bioquímica básica: normales. • IgA y Ac antitransglutaminasa: negativos. • Función tiroidea (T4L y TSH): normal. • IGF-1: 162 ng/mL (VN: 81-430); IGFBP3: 3,67 mg/L (VN: 2,47-5,92). • Edad ósea: 10 años para una edad cronológica de 10 años y 10 meses con predicción de talla final de 160 ± 5 cm. • Cariotipo: normal (46, XY). |


