|
| Temas de FC |
A. Rodríguez Sánchez*, M. Sanz Fernández*, M. Echeverría Fernández**
*Endocrinología Pediátrica Servicio Pediatría. Hospital General Universitario Gregorio Marañón de Madrid. **Servicio de Pediatría. Hospital Universitario Rey Juan Carlos I Móstoles, Madrid
| Resumen
La hiperplasia suprarrenal congénita (HSC) es una alteración hereditaria debida a un fallo en la esteroidogénesis suprarrenal. Se debe, hasta en el 90-95% de los casos, al déficit de 21-hidroxilasa (21OHD). |
| Abstract
Congenital adrenal hyperplasia (CAH) is a family of inherited disorders of adrenal steroidogenesis. Over 90-95% of CAH cases are due to 21-hydroxylase deficiency (21OHD). |
Palabras clave: Hiperplasia suprarrenal congénita; Pérdida salina; Genitales ambiguos
Key words: Congenital adrenal hyperplasia; Salt wasting; Ambiguous genitalia
Pediatr Integral 2015; XIX (7): 488-497
Hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa
Introducción
La hiperplasia suprarrenal congénita (HSC) es una enfermedad hereditaria, autosómica recesiva, que implica un fallo en la esteroidogénesis suprarrenal. Se debe, hasta en el 90-95% de los casos, al déficit de 21-hidroxilasa (21OHD, OMIM#201910).
El déficit de 21-hidroxilasa da lugar a una disminución de la síntesis de las hormonas situadas por debajo del bloqueo, los glucocorticoides y mineralocorticoides, un aumento de los productos previos a dicho bloqueo, como 17-hidroxiprogesterona (17OHP) y un aumento de síntesis de la vía metabólica no afectada, la de los andrógenos.
El déficit de cortisol produce un aumento de ACTH compensatorio y, con ello, una hiperplasia de la glándula. Probablemente, existen otras vías metabólicas alternativas que en presencia de elevadas concentraciones de 17OHP, favorecen la síntesis de andrógenos con gran potencia biológica, como dihidrotestosterona (DHT)(1,2).
Epidemiologia
En nuestro medio, la frecuencia de formas clásicas neonatales es 1:10.000-1:14.000, lo que implica una frecuencia de portadores de mutación grave de 1:50-1:60 en población general; de ahí, la importancia de realizar un adecuado consejo genético(3). Es más prevalente en algunos grupos étnicos.
Formas clínicas
La 21–hidroxilación suprarrenal es catabolizada por el citocromo P450c21, enzima que convierte progesterona en deoxicorticosterona y 17–hidroxiprogesterona en 11–deoxicortisol, lo que culmina en la síntesis de aldosterona y cortisol, respectivamente. La severidad del compromiso de la función del citocromo P450c21 es la que determina la clínica, que se manifiesta en grados variables de déficit de cortisol y aldosterona asociados a un exceso de andrógenos (androstendiona, dehidroepiandrosterona [Fig. 1])(1).
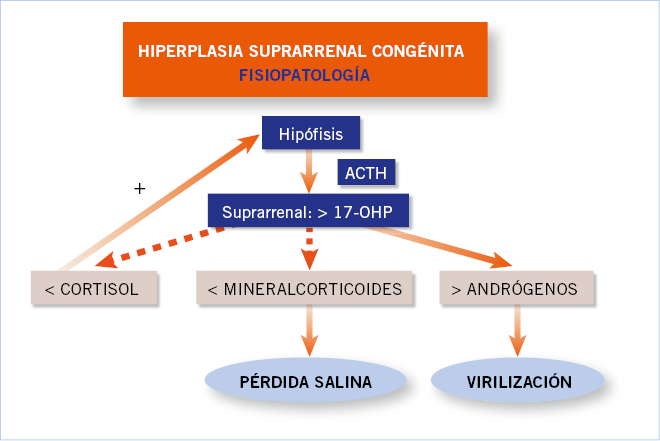
Figura. 1. Hiperplasia suprarrenal congénita.
Los pacientes con déficit grave de 21-hidroxilasa presentan no solo una falta de gluco y mineralocorticoides, sino también, una disminución en la síntesis de catecolaminas, en especial de adrenalina. Es debido a la íntima relación anatómica, vascular y bioquímica que existe entre la corteza y la médula adrenal(4).
Existen tres formas clínicas de presentación:
1. La forma clásica con pérdida salina, con una actividad enzimática del 0-1%.
2. La forma clásica sin pérdida salina, con una actividad enzimática del 1-2%.
3. La forma tardía, con una actividad enzimática hasta del 50%.
Además, algunos sujetos pueden presentar formas crípticas o ser portadores asintomáticos de la enfermedad.
Forma clásica con pérdida salina
Es la forma más grave de la enfermedad. El 75% de los casos de la forma clásica pueden presentar pérdida salina, como consecuencia de la deficiencia en la síntesis de mineralocorticoides.
Clínicamente, se caracteriza por un cuadro clínico, iniciado en los primeros días-semanas de vida, progresivo, con: anorexia, ausencia de ganancia ponderal, astenia, poliuria y vómitos. Puede evolucionar en poco tiempo a un cuadro severo de deshidratación hipotónica y shock hipovolémico de consecuencias letales. Cursa con: acidosis metabólica hiponatrémica e hiperpotasémica, natriuresis elevada, disminución de aldosterona, elevada actividad de renina plasmática (ARP) y cociente ARP/aldosterona elevado(5).
El exceso de secreción suprarrenal de andrógenos no afecta a la diferenciación de los genitales externos en el varón. Sin embargo, el hiperandrogenismo en las niñas produce una virilización de los genitales externos que lleva a la aparición de genitales ambiguos. En las mujeres afectas, cuando la suprarrenal fetal comienza a producir andrógenos en cantidades elevadas, el seno urogenital se encuentra en proceso de septación y los niveles aumentados de andrógenos pueden impedir la formación de vagina y uretra como estructuras separadas e independientes. Posteriormente, los andrógenos actuarán sobre sus receptores induciendo hipertrofia de clítoris, fusión de los labios mayores y migración rostral del orificio uretral-vaginal. El máximo grado de virilización dará lugar a un fenotipo masculino con hipertrofia del clítoris, hipospadias perineal y labios mayores escrotalizados, con ausencia de testes (Figs. 2a y b).
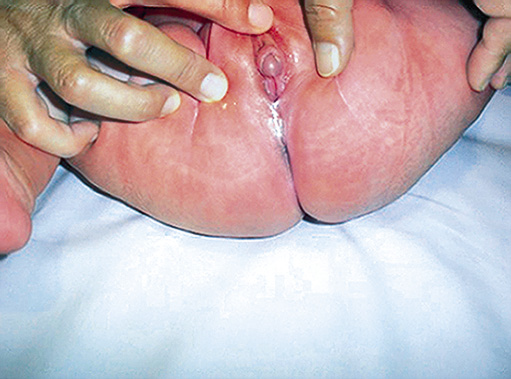
Figura 2 2a: Obsérvese la hipertrofia de clítoris y fusión de labios menores. (Estadio de Prader II).

Figura 2. 2b: Obsérvese el aspecto absolutamente virilizado que puede llevar a una signación errónea de sexo masculino (Estadio de Prader V).
Es muy útil para clasificar los diferentes grados de virilización genital, utilizar los estadios definidos por Prader (Fig. 3).
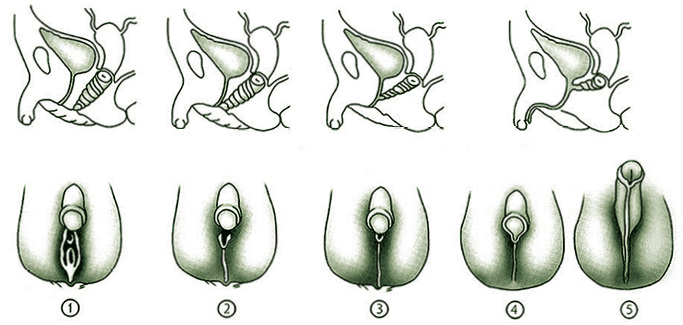
Figura 3. Estadios de Prader: 1: Hipertrofia de clítoris. Vulva pequeña. 2: Clítoris muy hipertrofiado. Seno urogenital. 3: Importante hipertrofia de clítoris, fusión de labios mayores y seno urogenital único. 4: Importante hipertrofia de clítoris con hipospadias perineal, fusión de labios mayores con apariencia escrotal. 5: Aspecto externo de genitales masculinos normales, ausencia de testículos en las bolsas.
Estas niñas muy virilizadas pueden ser erróneamente identificadas como varones con criptorquidia. Las estructuras derivadas del conducto de Wolf requieren concentraciones locales mucho más altas de testosterona que los genitales externos para lograr su diferenciación completa. Las estructuras mullerianas se desarrollan con normalidad, por lo que el desarrollo del útero, trompas y los 2/3 internos de la vagina son normales(1), 21OHD es la causa más frecuente de alteración de la diferenciación sexual en el recién nacido con cariotipo 46XX.
Forma clásica sin pérdida salina o virilizante simple
Esta forma clínica se presenta en el 25% de los casos de la forma clásica, y se caracteriza por un déficit en la síntesis de cortisol y un exceso en la producción de andrógenos suprarrenales desde la época fetal.
A diferencia de la forma con pérdida salina, la síntesis de aldosterona no está tan gravemente alterada, por lo que se mantiene la homeostasis del sodio. En algunos casos, los niveles de renina pueden estar elevados, debido a una depleción crónica de sodio. La diferencia entre esta forma clínica y la forma con pérdida salina no es nítida y existen distintos grados intermedios(6).
Estos pacientes presentan una virilización de grado variable, pero sin signos clínicos de pérdida salina. Las niñas son identificadas precozmente por la virilización de los genitales externos, pero las niñas con una virilización leve y los niños, suelen diagnosticarse más tardíamente, en la infancia, cuando se ponen de manifiesto los signos de hiperandrogenismo.
En la etapa postnatal, el exceso de andrógenos continúa virilizando los genitales y determina la aparición de una pseudopubertad precoz. Los signos de hiperandrogenismo incluyen: pubarquia, axilarquia, aumento del olor corporal, acné severo, crecimiento exagerado del pene, hipertrofia de clítoris, aceleración de la velocidad de crecimiento y, más aún, de la maduración ósea, con resultado de talla adulta baja. En ocasiones, si se activa el eje hipotálamo-hipófiso-gonadal, puede añadirse un cuadro de pubertad precoz central. Un mal control de la enfermedad puede dar lugar en las niñas a: acné, hirsutismo y disfunción ovárica(5).
Forma no clásica, parcial o tardía
Es una deficiencia enzimática parcial, con actividad de 21OH suficiente para la síntesis de mineralocorticoides y cortisol, que se acompaña de una hiperproducción de andrógenos.
Clínicamente, se manifiesta por un cuadro de hiperandrogenismo que puede hacerse evidente durante la infancia o la adolescencia o incluso comenzar en la edad adulta. Habitualmente, se produce en la segunda infancia o en edades peri o postpuberales. Generalmente, estos síntomas de hiperandrogenismo son poco marcados y coincidentes con el inicio de la adrenarquia. Pueden acompañarse de acné, oligomenorrea, alopecia de distribución masculina e incluso obesidad, intolerancia a los hidratos de carbono e hiperinsulinismo(7). No existe síndrome de pérdida salina ni virilización prenatal.
Forma críptica
Algunos pacientes, tanto varones como mujeres, pueden no manifestar síntomas de la enfermedad, aunque presenten alteraciones bioquímicas y genético-moleculares comparables a los que tienen síntomas. Generalmente, se detectan al realizar estudios de familiares afectos o en programas de cribado neonatal. El seguimiento de estos casos a menudo muestra que los signos de hiperandrogenismo aparecen posteriormente.
Portadores
Son aquellos pacientes con mutación en un solo alelo, detectados generalmente en el estudio de familiares afectos, en programas de detección neonatal o por signos de hiperandrogenismo. Se trata de personas sanas que no requieren ningún tratamiento específico.
Diagnóstico
Se sospechará 21OHD en los siguientes casos:
• Cualquier niño/a con clínica de pérdida salina en las primeras semanas de vida.
• Niñas virilizadas al nacimiento o inicio de virilización en la etapa postnatal, pubertad precoz o adrenarquia.
• Niños con inicio de virilización en la infancia.
El diagnóstico de 21OHD se basa en el análisis del esteroide previo al bloqueo enzimático: 17–hidroxiprogesterona (17OHP)(8).
Diagnóstico clínico
Diagnóstico en el recién nacido (Algoritmo 1)
El diagnóstico del déficit de 21OHD es el primero que se plantea ante un recién nacido con genitales ambiguos.
Sin embargo, un varón con hiperplasia suprarrenal en los primeros días de vida es indistinguible de un niño sano. La crisis de pérdida salina aparece a partir del 5º-10º día, cuando el niño está en su domicilio, y la clínica de astenia y vómitos, en ocasiones, es atribuida a un cuadro viral, estenosis hipertrófica de píloro o sepsis clínica. La importancia de este diagnóstico se refleja en la mayor prevalencia de mujeres en todas las series, lo que indica que fallecen varones sin diagnosticar.
En el periodo neonatal, en el recién nacido a término, se consideran normales valores de 17OHP <35 ng/ml en suero(9). Por encima de esta cifra, puede sospecharse 21OHD. Se deben excluir aquellos recién nacidos con valores transitoriamente elevados de 17OHP (bajo peso al nacimento, prematuridad, enfermedad grave en el periodo neonatal), que suelen normalizarse antes del año de edad(10).
Diagnóstico en el niño mayor/adolescente (Algoritmo 2)
Excluyendo el periodo neonatal, se consideran normales los valores basales inferiores a 2-3 ng/ml. Los niveles basales de 17OHP pueden no diferir de los valores normales, pero suelen estar elevados durante el pico diurno de producción de cortisol, por lo que los valores de la primera hora de la mañana suelen ser los más informativos. El test de estimulación con ACTH se recomienda realizarlo en el periodo prepuberal, ante la presencia de pubarquia prematura con aceleración del crecimiento y de la edad ósea, cuando la 17OHP basal es superior a 1 ng/ml, y en la adolescencia o periodo postpuberal, ante una 17OHP superior a 1,7-2 ng/ml en fase folicular. Se aconseja realizarlo también a los padres y familiares de cualquier paciente afecto de forma clásica o no clásica para detectar formas crípticas.
Programa de detección precoz: cribado neonatal
La detección precoz de 21OHD está recomendada internacionalmente con un nivel de evidencia 1/++(5). Sin embargo, en España solo en algunas comunidades autónomas se realiza cribado de esta enfermedad. En la Comunidad Autónoma de Madrid, el programa de detección precoz se inició en 1990(9).
El programa de detección precoz neonatal de 21OHD tiene los siguientes objetivos:
1. Anticiparse a la aparición de una crisis de pérdida salina grave y potencialmente mortal.
2. Evitar la incorrecta asignación de sexo en una niña con genitales externos virilizados.
3. Diagnosticar precozmente las formas virilizantes simples para evitar la hiperandrogenización durante la infancia.
4. La detección precoz de las formas no clásicas no es el objetivo de la detección precoz; pero, en ocasiones, pueden beneficiarse de este programa.
Se basa en la determinación de 17OHP, en una muestra de sangre capilar al 2º día de vida, simultánea con la detección precoz de hipotiroidismo y de otras enfermedades (“Prueba del talón”). Para analizar 17OHP, se debe tener en cuenta: sexo, edad gestacional y peso al nacimiento. En el prematuro, sobre todo si es menor de 30 semanas de edad gestacional, los valores de 17OHP pueden elevarse sin que presenten 21OHD. La guía de actuación ante un recién nacido positivo en el cribado neonatal se muestra en el algoritmo 3(9).
Diagnóstico genético molecular
El estudio molecular confirma la sospecha clínica y bioquímica. Este estudio se debe extender a los padres y hermanos, ya que pueden beneficiarse de consejo genético.
Es una enfermedad de carácter hereditario autosómico recesivo, debida a un déficit en la actividad de la enzima esteroide 21-hidroxilasa que es causado por mutaciones presentes en el gen que la codifica: CYP21A2 (antes denominado CYP21B)(11).
El gen CYP21A2 se localiza en el complejo mayor de histocompatibilidad HLA, en el brazo corto del cromosoma 6 (6p21.3), junto al gen del factor 4 del complemento.
La base molecular de la 21OHD reside fundamentalmente en un grupo de mutaciones recurrentes de las que se conoce el efecto funcional y repercusión clínica. Su caracterización puede apoyarse en un cribado básico de un grupo de mutaciones puntuales frecuentes que ha de ser completado, no solo por la secuenciación completa de los alelos no caracterizados, sino también por el análisis de deleciones, conversiones y duplicaciones del gen para una caracterización adecuada de los alelos deficientes.
Las gravedad de la presentación clínica se correlaciona muy directamente con la severidad de las mutaciones y es muy fuerte, aunque no total, la correlación genotipo/fenotipo, tanto a nivel clínico como bioquímico(5,12) (Tabla I).

Debido a la prevalencia de alelos 21OHD en población general, los afectos de la deficiencia son muy frecuentemente heterozigotos compuestos (mutaciones distintas en los dos alelos) y la homozigosis para las mutaciones más frecuentes puede presentarse en ausencia de consanguineidad. Sin embargo, la homozigosis para las mutaciones raras es generalmente debida a consanguinidad, a veces, ancestral y no conocida(12).
Tratamiento
El tratamiento de esta entidad dependerá del grado de afectación enzimática y de las manifestaciones clínicas de la enfermedad. El objetivo terapéutico es reemplazar la secreción fisiológica de los glucocorticoides y mineralocorticoides para evitar la pérdida salina, la corrección quirúrgica de los genitales externos en las niñas afectas, controlar los signos de hiperandrogenismo y mejorar las consecuencias que esta enfermedad puede originar en la vida adulta.
Ello requiere un seguimiento individualizado y un abordaje multidisciplinar, en el que es necesaria la implantación de un programa bien estructurado de intervención y seguimiento(13).
Como ocurre en otras patologías poco frecuentes y complejas, estos pacientes deben ser atendidos en Centros Clínicos de Referencia, para evitar que reciban una atención fragmentada o inadecuada. Sin embargo, en muchas ocasiones, esto no es posible(14).
El objetivo del tratamiento de 21OHD es diferente según la edad del paciente. En los niños, el objetivo primordial es la sustitución gluco y mineralocorticoide para evitar las crisis de pérdida salina y, además, disminuir la secreción suprarrenal de andrógenos para evitar los síntomas de virilización y obtener un crecimiento normal. El debut de 21OHD en el recién nacido debe considerarse una crisis de insuficiencia suprarrenal aguda y tratarse como tal(13).
Tratamiento con glucocorticoides
La hidrocortisona es el fármaco de elección en los niños, debido a su potencia biológica superponible a la del cortisol endógeno y a que su vida media es corta. Otros glucocorticoides, como la prednisolona o la dexametasona, tienen mayor repercusión sobre el crecimiento y otros sistemas y no se deben administrar en la infancia. Es preferible la utilización de hidrocortisona en comprimidos y no en suspensión, ya que la distribución del fármaco en el líquido es más irregular e inestable que en comprimidos(5).
Es preciso individualizar la dosis necesaria para cada paciente, ya que está influenciada por múltiples factores. El objetivo es tratar con la mínima dosis eficaz que permita un equilibrio entre el crecimiento y desarrollo puberal normal, con una supresión adecuada de los andrógenos suprarrenales. La infradosificación podría dar lugar a crisis de pérdida salina y aumento de la síntesis de hormonas sexuales de origen adrenal, virilización con cierre prematuro de epífisis y talla baja en el adulto, además, puede asociarse una pubertad precoz dependiente de gonadotropinas. El tratamiento excesivo con glucocorticoides da lugar a un síndrome de Cushing, con enlentecimiento del crecimiento, adiposidad central y supresión de la síntesis de hormonas sexuales de origen central.
Fuera del periodo neonatal y el primer año de edad, la dosis diaria de hidrocortisona recomendada es 10-20 mg/m2/día dividido en 3 dosis equivalentes(5,15).
El tratamiento debe ser cuidadosamente monitorizado. La variable clínica más importante es mantener una adecuada velocidad de crecimiento con una normalidad en el peso y tensión arterial. Entre los parámetros bioquímicos, 17OHP, androstendiona y testosterona son los mejores indicadores de un adecuado tratamiento glucocorticoideo en pacientes prepuberales. Los cambios de tratamiento deberán realizarse en el contexto clínico de cada paciente y no solo basándose en los parámetros analíticos.
Las indicaciones para iniciar tratamiento en un paciente con forma no clásica, incluyen: pacientes con síntomas importantes de hiperandrogenismo con una repercusión negativa sobre el crecimiento, maduración ósea o la función gonadal. En adolescentes, puede iniciarse tratamiento si existen síntomas como: hirsutismo, oligoamenorrea y acné severo. El tratamiento se realiza con hidrocortisona a dosis de 8-10 mg/m2/día. Si se ha finalizado el crecimiento, se puede utilizar prednisona a dosis de 5 mg/día, pudiendo asociarse un anovulatorio con acción antiandrogénica, aunque para el control efectivo del hirsutismo se requieren tratamientos prolongados(7).
Tratamiento con mineralocorticoides
El mineralcorticoide a emplear es la 9a-fluorhidrocortisona oral. Los lactantes necesitan dosis mayores en los primeros meses de vida, generalmente 0,1-0,15 mg/día, mientras que los lactantes mayores y los niños se mantienen habitualmente con 0,05-0,1 mg/día. Para favorecer el efecto mineralcorticoide, se administran suplementos de cloruro sódico oral (4 mEq/kg/día) hasta que inicien la alimentación complementaria.
El control de la idoneidad del tratamiento mineralcorticoide lo da la ausencia de síntomas, tensión arterial y frecuencia cardiaca normales, normalidad electrolítica y del nivel de actividad de renina plasmática. El mantenimiento del balance de sodio reduce la vasopresina y la ACTH, contribuyendo a disminuir la dosis de glucocorticoides(16).
Tratamiento en las situaciones de estrés
Los pacientes con las formas graves de 21OHD no pueden producir suficiente cortisol en respuesta a una situación de estrés, por lo que deberemos aumentar la dosis de glucocorticoides entre 2 y 10 veces la dosis de mantenimiento en función del grado de estrés. De no hacerlo, se puede desencadenar una crisis de insuficiencia suprarrenal(5,15).
Ante una situación de estrés leve, como infecciones intercurrentes, se recomienda duplicar la dosis de hidrocortisona durante los días que dure el proceso. Es frecuente que en estas situaciones, sobre todo los lactantes y preescolares, vomiten, por lo que es aconsejable administrar hidrocortisona i.m., lo que asegura una cobertura esteroidea de 6-8 horas. La dosis aproximada es, en menores de 2 años, de 25 mg i.m.; de 2-12 años, de 50 mg i.m.; y a partir de 12 años y adultos, 100 mg i.m.
En casos de estrés intenso, como: cirugía, infecciones graves o procesos gastrointestinales que impiden la administración/absorción de la medicación oral, será preciso un tratamiento más específico, que se anteponga/trate la crisis suprarrenal:
• De sostén: corregir, si existe, el estado de hipovolemia, deshidratación, hiponatremia, hiperpotasemia, acidosis metabólica e hipoglucemia.
• Etiológico: hidrocortisona i.v. (100 mg/m2/día) por vía i.v.
No es necesario aumentar la dosis de hidrocortisona en caso de estrés emocional, previo a la realización de ejercicio físico o en pacientes con la forma no clásica, a menos que su función adrenal sea subóptima o esté yatrogénicamente suprimida.
Tratamiento quirúrgico
Se realizará corrección quirúrgica de las malformaciones de los genitales externos de la niña afecta hacia el sexo femenino. Siempre deberá ser valorada en el periodo neonatal por un cirujano pediátrico especialista y diseñar la secuencia de intervenciones, que deberán realizarse en centros con experiencia en este tipo de patologías(17,18).
Se recomienda que se realice si es posible en un único tiempo quirúrgico y precozmente, antes de los 12-15 meses de edad, para que la niña pueda establecer un esquema corporal adecuado.
Los resultados de la cirugía deberán ser reevaluados en la adolescencia y edad adulta para asegurar la morfología normal de los genitales y poder tratar las posibles complicaciones postoperatorias, como fístulas uretro-vaginales y estenosis vaginales.
Complicaciones a largo plazo
Talla baja
La mayoría de los pacientes con déficit de 21ODH alcanzan una talla adulta menor que su talla diana. Varios factores contribuyen a ello: los patrones de secreción de hormona de crecimiento, la edad al diagnóstico e inicio de tratamiento, el control hormonal y la dosis de glucocorticoides utilizada, aunque parece que el curso natural de la enfermedad con hiperproducción de andrógenos y dosis suprafisiológicas de glucocorticoides son los factores que más afectan a la talla adulta(5).
Los pacientes con 21OHD pueden asociar en su evolución pubertad precoz central. En estos casos, el tratamiento es mantener el tratamiento hormonal sustitutivo y asociar análogos inhibidores de GnRH(5,13).
Restos ectópicos adrenales
El aumento de ACTH puede producir una hiperplasia de los tejidos sensibles a esta, como las glándulas suprarrenales y otras zonas en las que pueden hallarse restos de células adrenales, como en los testículos y ovarios. No es un tumor maligno. Su importancia radica en la producción de infertilidad y disfunción gonadal. El diagnóstico se basa en la realización de ecografías periódicas.
El tratamiento requiere intensificar el tratamiento con glucocorticoides y, en ocasiones, tratamiento quirúrgico.
Fertilidad
Las mujeres pueden presentar disminución de fertilidad, debido a las alteraciones hormonales que derivan de un control inadecuado de la enfermedad. Pueden presentar dispareunia por estenosis del introito vaginal y alteraciones anatómicas derivadas de la cirugía. La androgenización cerebral durante el periodo fetal podría tener consecuencias en la conducta sexual en etapas posteriores.
Durante el embarazo, la paciente con forma clásica de 21OHD debe continuar el tratamiento previo de gluco y mineralcorticoides. Durante el parto, se debe administrar hidrocortisona parenteral y considerarlo una situación de estrés severo.
Es fundamental realizar consejo genético previo a gestación.
Los varones con formas clásicas también pueden presentar fertilidad reducida por hipogonadismo hipogonadotropo asociado, aumento de andrógenos de origen suprarrenal que suprimen la FSH, disminuyendo la producción de espermatozoides, o por restos ectópicos adrenales testiculares.
Alteraciones metabólicas
El tratamiento crónico con corticoesteroides es un factor de riesgo para la osteoporosis. Se deberá realizar profilaxis en pacientes jóvenes con 21OHF con ejercicio físico y suplementos de calcio y vitamina D.
La obesidad, hipertensión arterial, resistencia insulínica e intolerancia a los hidratos de carbono son efectos secundarios a prevenir y tratar si es preciso(7).
Diagnóstico y tratamiento prenatal
El diagnóstico prenatal se plantea en el feto que presenta riesgo de padecer la enfermedad severa: ambos progenitores portadores de mutaciones severas, uno de los progenitores presenta la enfermedad o ya ha habido un miembro diagnosticado en la familia. Es necesario realizar el estudio completo, bioquímico y genético molecular antes de programar un embarazo en las familias con caso índice afecto.
El objetivo es prevenir la virilización de los genitales en el feto femenino afectado de 21OHD. Está basado en la administración precoz, de dexametasona a la madre. La dexametasona atraviesa la barrera placentaria y es capaz de disminuir la hiperproducción de andrógenos de la glándula suprarrenal del feto. Es un tratamiento eficaz para evitar la virilización de los genitales del feto femenino, si se realiza a dosis adecuadas y antes de la semana 8, cuando empieza la diferenciación genital(19).
Sin embargo, obliga a exponer innecesariamente a un tratamiento con dexametasona a 7 de cada 8 fetos, (3 de 4 niñas y 4 de 4 varones).
Actualmente, se debe considerar un tratamiento experimental, ya que se desconocen los efectos a largo plazo que la administración de corticoides en época prenatal puede tener sobre el recién nacido y por los potenciales efectos secundarios de la administración de dexametasona sobre la mujer gestante.
Se debe realizar en centros con experiencia, después de informar detalladamente de la relación riesgo/beneficio que entraña(5,19,20).
El diagnóstico preimplantacional no se encuentra todavía muy extendido y no está disponible en los hospitales públicos de nuestro medio. Debe tenerse en consideración, que se trata de un locus complejo con un pseudogén, en el que preexisten las mutaciones, lo que dificulta su estudio genético-molecular.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.** Rodríguez Sánchez A, Rodríguez Arnao J, Dabon Westphal P, Míguez Navarro C, Rodríguez Arnao MD. Hiperplasia suprarrenal congénita por defecto de la 21-hidroxilasa. Acta Pediátrica Española. 2001; 59: 497-510.
Visión general con énfasis en los aspectos clínicos de 21OHD.
2.* WL Auchus RJ, Miller: “Congenital adrenal hiperplasia- More dogma bites the dust”. J Clin Endocrinol Metab. 2012; 97(3): 772-5.
Resume de una forma sencilla, el complejo mecanismo de la vía alternativa o “puerta de atrás” de la síntesis de andrógenos en situaciones patológicas como 21OHD.
3.** Ezquieta B, Ruano MF, Dulin E, Arnao DR, Rodríguez A. (Prevalence of frequent recessive diseases in the Spanish population through DNA analyses on samples from the neonatal screening). Med Clin. 2005; 125: 493-5.
Con amplia experiencia en el estudio genético molecular de 21OHD, los autores describen la frecuencia de la enfermedad en nuestro medio.
4.* Kim MS R-LA, Bali B, Lane CJ, Park AH, Hall S. Decreased adrenomedullary function in infants with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2014; 99: E1597-601.
En este artículo, se describe la íntima relación entre la síntesis de glucocorticoides y adrenalina en la glándula suprarrenal y cómo en las formas graves de 21OHD, esta síntesis está disminuida.
5.*** Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010; 95: 4133-60.
Guía internacional de consenso sobre la fisiopatología, clínica, diagnóstico y tratamiento de 21OHD.
6.** Nimkarn S, Lin-Su K, Berglind N, Wilson RC, New MI. Aldosterone to renin ratio as a marker for disease severity in 21-hydroxilase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007; 92: 137-42.
Evaluación de la diferente y progresiva afectación de la síntesis de mineralcorticoides en 21OHD, según la gravedad de la enfermedad.
7.** Witchel SF. Non –classic congenital adrenal hyperplasia. Steroids. 2013; 78: 747-50.
Visión general de la clínica, diagnóstico y tratamiento de las formas no clásicas o de inicio tardío de 21OHD.
8.* Rodríguez Arnao MD, Sanz Fernández M, Rodríguez Sánchez A: “Hiperplasia adrenal congénita”. Manual del residente de Endocrinología y Nutrición. SEEN, 2015; capítulo 7. www.seen.es.
Resumen muy práctico del diagnóstico y tratamiento de 21OHD.
9.** Rodríguez Arnao MD, Rodríguez Sánchez A, Dulín Iñiguez: “Detección precoz de alteraciones endocrinas”. Rev Esp Endocrinol Pediatr. 2013; (Suppl): 59-71.
Guía de actuación en 21OHD diagnosticada mediante cribado neonatal en España.
10.** Huidobro Fernández B. Echeverría Fernández B, Dulín Íñiguez E, Roldán Martín B, Rodríguez Arnao MD, Rodríguez Sánchez A. Neonatal screening for congenital adrenal hyperplasia: Transitory elevation of 17-hydroxyprogesterona. J Pediatric Endocrinol Metabol. 2011; 24(3-4): 155-62.
Estudio sobre los recién nacidos que pueden presentar cifras elevadas de 17OHP en el período neonatal, pero que no se corresponden con pacientes con 21OHD. Muy útil para evitar el diagnóstico erróneo de 21OHD en estos niños, que normalizan sin ningún tratamiento este dato analítico.
11.** Ezquieta B, Cueva E, Oyarzabal M, Oliver A, Varela JM, Jariego C. Gene conversion (655G splicing mutation) and the founder effect Gln318Stop) contribute to the most frequent severe point mutations in congenital adrenal hyperplasia (21-hydroxylase deficiency) in the Spanish population. Clin Genet. 2002; 62: 181-8.
Estudio genético molecular de 21OHD en pacientes españoles, realizado en una amplia muestra.
12.*** New MI, Abrahan M, González B, Dumic M, Razzaghy M, Chitayat D, Sun L, Zidi M, Wilson RC, Yuen T. Genotype-phenotype correlation in 1.507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. PNAD. 2013; vol. 110, no 7: 2611-6.
Estudio descriptivo que correlaciona la forma clínica de 21OHD con la gravedad de la mutación genética que presenta, realizado en un elevado número de pacientes.
13.** Speiser PW. Medical treatment of classic and nonclassic congenital adrenal hyperplasia. Adv Exp Med Biol. 2011; 707: 41-5.
Visión general y práctica de las pautas de tratamiento con gluco y mineralcorticoides en 21OHD.
14.*** Auchus RJ, Witchel SF, Leight KR, Aisenberg J, Azziz R, Bacheg T, Baker LA et als: Guidelines for the development of comprehensive care centers for congenital adrenal hyperplasia: Evidence form de CARES foundation initiative. International Journal of Pediartics Endocrinology. 2010; Article ID 275213: 1-17; doi: 10.1155/2010/275213.
Guía de consenso por un grupo internacional de expertos donde recomiendan el seguimiento de estos pacientes en centros clínicos especializados.
15.** Trapp CM, Speiser PW, Oberfield SE: “Congenital adrenal hyperplasi: an update in children”. Curr Opin Endocrinol Diabetes Obes. 2011; 18(3) 166-70.
Resumen del tratamiento de 21OHD, en especial en niños que no han finalizado su crecimiento, hace hincapié en la dificultad de la optimización individulizada del tratamiento.
16.* Padidela R, Hindmarsh PC. “Mineralocorticoid deficiency and treatment in congenital adrenal hyperplasia”. Int J Pediatr Endocrinol. 2010; 56: 656-9.
Descripción detallada del tratamiento con mineralcorticoides en pacientes con 21OHD y defecto de aldosterona.
17.** Vidal I, Gorduza, DB, Haraux E, Gay C, Chatelain P, Nicolino M. Surgical options in disorders of sex development with ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010; 24: 311-24.
Valoración de los distintos tratamientos quirúrgicos en recién nacidos con ambigüedad genital, como en niñas afectas de forma virilizante de 21OHD.
18.* Rodríguez Sánchez A, Moreno M, Rodríguez Arnao MD, Ezquieta B, Molina E, Vázquez J: Resultados de la reconstrucción genital de la niña afecta de hiperplasia suprarrenal congénita por defecto de la 21-hidroxilasa. An Pediat. 2003; 58 Suppl 2: 156-8.
Descripción de la técnica quirúrgica empleada, resultados funcionales y anatómicos en una serie de niñas con 21OHD tratadas en un centro español con amplia casuística.
19.** Miller WI, Witchel SF: Prenatal treatment of congenital adrenal hiperplasia: risks outweigh benefits. Am J Obst Gynecology. 2013; 354-59.
Valoración del tratamiento y diagnóstico prenatal de mujeres embarazadas con riesgo de presentar fetos afectos de 21OHD.
20.* A. Rodríguez, B. Ezquieta, JM Varela, M Moreno, E. Dulin, MD Rodríguez Arnao: “Diagnóstico genético molecular y tratamiento prenatal de la hiperplasia adrenal por déficit la 21-hidroxilasa”. Med Clin (Barc). 1997; 109: 669-2.
Descripción de la pauta de tratamiento y diagnóstico de la primera niña tratada prenatalmente en España afecta de 21OHD evitando la virilización genital.
| Caso clínico |
|
Motivo de ingreso: varón con dificultad en la alimentación. Varón de 45 días de vida que desde los 6 o 7 días presenta un cuadro progresivo de decaimiento, avidez por el alimento y agua, vómitos y pérdida de peso. No presenta fiebre, diarrea ni otros datos de interés. Lactancia natural exclusiva. No recibe ningún tratamiento. Antecedentes obstétricos Embarazo controlado y normal. No factores de riesgo infeccioso. Parto: eutócico a la 39 semanas. Peso: 3.460 g; Longitud: 49 cm; Apgar: 9/10, no REA. Antecedentes familiares Padres no consanguíneos. Madre 34 años. G2A1V2 sana, menarquia a los13 años. Talla: 162 cm. Padre: 39 años. Sano, Talla: 177 cm. Exploración física Peso: 3.100 g, regular estado general, deshidratación 5%. Leve hiperpigmentación de mamilas y de genitales externos. Auscultación cardíaca rítmica, sin soplos. Auscultación pulmonar: buena ventilación bilateral. No signos de dificultad respiratoria. Clavículas y paladar íntegros. Pene de 4 cm de longitud, ambos testes escrotales. Pruebas complementarias EAB: pH: 7,00; HCO3: 7 mmol/L; EB: -10 mmol/L. Bioquímica destaca: Na: 120 mEq/L; K: 8,5 mEq/L. 17-OHP: 1.204 mcg/L (N: <30). Tratamiento seguido y evolución Ante la sospecha diagnóstica de 21OHD, se inició tratamiento con fluidoterapia iv, corrección hidroelectrolítica e hidrocortisona iv hasta la estabilización clínica. Disminución progresiva de la dosis de hidrocortisona y administración vía oral asociada a fluorhidrocortisona. Confirmación diagnóstica de la enfermedad con análisis genético molecular compatible con 21OHD, con mutación grave en ambos alelos: heterocigota compuesto. En su alelo paterno, presenta una deleción/conversión que incluye la mutación del procesamiento del intron 2 (C.293-132Ao C>G) y la deleción c.332-339de; y en el aleo materno, una “conversión grande” del gen detectada por MLPA. Este genotipo da lugar a c.293-13AoC>G hemizigosis y c.332-339del.
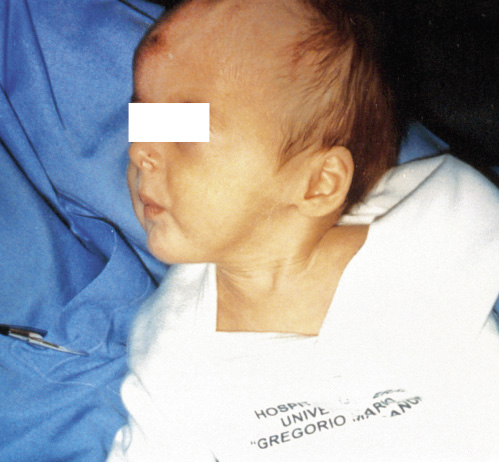
Aspecto característico del paciente descrito después de haber recibido el tratamiento inicial de la crisis de pérdida salina. Pese a esto, todavía se observa el estado de deshidratación y fallo de medro característico.
|