|
| Temas de FC |
J. Costa Sarri![]()
Pediatra y neuropediatra. Barcelona
| Resumen
Las alteraciones del perímetro craneal (PC) son uno de los muchos aspectos que el pediatra debe conocer y supervisar a lo largo del desarrollo del niño menor de dos años. Es en esta fase de la vida cuando hemos de ser más cuidadosos en la vigilancia del crecimiento y la forma del cráneo. Se abordan los conceptos de macrocefalia y microcefalia. Las alteraciones del crecimiento cerebral pueden ser de causa congénita o adquirida y pueden presentarse clínicamente aisladas o constituir parte de un síndrome congénito. El dismorfismo craneal es una anomalía que ha de detectarse desde Atención Primaria (AP). Su incidencia ha ido en aumento a causa de la implantación de la campaña para evitar el síndrome de muerte súbita del lactante (SMSL) a principios de los años 90. Este dismorfismo es causa de derivación especializada, pues, a menudo, hay dificultad para diferenciar la causa postural de la causa sinostótica de la deformidad. El tratamiento y el pronóstico de los dismorfismos craneales varían en función de la etiología y la gravedad de la deformidad. |
| Abstract
Changes in head circumference (HC) are one of the many aspects that pediatricians must be aware of and monitor throughout the development of children under two years of age. It is during this stage of life that we must be most careful in monitoring the growth and shape of the skull. The concepts of macrocephaly and microcephaly are discussed. Brain growth abnormalities can be congenital or acquired and may occur clinically in isolation or as part of a congenital syndrome. Cranial dysmorphism is an abnormality that must be detected in primary care (PC). Its incidence has been increasing due to the implementation of the campaign to prevent sudden infant death syndrome (SIDS) in the early 1990s. This dysmorphism is a cause for referral to a specialist, as it is often difficult to differentiate between the postural cause and the synostotic cause of the deformity. Treatment and prognosis of cranial dysmorphisms vary depending on the etiology and severity of the deformity. |
Palabras clave: Macrocefalia; Microcefalia; Plagiocefalia; Craniosinostosis.
Key words: Macrocephaly; Microcephaly; Plagiocephaly; Craniosynostosis.
Pediatr Integral 2025; XXIX (7): 484 – 493
OBJETIVOS
• Conocer los conceptos de macrocefalia y microcefalia para poder orientar, con valoración de la historia clínica, el plan a seguir: si control en pediatría o derivación a neuropediatría.
• Orientación inicial de la dismorfia craneal: si postural o sinostótica. Planificar el seguimiento de la misma en pediatría o derivación a neurocirugía.
• Aplicación del cefalómetro como herramienta útil en el seguimiento de los casos de dismorfia craneal.
• Hacer una buena anamnesis, exploración y derivación a neuropediatría o neurocirugía, según el caso, lo mejor orientada posible. No olvidar nunca medir el perímetro craneal a los progenitores y valorar la etnia del paciente (sobre todo en los casos de macro y microcefalia).
Alteraciones del perímetro y forma craneal
https://doi.org/10.63149/j.pedint.78
Alteraciones del perímetro craneal
Introducción
En la consulta de pediatría es tan importante la medición del perímetro craneal (PC) como la del peso y la talla. Todos los pacientes han de tener su gráfica de PC, al menos, hasta los dos años.
Desde que el pediatra inicia su labor con el recién nacido (RN) y su familia, ha de explorar siempre al paciente de una forma sistemática y no puede olvidarse de palpar el cráneo, las suturas y las fontanelas. Hay que observar la evolución del cráneo y su tamaño, elaborando una gráfica continuada del PC, al menos hasta los dos años (algunos autores lo alargan hasta los tres años; y ante un seguimiento con anomalías en los límites superior o inferior de la gráfica de crecimiento del PC, se aconseja continuarla durante toda la infancia o hasta que desaparezca la sintomatología)(1).
Modo de medir el PC: ha de utilizarse una cinta flexible, pero que no se pueda distender. Es importante buscar siempre la mayor circunferencia posible desde la frente hasta el occipucio (circunferencia fronto-occipital). Es frecuente que se puedan cometer errores de medida; por lo que, en caso de discrepancias, se aconseja medir hasta tres veces para asegurar el valor obtenido(2). Procurar siempre que mida la misma persona al mismo paciente y con la misma cinta, a ser posible.
El PC es una medida variable dentro de la población general; no existe un punto de corte universal para considerar que un PC es anormal, sino que la definición de micro y macrocefalia depende de otros factores como:
• La evolución dinámica del mismo a lo largo de la edad: no será lo mismo un paciente a +2,5 DS (desviación standard), que previamente estuviera en -1DS, pues hace más de dos saltos de DS, que otro que subiera desde +1,5 DS, que hace solo un salto de DS. A mayor salto en DS, mayor probabilidad de patología.
• El resto de las medidas antropométricas.
• La evolución del neurodesarrollo.
• Los antecedentes médicos y neurológicos del paciente.
• El PC de los progenitores, etc.
Según la fuente bibliográfica o la etnia del paciente, se utilizarán diferentes percentiles o diferentes valores de DS, para la catalogación de macro o microcefalia. En general, se pueden establecer una serie de principios a la hora de interpretar un valor de PC:
• Entre los pacientes con microcefalia, aquellos con un valor de PC entre -2DS y -3DS son los que tienen menor riesgo de presentar una alteración neurológica (sobre todo entre -2 y -2,5 DS).
• Los pacientes tienen mayor probabilidad de presentar una alteración neurológica cuanto mayor sea la desviación de la normalidad. Entre +3DS y -3DS no hay una relación directa entre el perfil cognitivo y la DS cefálica, pero a partir de estas cifras, el riesgo de dificultades de aprendizaje graves aumenta según aumenta la desviación.
• Las mediciones de los primeros meses tienen menor valor pronóstico, pues es, en el primer año de vida, cuando se va adquiriendo el valor genético de PC (valor de PC acorde con el de los padres). Siempre hay que valorarlo en su contexto global.
• Tener en cuenta que el PC es una aproximación a lo que desearíamos poder medir, que es el volumen cefálico (sobre todo en su componente encefálico). Un perímetro redondo, por ejemplo, tiene mayor volumen que uno elíptico con el mismo valor de PC(2).
• En los pacientes prematuros hemos de elaborar su gráfica de PC teniendo en cuenta su edad corregida.
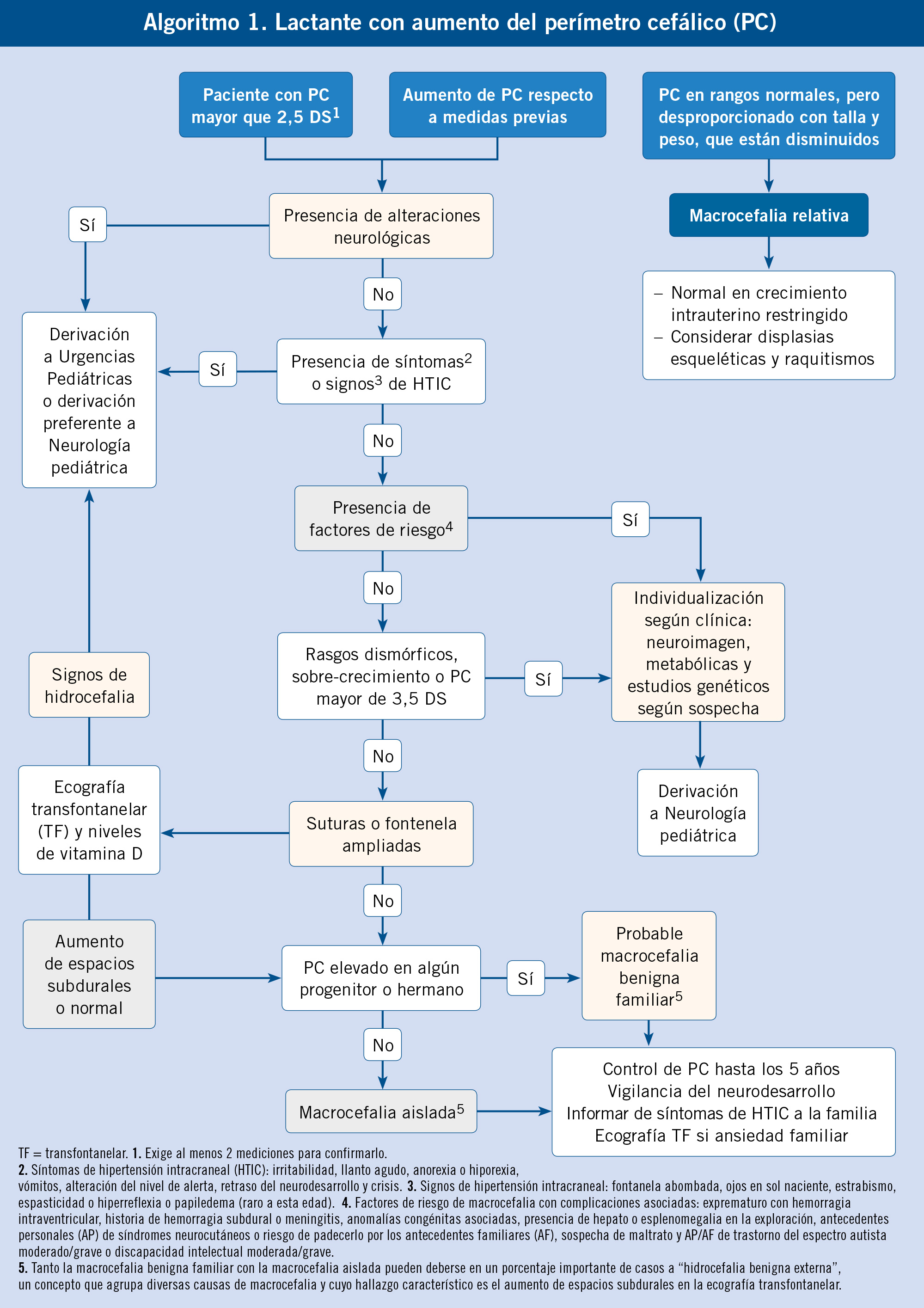
Macrocefalia (Tablas I, II y III)



Se considera macrocefalia un PC de más de 2,5 DS. A tener en cuenta, el concepto de “macrocefalia relativa”, que son aquellos pacientes que tienen un PC en rango normal, pero desproporcionado a su peso y talla, que están disminuidos.
La macrocefalia relativa puede ser normal en caso de crecimiento intrauterino restringido, pero sin este antecedente, pensar en las posibles causas de la misma (Tabla I).
Causas más frecuentes de macrocefalia
• La hidrocefalia externa benigna o derrame pericerebral, de la que se hablará más adelante.
• La hipertensión endocraneal (HTEC) o hipertensión intracraneal (HIC), que representa una emergencia más o menos importante según la velocidad y la importancia de su instalación. Hay diferentes posibles etiologías de la misma(2):
– Hemorragia (intraventricular, epidural, subdural, subaracnoidea o parenquimatosa).
– Tumor de fosa posterior (FP).
– Estenosis de acueducto de Silvio.
– Otras malformaciones de FP: síndrome de Dandy-Walker, malformación de Chiari, síndrome de Klippel-Feil y síndrome de Walker-Warburg.
– Malformación de la vena de Galeno.
– Malformación arteriovenosa.
– Absceso.
– Causa postinfecciosa.
Principales mecanismos y causas de macrocefalia(1-3)
• Por aumento del parénquima cerebral (megalencefalia): puede ser debido a una malformación primaria, con grandes circunvoluciones, con un patrón simple o con polimicrogiria. También puede ser esporádica o asociarse a diferentes patologías como: neurofibromatosis, esclerosis tuberosa, acondroplasia, gigantismo hipofisario, gigantismo cerebral, depósito anormal de productos metabólicos, como la forma infantil de idiocia amaurótica, el gargolismo, la degeneración esponjosa de la sustancia blanca y algunas leucodistrofias.
– Megalencefalia anatómica: no sindrómica: macrocefalia aislada, macrocefalia familiar benigna; y sindrómica: síndromes neurocutáneos, acondroplasia, síndrome de Sotos, síndrome X-frágil, síndrome de Proteus, alteraciones de la vía PL3K/AKT/mTor y de la vía RAS-MAPK.
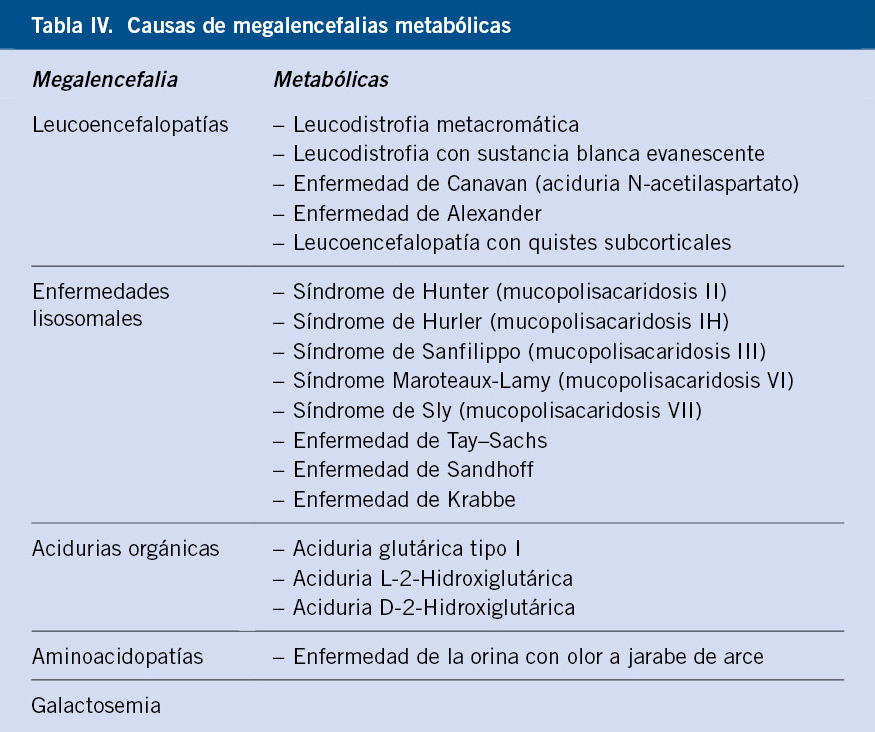
– Megalencefalia metabólica: leucoencefalopatías; enfermedades lisosomales; acidurias orgánicas; aminoacidopatías; y galactosemia.
• Por hidrocefalia:
– Hidrocefalias comunicantes: papiloma y síndrome de Arnold Chiari II.
– Hidrocefalias obstructivas: estenosis de acueducto de Silvio y lesiones con efecto masa.
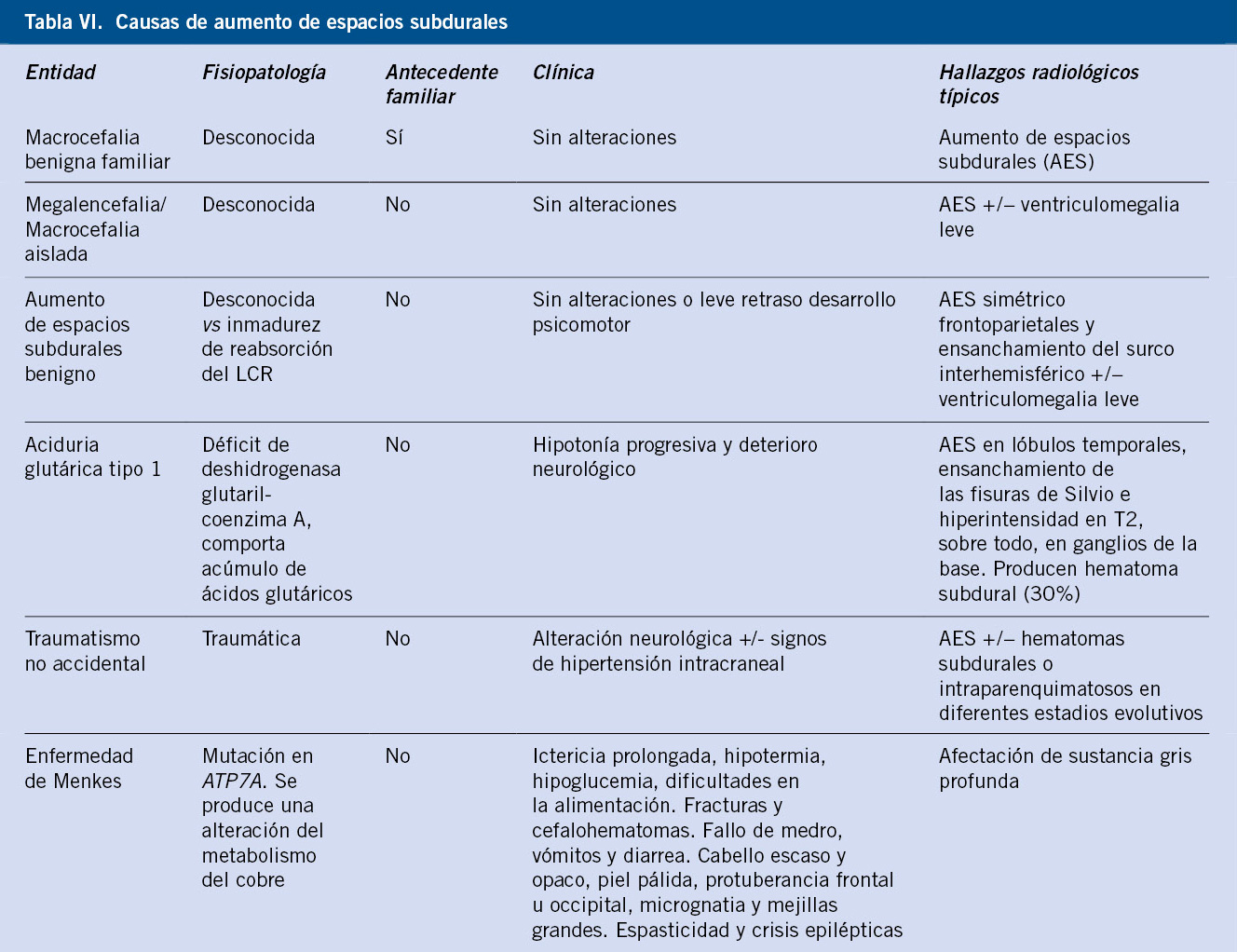
– Hidrocefalia externa benigna o derrame pericerebral: muy frecuente y, en la mayoría de las ocasiones, sin consecuencias sobre el neurodesarrollo. Se observa generalmente una historia familiar idéntica con macrocefalia benigna en uno de los padres. Esta es una situación que puede favorecer la aparición de un hematoma subdural de forma espontánea o tras un traumatismo menor, sobre todo durante los 6 primeros meses.
• Por aumento de sangre o tejido vascular: casos de hemorragia o malformación vascular.
• Por aumento del componente óseo: casos de talasemia, osteopetrosis, raquitismo, hipofosfatasia y osteogénesis imperfecta.
• Lesiones ocupantes de espacio: quistes, abscesos o tumores
Los pacientes con macrocefalia, pero con desarrollo psicomotor y examen neurológico normal y sin antecedentes personales reseñables: medir el PC en los progenitores y valorar la gráfica de evolución del PC, considerando, como primera opción, el diagnóstico de macrocefalia benigna familiar, principalmente secundaria a un derrame pericerebral (hidrocefalia externa benigna). Ante cualquier duda en la evolución, hacer consulta con neuropediatría.
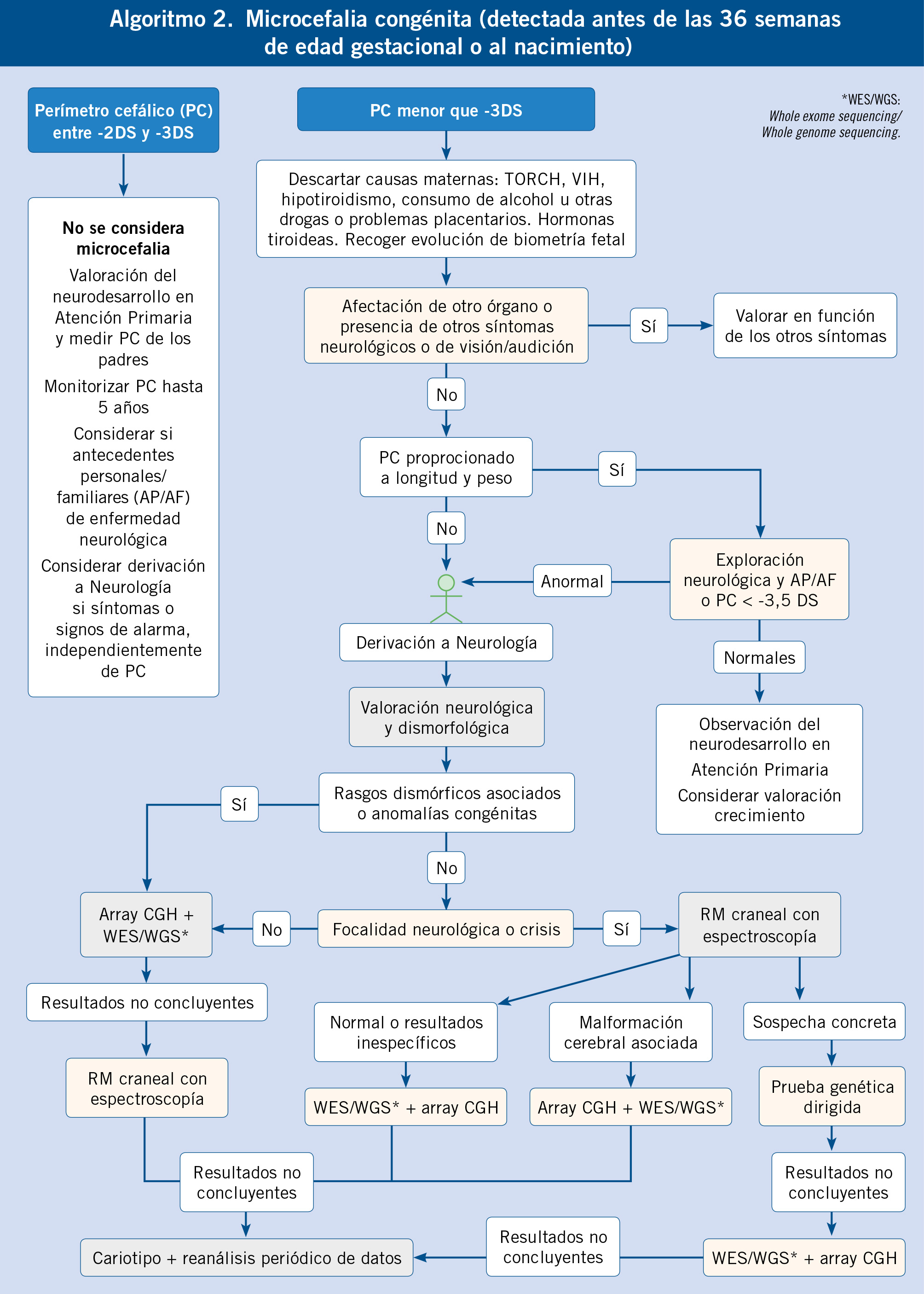
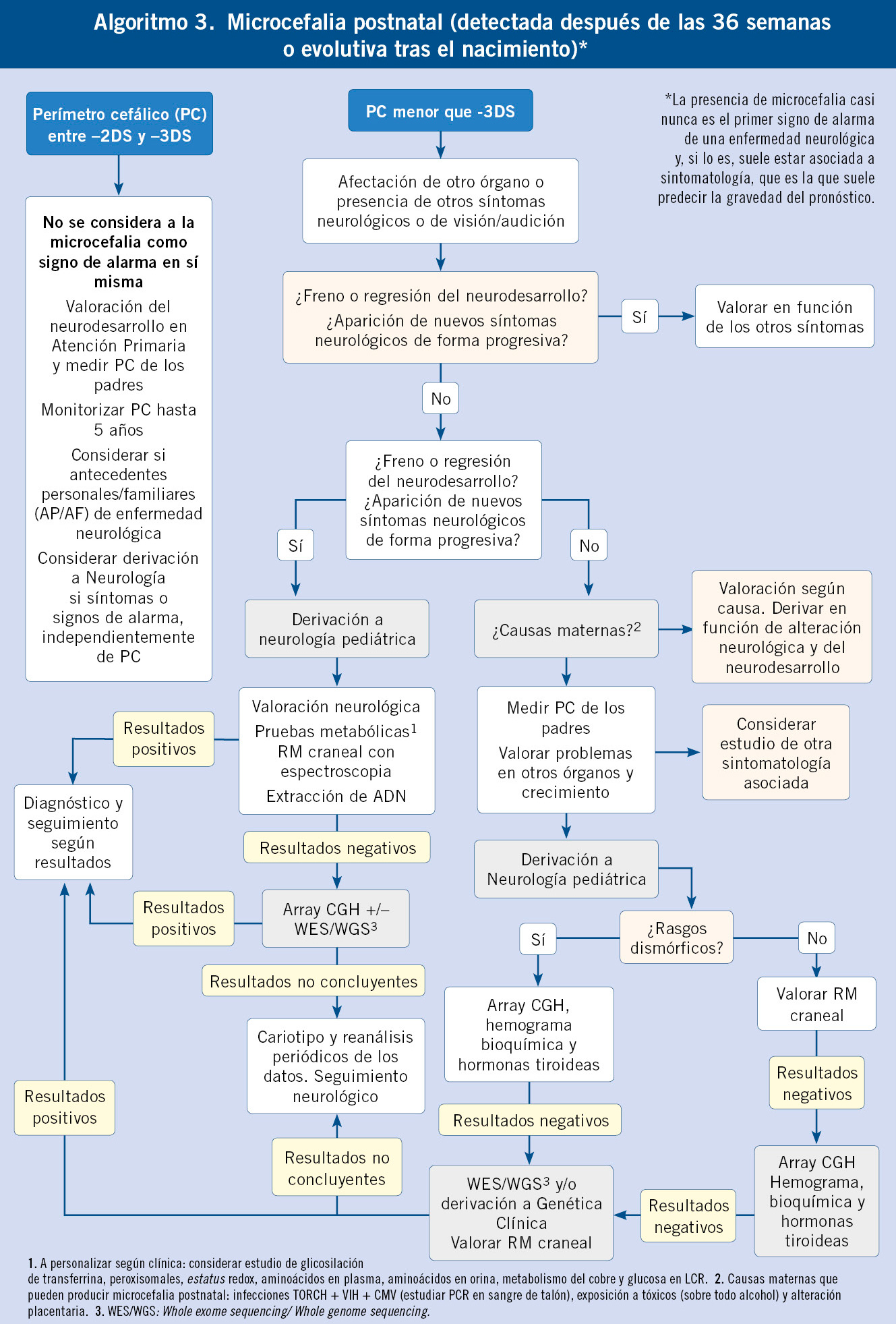
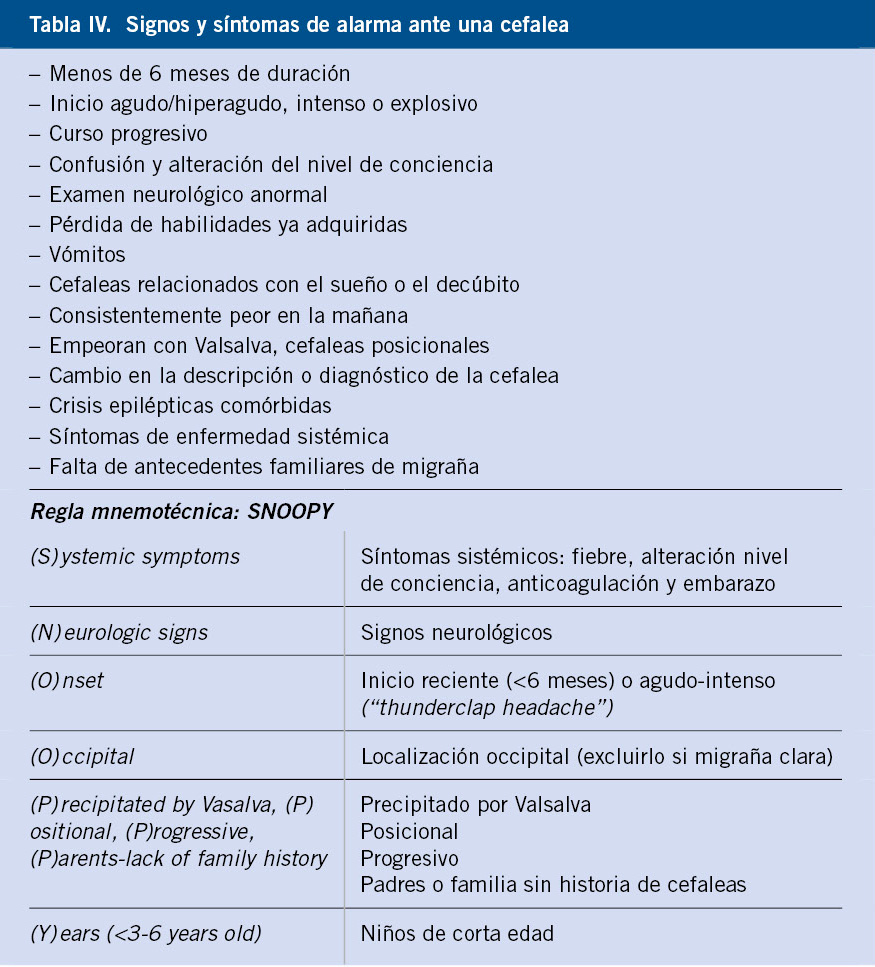
Microcefalia (Tabla IV)

Definición
Los autores no se ponen de acuerdo en el valor a partir del cual se considera una medida del PC patológica (a partir de -2,5… -3DS…), puesto que hay muchas personas con estas cifras de PC y un desarrollo completamente normal. No obstante, tanto la American Academy of Neurology (AAN) como la Asociación Española de Pediatría (AEP) han indicado unos valores, a partir de los cuales se habla de microcefalia, independientemente de si tienen o no repercusión clínica: por debajo de 2 DS la AAN y por debajo de 3 DS la AEP(1).
El Dr. Jean Aicardi en su libro: “Diseases of the Nervous System in Childhood”, en su 1ª edición del año 1992, hablando de la microcefalia, dice: “la microcefalia es difícil de definir; algunos autores lo refieren a un PC inferior o igual a -2DS o al percentil (Pc) 3. Todos los autores, no obstante, están de acuerdo en que pacientes con «cabezas pequeñas» (entre -2DS y -3DS) pueden no tener ninguna patología; aunque también es cierto que, estadísticamente, hay una mayor prevalencia de microcefalia leve en niños con dificultades en el aprendizaje”.
Clasificación de la microcefalia
• Microcefalia congénita: pacientes que desde las 36 semanas de edad postconcepcional ya presentan la alteración (o desde el nacimiento, si no tenemos esa medida previa).
• Microcefalia postnatal: pacientes que no tienen microcefalia al nacer y la alteración va progresando con el paso del tiempo.
La microcefalia congénita ocurre principalmente por dos fenómenos:
1. Por fallo o reducción de la neurogénesis en épocas precoces del desarrollo.
2. Por alteración de elementos del contenido encefálico, sobre todo de la sustancia blanca, o por un fenómeno disruptivo (por infecciones, isquemia intrauterina o por exposición a tóxicos), o por un fenómeno neurodegenerativo muy precoz (p. ej., el síndrome de Aicardi-Goutières) u otras enfermedades genéticas.
El pronóstico depende de muchos factores (momento de incidencia de la noxa, su intensidad… y otros factores que puedan coadyuvar); por lo tanto, una misma noxa no ha de dar necesariamente la misma clínica(2).
Posibilidades de clasificación de la microcefalia, desde un punto de vista descriptivo:
• Congénita: presente al nacimiento.
• Postnatal: no presente al nacimiento.
• Genética: causa intrínseca.
• Ambiental: causa extrínseca.
• Estática: evoluciona por el mismo percentil.
• Progresiva: disminución de percentiles con el tiempo.
• Proporcionada: al peso y la talla del paciente.
• Desproporcionada: con peso y talla normales.
• Sindrómica: asociada a otras alteraciones.
• Aislada: sin otras alteraciones asociadas.
Microcefalia congénita(4,5)
• Microcefalia congénita secundaria a una causa adquirida prenatal: pacientes que desde las 36 semanas de edad gestacional, o desde el nacimiento, ya presentan la alteración, de causa adquirida:
– Infecciones congénitas (TORCH, VIH, Zika).
– Exposición a tóxicos (alcohol, cocaína, etc.)
– Exposición a fármacos, especialmente fármacos antiepilépticos.
– Otros factores maternos que condicionen insuficiencia placentaria, anemias o malnutrición graves.
• Microcefalia congénita primaria: posibilidades:
– Con anomalías congénitas en otros órganos y/o rasgos dismórficos (microcefalias sindrómicas): orientan hacia síndromes de microdeleción/microduplicación o síndromes monogénicos con afectación multisistémica.
– Con talla baja: grupo heterogéneo que agrupa displasias esqueléticas y enfermedades con alteración en la reparación del ADN.
– Con malformación del desarrollo cortical y/o de la fosa posterior asociada.
– Con holoprosencefalia asociada: etiología compleja, con implicación de factores ambientales (p. ej., diabetes materna), cromosomopatías (p. ej., trisomía 13), enfermedades monogénicas o síndromes de microdeleción/microduplicación.
– Aisladas: con patrón giral simplificado y/o aumento leve del tamaño ventricular: grupo de enfermedades de herencia autosómica recesiva, cuyo evento patogénico es la reducción de la proliferación de neuroblastos. Tradicionalmente, conocidas como microcefalia vera, actualmente se prefiere el término de microcefalia primaria hereditaria. Habitualmente, no suelen presentar problemas neurológicos extremadamente graves, más allá de: discapacidad intelectual, epilepsia (habitualmente, no farmacorresistente y leve), signos piramidales sin problemas para la marcha autónoma… Existe, no obstante, una importante variabilidad fenotípica asociada a los distintos genotipos, por lo que es recomendable ser reservado desde el punto de vista pronóstico.
Microcefalia postnatal(5,6)
Pacientes que tienen una disminución del PC detectada después de las 36 semanas postconcepción o después del nacimiento.
Hay que separar las causas en las que existe una lesión estática que destruya parte del parénquima cerebral y/o bloquee el crecimiento normal del sistema nervioso, de aquellas que producen una lesión progresiva(7). La lesión estática incluye causas adquiridas (encefalopatía hipóxico-isquémica, lesiones adquiridas en relación con la prematuridad, infecciones congénitas, etc.) y causas genéticas. La lesión progresiva generalmente produce más manifestaciones clínicas que la simple microcefalia. Algunas de las causas más relevantes son: enfermedades metabólicas, síndrome de Rett y síndrome de Angelman, pacientes con mutaciones genéticas que cumplan criterios clínicos de síndrome de Rett o de Angelman, síndrome de Aicardi-Goutières, etc.
Conclusiones
Las causas más frecuentes de macrocefalia son el derrame pericerebral (hidrocefalia externa benigna), frecuentemente responsable de una macrocefalia familiar benigna, y la HTEC. Según la clínica, considerar la posibilidad de una macrocefalia familiar benigna, mientras no se demuestre lo contrario. Valoración de los casos de macrocefalia relativa, más allá de los valores absolutos del PC.
El concepto de microcefalia no está plenamente consensuado, aunque sí hay que valorar un riesgo importante por debajo del Pc 3, sobre todo desde el punto de vista pronóstico, que depende principalmente de la causa. Con respecto a las causas genéticas y metabólicas de macro-microcefalia, me remito a la referencia bibliográfica nº 2 de los doctores M. Alvarez Molinero y D. Gómez Andrés, donde están detalladas y esquematizadas.
El dismorfismo craneal (Figs. 1 y 2)

Figura 1. Huesos, suturas y fontanelas. Fuente: https://www.stanfordchildrens.org/es; y referencia(21).

Figura 2. Cierre aproximado en meses de suturas y fontanelas.
Fuente: https://www.adam.com/; y referencia(21).
Introducción
Las alteraciones en la forma del cráneo se encuentran frecuentemente en la revisión del niño sano. Ha habido un aumento importante de la incidencia de esta patología a partir de la campaña “back to sleep”, de la década de los 90 del siglo pasado. Así como las alteraciones en el tamaño del cráneo (micro y macrocefalia) se detectan precozmente con la medida del PC de forma sistemática, las alteraciones en la forma del cráneo pueden pasar más desapercibidas.
El cráneo está formado por siete huesos planos mayores (un hueso occipital, dos huesos parietales, dos huesos temporales y dos huesos frontales), separados entre sí por seis suturas craneales mayores (una metópica, una sagital, dos coronales y dos lambdoideas) y suturas menores bilaterales (fronto-nasales, fronto-esfenoidales y escamosas del temporal). Esto permite el moldeo de la cabeza para poder pasar por el canal del parto y su crecimiento progresivo posterior. Las suturas craneales se fusionan con posterioridad en diferentes tiempos (véanse los datos aproximados de su cierre en la figura 2).
Los dismorfismos craneales pueden ser de causa postural o de causa sinostótica. Detectar estas dismorfias, de un modo precoz, puede cambiar de manera significativa el pronóstico y reducir los costes del tratamiento.
• De causa postural: se deben a cambios en la forma de los huesos craneales en función de las zonas de decúbito.
• De causa sinostótica: secundarios a la fusión prematura de las suturas craneales.
Un diagnóstico acertado y precoz permitirá llevar a cabo una actitud terapéutica conservadora, eficaz en los casos de deformidades craneales secundarias a moldeamientos externos (plagiocefalia posicional), y diferenciarlos de aquellos pacientes afectados de una craneosinostosis verdadera, que requerirán una corrección quirúrgica temprana y un seguimiento estricto, para descartar el riesgo de complicaciones(8).
En la plagiocefalia postural, las suturas permanecen abiertas y, por tanto, estos pacientes no presentan un riesgo aumentado de HIC. La HIC sí puede suceder en la craneosinostosis, pues, en estos casos, se compromete el crecimiento craneal.
Deformidades posicionales
Las deformidades posicionales pueden producirse en el periodo intrauterino y encontrarse presentes en el momento del parto o aparecer más tardíamente, en las primeras semanas de vida.
Periodo intrauterino(8)
• Dolicocefalia del RN pretérmino.
• Plagiocefalias posturales anteriores.
Periodo postnatal(8)
Plagiocefalia occipital postural, de alta incidencia a partir de la campaña “back to sleep” para la prevención del SMSL.
El vocablo plagiocefalia proviene del griego: Plagio (oblicuo) y Kephalos (cabeza); se refiere a los varios tipos de deformidad craneal, caracterizados por la asimetría u oblicuidad de la cabeza. Se ha de diferenciar la postural o posicional de la sinostótica.
Las plagiocefalias posicionales generalmente afectan la parte posterior del cráneo; muy raramente se producen plagiocefalias posicionales anteriores. La plagiocefalia occipital posicional recibe también otros nombres: plagiocefalia por moldeamiento, deformativa, sin craneosinostosis, postural y funcional(8).
Plagiocefalia postural
En el año 1992, la Academia Americana de Pediatría (AAP) inicia la campaña “Back to sleep”, y se observa una reducción del SMSL hasta en un 40 %. Al mismo tiempo, se observa un aumento de casos de plagiocefalia postural, llegando a ser la demanda más frecuente en una consulta de neurocirugía infantil(8). Según estudios en EE.UU. e Italia, la incidencia varía entre el 22,1 % y el 37 % de recién nacidos(9,10). La máxima prevalencia es a las siete semanas de vida, y disminuye con la edad: hasta un 3,3 % de la población hacia los 2 años, y hasta un 2 % en la adolescencia(11,12).
La incidencia de craneosinostosis occipital en la literatura es mucho menor: en torno a 3:100.000 RN(13).
La plagiocefalia postural es de carácter “externo”, producida por fuerzas mecánicas extrínsecas que actúan sobre la sutura lambdoidea, bien durante la etapa intrauterina o, más frecuentemente, en el periodo postnatal.
Diversos factores pueden actuar sobre la cabeza fetal, produciendo un fenómeno de moldeamiento craneal en etapa intrauterina: oligohidramnios, bandas amnióticas, embarazos múltiples, encajamiento fetal prematuro, anomalías uterinas (útero bicorne, tabicamientos…), macrocefalia, fetos macrosómicos y partos instrumentados.
Otras causas postnatales también lo pueden producir: posición de “bienestar” del lactante (siempre la misma), utilización abusiva de carritos y sillas, apoyando el bebé su cabeza de la misma forma, durante periodos largos de tiempo, tortícolis congénita, hipotonía, estrabismo, malformación de la unión occípito-cervical. Todos estos factores influyen en la disminución de la motilidad espontánea del bebé.
También puede haber causas mixtas, que incluyan tanto factores antenatales como postnatales.
El diagnóstico es fundamentalmente clínico. La morfología craneal y los antecedentes clínicos permiten un diagnóstico de certeza en un alto porcentaje de casos. Clínicamente, se observa una antepulsión frontal del lado de la plagiocefalia postural con una cabeza que tiene forma de paralelogramo (se acompaña un esquema más adelante)(14).
Dolicocefalia del prematuro
La dolicocefalia, como deformidad postural, aparece normalmente en el RN pretérmino de bajo peso: las cabezas son alargadas y estrechas, tienen macrocefalia relativa y ausencia de tono en la musculatura cervical, con lo que la cabeza tiende a apoyarse lateralmente y ese peso de la cabeza sobre un hueso muy fino y débil produce ese aspecto característico. La sutura sagital en este caso no se encuentra fusionada, y la deformidad se corrige espontáneamente en torno a los 3-4 meses de edad, siempre que el desarrollo psicomotor sea normal. Fenotípicamente, se asemeja a la escafocefalia(8).
Craneosinostosis
Puede ser de causa primaria por defectos de la osificación de origen genético (principalmente los genes FGFR [fibroblast growth factor receptors]) o de causa secundaria a diferentes patologías (p. ej.: enfermedades hematológicas o metabólicas –raquitismo, hipotiroidismo–), aunque la mayor parte de los casos son de causa idiopática.
Se han descrito factores predisponentes a presentar craneosinostosis, como son: el crecimiento intrauterino restringido, la posición anormal intraútero, el oligohidramnios o la exposición prenatal a teratógenos, como el ácido valproico o la fenitoína(15).
La craneosinostosis ha mantenido su prevalencia constante y ha afectado a uno de cada 2.100-2.500 niños(16,17).
La más frecuente es la escafocefalia, por sinostosis de la sutura sagital y, la menos frecuente, la plagiocefalia posterior por craniosinostosis de la sutura lambdoidea(18).
Por el contrario, el dismorfismo craneal predominante es la plagiocefalia postural o deformidad sin sinostosis, hecho importante, ya que implica un manejo y un pronóstico significativamente mejor(18,19).
Valoración clínica
• Exploración sistemática del tamaño y la forma craneal en las visitas de seguimiento del paciente. Determinar en la anamnesis aspectos como la etnia familiar, así como la morfología craneal de los progenitores, pues puede haber morfologías craneales correspondientes a variantes de la normalidad, sin traducción patológica(8,15).
¿Cómo orientarnos delante de un dismorfismo craneal y sospechar si es una plagiocefalia postural o nos encontramos ante una craneosinostosis? Es bueno hacerse las siguientes preguntas:
– ¿Estaba presente en el momento del nacimiento?
– ¿Tiene una posición cérvico-cefálica preferente para dormir?
– ¿Mejora o empeora con el paso del tiempo?
El dismorfismo craneal presente desde el nacimiento, junto con la tendencia del paciente a mantener una misma posición en decúbito y la mejoría observada al reducir el tiempo en dicha postura, nos orientará hacia el diagnóstico de dismorfia craneal de causa postural(20).
Hay una asociación entre la plagiocefalia postural y la tortícolis congénita, por lo que siempre habrá que descartarla. Podría ser la causa de que se mantenga el cráneo en una misma posición.
Se ha de hacer una exploración craneofacial detallada, inspeccionando el cráneo en diferentes proyecciones: frente, perfil y coronal, y responder las preguntas siguientes:
– ¿Protruye un frontal más que el otro?
– ¿Tienen la misma dimensión las fisuras palpebrales?
– ¿Los arcos ciliares se encuentran a la misma altura?
– ¿Protruye más una órbita que la otra?
– ¿Sobresale más un arco zigomático que el otro?
Seguidamente, palparemos las suturas y fontanelas. Si encontramos alguna o algunas suturas aparentemente cerradas, sospecharemos una craneosinostosis. Si las encontramos abiertas, pensaremos en causas posicionales(21).
• Medición de los valores antropométricos, utilizando el cefalómetro (Fig. 3) para determinar el índice cefalométrico (IC) y el índice de plagiocefalia (IP)(15,21).

Figura 3. Utilización del cefalómetro.
– Índice cefalométrico (Fig. 4): permite valorar numéricamente el grado de alargamiento o aplanamiento del cráneo, midiendo el diámetro longitudinal y el mayor diámetro transversal.

Figura 4. Índice cefalométrico: permite valorar numéricamente el grado de alargamiento o aplanamiento del cráneo. Se calcula multiplicando por 100 el resultado del cociente entre la distancia biparietal máxima y la distancia anteroposterior tomada en la línea media.
– Índice de plagiocefalia (Fig. 5): determina el grado de dismetría entre las diagonales mayor y menor del cráneo.

Figura 5. Índice de plagiocefalia: es el valor en milímetros que se obtiene al restar la diagonal craneal menor de la diagonal craneal mayor, indicando el grado de asimetría craneal.
Estos valores se pasan a la gráfica adjunta y nos orientarán en el pronóstico y seguimiento de la plagiocefalia.
El PC no suele estar alterado en las dismorfias craneales de causa postural.
Exámenes complementarios
La radiología convencional del cráneo puede dar imágenes de difícil interpretación; no es sencillo distinguir “fusiones puntuales” de la sutura, puentes óseos, estenosis o esclerosis de los bordes suturales, que permitan diferenciar una sinostosis lambdoidea de una plagiocefalia postural(8).
Actualmente, en lactantes menores de 8-12 meses, se recomienda el uso de ecografías hechas por radiólogos expertos. El diagnóstico definitivo será mediante una tomografía computarizada (TC) con reconstrucción tridimensional, en el ámbito hospitalario, para el diagnóstico de craneosinostosis y para determinar el manejo prequirúrgico si procede(8,22,23).
Morfología de las craneosinostosis
Las craneosinostosis pueden ser simples o múltiples, según el número de suturas afectadas(20).
• Simples:
– Escafocefalia: por fusión de la sutura sagital.
– Plagiocefalia anterior sinostótica: por fusión unilateral de la sutura coronal.
– Plagiocefalia posterior sinostótica: por fusión unilateral de la sutura lambdoidea.
– Trigonocefalia: por fusión de la sutura metópica.
• Múltiples:
– Braquicefalia anterior: por fusión de ambas suturas coronales.
– Braquicefalia posterior: por fusión de ambas suturas lambdoideas.
– Síndrome de Mercedes-Benz: por fusión de la porción posterior de la sutura sagital y de ambas suturas lambdoideas.
– Oxicefalia: por fusión de todas las suturas (fusión universal).
• Escafocefalia: corresponde al 50 % de todas las craneosinostosis y es más prevalente en el sexo masculino(16). La fusión precoz de la sutura sagital ocasiona un cráneo alargado y estrecho. Aunque el aspecto puede recordar la dolicocefalia del prematuro, de la que se ha hablado anteriormente, la escafocefalia se acompaña habitualmente de abombamientos frontales y occipitales compensadores y no mejora con el paso del tiempo(8). Pueden presentar una cresta sagital, frontal anterior, que puede ser palpable y/o visible. Hay riesgo de HTEC si no se trata, lo que obliga a un seguimiento estrecho y controles por neurocirugía y oftalmología. Si se produce la fusión interna de la sutura sagital en su tercio medio, observaremos lo que se conoce como “deformidad en silla de montar”.
• Plagiocefalia anterior sinostótica: afectación de una o de ambas suturas coronales. Es la segunda forma más frecuente de craneosinostosis (aproximadamente de un 25 %). Es más prevalente en el sexo femenino(16). Afecta aproximadamente a 1:3.000 RN. Es una craneosinostosis compleja, que incluye el cierre unilateral de una sutura coronal y de otras suturas relacionadas de la base del cráneo (frontoesfenoidal, frontoetmoidal y esfenozigomática), lo que da una alteración importante de la zona frontoorbitaria; se oblicua el eje nasal y se desvía la raíz nasal. El peñasco temporal se encuentra mal posicionado, lo que conlleva que el pabellón auricular se encuentre adelantado y descendido. Frecuentemente, hay un estrabismo secundario, que suele requerir tratamiento quirúrgico(8).
• Plagiocefalia posterior sinostótica: sinostosis de una sutura lambdoidea. Es la forma menos frecuente de craneosinostosis; corresponde al 2 % de todas ellas(15). Puede asociarse a malformación de Chiari I y a estenosis del foramen yugular, que provocará hipertensión venosa y HTEC(20).
El diagnóstico diferencial con la plagiocefalia occipital posicional atenderá a criterios meramente clínicos:
– Desde una visión cenital, en la plagiocefalia posicional (Fig. 6) se observa el aplanamiento de la región posterior del cráneo producido por el apoyo continuado del lactante sobre esta zona; se asocia habitualmente a un abombamiento compensador del occipital contralateral y del hueso frontal del mismo lado del aplanamiento; el pabellón auricular está desplazado hacia adelante y la cabeza tiene forma de paralelogramo.

Figura 6. A. Plagiocefalia posicional. B. Sinostosis lambdoide. Fuente: referencia(8).
– Por el contrario, en la sinostosis lambdoidea, el aplanamiento posterior se asocia a un abombamiento parietal contralateral. En la región anterior existe un mínimo abombamiento frontal y el pabellón auricular del lado de la sinostosis se encuentra en un modo normal o bien desplazado posteriormente. Vista desde el zénit, la cabeza tiene forma trapezoide. También ayuda la observación de la línea bimastoidea (Fig. 7) de la base de cráneo(8).

Figura 7. Línea bimastoidea desviada. Fuente: referencia(8).
• Trigonocefalia: es la craneosinostosis más frecuentemente asociada a malformaciones cerebrales y a retardo mental. Los signos clínicos característicos son: frente en quilla, con disminución del diámetro bitemporal, aumento del diámetro biparietal e hipotelorismo (cráneo en forma de triángulo). Tiene una amplia variedad fenotípica. La sutura metópica es la primera que desaparece; su cierre se produce hacia el segundo año de vida, pero en algunos lactantes puede encontrarse cerrada antes de los 10 meses de vida, dando lugar a una dismorfia significativa, con la observación de una cresta frontal medial con retrusión fronto-orbitaria, aplanamiento temporal y de ambos frontales e hipotelorismo, con lo que hay que pensar en una cirugía, por la posibilidad de desarrollar un aumento de la presión intracraneana y/o un trastorno del desarrollo cognitivo(8).
Ocasionalmente, un lactante puede desarrollar una cresta metópica visible o palpable en la región frontal, pero manteniendo la morfología craneal normal; esta anomalía no da alteraciones cognitivas o HTEC, y ha de considerarse la intervención por causa estética. Entre estos extremos se encuentran los pacientes con variantes menores de esta patología, asociada a formas más leves de trigonocefalia; en estos casos, la indicación de intervención quirúrgica es más discutible(8).
Algunos lactantes pueden desarrollar una unión precoz de la sutura metópica, pudiéndose observar la cresta metópica, sin ser una sinostosis definitiva(24).
• Braquicefalia anterior por craneosinostosis de las dos suturas coronales, que da el aspecto a una cabeza ancha y aplanada en su parte posterior. Es la dismorfia que más frecuentemente se asocia a las craneosinostosis sindrómicas, como, por ejemplo, los síndromes de Crouzon, Muenke y Apert(25).
Tratamiento
En el tratamiento de las dismorfias craneales de causa sinostótica, siempre habrá que hacer una valoración neuroquirúrgica. En las no sinostóticas habrá que individualizar según la etiología y la gravedad de cada caso; la mayoría de ellas serán corregidas sin necesidad de derivación a neurocirugía, pero habrá que hacerla en caso de mala evolución clínica.
El tratamiento de la plagiocefalia postural incluye una serie de medidas que, en opinión de muchos autores, deben ser escalonadas: reposicionamiento, rehabilitación, técnicas de ortesis craneal y, en último y excepcional lugar, reconstrucción quirúrgica.
Escalonamiento del tratamiento de las plagiocefalias posturales:
1. Cambios de posición de la cabeza mientras el niño duerme.
2. Cuando esté despierto, que practique movimientos de cabeza y cuello y ejercicios sobre superficies duras.
3. Juegos en decúbito prono y posición de gateo a partir de 4,5-5 meses (Tummy time), siempre bajo supervisión.
4. Tratamiento de la tortícolis, si existe, con fisioterapia adecuada.
5. Tratamiento con ortesis craneal (casco) en los casos refractarios a las primeras medidas. La respuesta más idónea se obtiene a partir de los 4,5-5 meses de edad, y hasta un límite de 12 meses.
La indicación quirúrgica estará reservada para los casos con craneosinostosis y, también, para aquellas dismorfias craneales sin sinostosis, que tengan una deformidad severa, persistente, y en los que los tratamientos conservadores y la banda/casco ortésico no hayan tenido el efecto deseado(8).
Pronóstico
Los pacientes con plagiocefalia postural presentan, en general, muy buen pronóstico; la mayoría de casos se resuelven con medidas posturales y no presentan alteraciones cognitivas a largo plazo(8). Las Guías de Práctica Clínica (GPC) del NICE (National Institute for Health and Care Excellence), Cochrane y otros estudios no encuentran evidencia para sugerir que la plagiocefalia posicional se asocie a retardo mental o psicomotor en la edad adulta y que la mayoría de los casos se resuelven a los dos años de edad, sin secuelas(26-28).
No se ha encontrado evidencia tampoco en la asociación con estrabismo, excepto en la plagiocefalia frontal y la asociada a craneosinostosis(29).
Las craneosinostosis no asociadas a síndromes genéticos suelen tener también un buen pronóstico, y las intervenciones, que han de ser precoces, evitan sus consecuencias, que pueden ser importantes, como el estrabismo y otros defectos visuales, alteraciones psicológicas secundarias a la deformidad o la HIC(15,25).
A nivel cognitivo, los pacientes con craneosinostosis de una única sutura (coronal o lambdoide) son más propensos a presentar retardo en el aprendizaje en la educación primaria. Los pacientes con mejor pronóstico son los que presentan escafocefalia, pero también presentan una incidencia más alta de retardo en el aprendizaje con respecto a la población general(30,31).
La trigonocefalia (sinostosis metópica) se presenta con una amplia variedad fenotípica; solo las formas más severas requieren corrección quirúrgica. Los niños con trigonocefalia tienen un riesgo especial de presentar problemas oftalmológicos, como la hipermetropía, y presentan una mayor prevalencia de problemas de conducta, como el trastorno del espectro autista y el trastorno por déficit de atención con hiperactividad.
Conclusiones
Un diagnóstico diferencial acertado es fundamental para distinguir las deformidades posturales de las verdaderas craneosinostosis.
La dolicocefalia del prematuro tiene un manejo conservador, a diferencia de la sinostosis sagital.
La plagiocefalia occipital raramente se debe a una sinostosis lambdoidea; la mayoría de los casos es consecuencia de un moldeamiento de origen externo. Un manejo conservador apropiado que incida fundamentalmente en la reeducación postural, obtiene generalmente resultados muy satisfactorios.
Cualquier paciente con un diagnóstico de sospecha de craneosinostosis debe remitirse precozmente a un servicio de neurocirugía pediátrico con experiencia en estas patologías(8).
Función del pediatra de Atención Primaria
La función que se espera del pediatra de Atención Primaria es, en los casos de macrocefalia y microcefalia, un seguimiento cercano para la detección precoz de anomalías en el crecimiento del cráneo y posibles alteraciones en el desarrollo psicomotor del lactante y niño pequeño. Así mismo, en los casos de una dismorfia craneal, la orientación hacia una causa postural o una causa sinostótica de la anomalía; para, en este segundo caso, la derivación lo antes posible a neurocirugía.
Agradecimientos
• A los profesores A. Macaya y F. Rivier, a los que tengo en gran estima, que han leído previamente el trabajo y cuyos consejos me han sido de mucha utilidad para mejorarlo.
• Prof. François Rivier. Service de Neurologie Pédiatrique CHU de Montpellier. Université de Montpellier. Francia.
• Dr. Alfons Macaya. Servicio de Neurología Pediátrica. Hospital Infantil Universitario Vall d’Hebron, UAB. Barcelona.
Conflicto de intereses
No hay conflicto de interés en la elaboración del presente manuscrito ni fuente de financiación.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1.** Canelo Torres E, Gómez Gosálvez F, Jadraque Rodríguez R. Alteraciones en el perímetro craneal. Hospital General Alicante (HGUA). 2022.
2.*** Álvarez Molinero M, Gómez Andrés D. Alteraciones del perímetro cefálico: macrocefalia y microcefalia. Pediatr Integral. 2020; 7: 357-66. Disponible en: https://www.pediatriaintegral.es/publicacion-2020-10/alteraciones-del-perimetro-cefalico-macrocefalia-y-microcefalia/.
3. Rudolph CD, Rudolph AM, Hostetter MK, Lister G, Siegel NJ. Pediatría de Rudolph. 21 ed. 2004. Ed: Mc Graw-Hill, Interamericana.
4. Pirozzi F, Nelson B, Mirzaa G. From microcephaly to megalencephaly: determinants of brain size. Dialogues Clin Neurosci. 2018; 20: 267-82.
5.* Woods CG, Parker A. Investigating microcephaly. Arch Dis Child. 2013; 98: 707-13.
6. Seltzer LE, Paciorkowski AR. Genetic disorders associated with postnatal microcephaly. Am J Med Genet C Semin Med Genet. 2014; 166c: 140-55.
7.* Inder TE, Volpe JJ, Anderson PJ. Defining the neurologic consequences of preterm birth. N Engl J Med 2023; 389: 441-53.
8.*** Hinojosa Mena-Bernal J, Pascual B. Trastornos del tamaño y la forma del cráneo. Pediatr Integr. 2015; 9: 591-9. Disponible en: https://www.pediatriaintegral.es/publicacion-2015-11/trastornos-del-tamano-y-la-forma-del-craneo/.
9.* Ballardini E, Sisti M, Basaglia N, Benedetto M, Baldan A, Borgna-Pignatti C, et al. Prevalence and characteristics of positional plagiocephaly in healthy full-term infants at 8-12 weeks of life. Eur J Pediatr. 2018; 177: 1547-54.
10.* Biggs WS. Diagnosis and management of positional head deformity. Am Fam Physician. 2003; 67: 1953-6.
11. Bialocerkowski AE, Vladusic SL, Wei Ng C. Prevalence, risk factors, and natural history of positional plagiocephaly: A systematic review. Dev Med Child Neurol. 2008; 50: 577-86.
12. Roby BB, Finkelstein M, Tibesar RJ, Sidman JD. Prevalence of positional plagiocephaly in teens born after the “back to sleep” campaign. Otolaryngol – Head Neck Surg. 2012; 146: 823-8.
13. Rekate HL: Occipital plagiocephaly: a critical review of the literature. J Neurosurg. 1998; 89: 24-30.
14.* Esparza J, Hinojosa J, Muñoz MJ, Romance A, García Recuero I, Muñoz A. Diagnóstico y Tratamiento de la Plagiocefalia Posicional. Protocolo para un Sistema Público de Salud. Neurocirugía. 2007; 18: 457-67.
15.* Kajdic N, Spazzapan P, Velnar T. Craniosynostosis: recognition, clinical characteristics, and treatment. Bosn J of Basic Med Sci. 2018; 18: 110-6.
16.* Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of scaphocephaly. Am J Med Genet. 1996; 62: 282-5.
17. Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. Am J Med Genet Part A. 2008; 146: 984-91.
18.* Birgfeld CB, Heike C. Distinguishing between lambdoid craniosynostosis and deformational plagiocephaly: a review of this paradigm shift in clinical decision-making and lesson for the future. Craniomaxillofac Trauma Reconstr. 2020; 13: 248-52.
19. Branch LG, Kesty K, Krebs E, Wright L, Leger S, David LR. Deformational plagiocephaly and craniosynostosis: Trends in diagnosis and treatment after the “Back to Sleep” campaign. J Craniofac Surg. 2015; 26: 147-50.
20.*** Tresanchez-Lacorte B, Puerta-Roldán P, Vila-Soler J, Carsi-Durall A, López-Torres S, Rello-Saltor V. El pediatre davant un lactant amb dismorfisme cranial. Pediatr Catalana. 2022; 82: 139-44.
21.** Bosch Hugas J, Maria Costa Clara J. La plagiocefàlia posicional: una tasca d’Atenció Primària Pautes de diagnòstic, prevenció, tractament, seguiment i derivació des d’Atenció Primària. Barcelona. Generalitat de Catalunya, Departament de Salut. 2017.
22. Proisy M, Bruneau B, Riffaud L. How ultrasonography can contribute to diagnosis of craniosynostosis. Neurochirurgie. 2019; 65: 228-31.
23. Massimi L, Bianchi F, Frassanito P, Calandrelli R, Tamburrini G, Caldarelli M. Imaging in craniosynostosis: when and what? Childs Nerv Syst. 2019; 35: 2055-69.
24. Protocol d’activitats preventives i de promoció de la salut a l’edat pediàtrica. Catalunya. Generalitat de Catalunya, Departament de Salut. 2008.
25.* Governale LS. Craniosynostosis. Pediatr Neurol. 2015; 53: 394-401.
26.* Moulding helmets/cranial banding for plagiocephaly. NICE (National Institute for Clinical Excellence). Disponible en: https://www.nice.org.uk/.
27. Bialocerkowski AE, Vladusic SL, Howell SM. Conservative interventions for posicional plagiocephaly: a systematic review. Dev Med Child Neurol. 2005; 47: 563-70.
28. Hutchison BL, Hutchison LA, Thompson JM, Mitchell EA. Plagiocephaly and brachycephaly in the first two years of life: a prospective cohort study. Pediatrics. 2004; 114: 970-80.
29. Gupta PC, Foster J, Crowe S, Papay FA, Luciano M, Traboulsi EI. Ophthalmologic findings in patients with nonsyndromic plagiocephaly. J Craniofac Surg. 2003; 14: 529-32.
30.* Speltz ML, Collett BR, Wallace ER, Starr JR, Cradock MM, Buono L, et al. Intellectual and academic functioning of school-age children with single-suture craniosynostosis. Pediatrics. 2015;135(3):615-23.
31.* Magge SN, Westerveld M, Pruzinsky T, Persing JA. Long-term neuropsychological effects of sagittal craniosynostosis on child development. J Craniofac Surg. 2002;13(1):99-104.
Bibliografía recomendada
– Álvarez Molinero M, Gómez Andrés D. Alteraciones del perímetro cefálico: macrocefalia y microcefalia. Pediatr Integral. 2020; 7: 357-66. Disponible en: https://www.pediatriaintegral.es/publicacion-2020-10/alteraciones-del-perimetro-cefalico-macrocefalia-y-microcefalia/.
Artículo de actualización exhaustiva de las macro y microcefalias desde el punto de vista clínico y la descripción de una amplia patología al respecto.
– Hinojosa Mena-Bernal J, Pascual B. Trastornos del tamaño y la forma del cráneo. Pediatr Integr. 2015; 9: 591-9. Disponible en: https://www.pediatriaintegral.es/publicacion-2015-11/trastornos-del-tamano-y-la-forma-del-craneo/.
Artículo que contiene una interesante descripción y plan a seguir ante una dismorfia craneal y la alteración en el tamaño del cráneo. Una buena iconografía y patologías al respecto. Asimismo, contiene pautas claras sobre la derivación precoz a neurocirugía.
– Tresanchez-Lacorte B, Puerta-Roldán P, Vila-Soler J, Carsi-Durall A, López-Torres S, Rello-Saltor V. El pediatre davant un lactant amb dismorfisme cranial. Pediatr Catalana. 2022; 82: 139-44.
Artículo sobre la dismorfia craneal del lactante, que hace especial énfasis en el diagnóstico de las causas posturales y las sinostóticas. Expone también de una forma muy didáctica los distintos tipos de craneosinostosis.
– Bosch Hugas J, Maria Costa Clara J. La plagiocefàlia posicional: una tasca d’Atenció Primària Pautes de diagnòstic, prevenció, tractament, seguiment i derivació des d’Atenció Primària. Barcelona. Generalitat de Catalunya, Departament de Salut. 2017.
Monografía muy detallada de la plagiocefalia posicional, su exploración, la prevención primaria, el tratamiento y las pautas de seguimiento y derivación desde Atención Primaria.
| Caso clínico |
|
Lactante de 3 meses de edad, nacido a las 41 semanas de gestación. Antropometría al nacimiento de: peso: 2.900 g; longitud: 48 cm; PC: 34,5 cm, de sexo masculino. Primer hijo de padres sanos, no consanguíneos, sin antecedentes obstétricos de interés, excepto un parto prolongado. Presentación cefálica. Apgar: 7-9-10. Screening auditivo neonatal normal. Hace su seguimiento habitual, sin fallos, en pediatría del Centro de Atención Primaria, controlado siempre por el mismo pediatra. Controles sin nada a destacar. Evolución antropométrica que sigue sin pasar de +1 DS en peso y talla y PC en -0,5 DS. Al tercer mes de vida, el examen clínico es el siguiente: lactante de 3 meses con buen estado general, buena coloración, buena motilidad espontánea. Test de Pull to sit 3/4. Buen sostén cefálico, pero con claudicación cefálica inconstante. Motilidad correcta y simétrica de las cuatro extremidades. Buena fijación visual y seguimiento correcto hacia los cuatro puntos cardinales. Sonrisa franca. Emite sonidos. No se observan dismorfias o asimetrías faciales a destacar. En el examen del cráneo, se observa una dismorfia con aplanamiento occipital derecho y abombamiento frontal derecho, con el pabellón auricular derecho avanzado. A la inspección desde el zénit, el cráneo tiene aspecto de paralelogramo. La utilización del craniómetro nos da unas cifras de índice craniométrico del 85 % (valor normal) y del índice de plagiocefalia de 15 m/m. En el examen del cuello, no se objetivan limitaciones motrices claras. En el interrogatorio a los padres, explican que es un niño tranquilo y que, cuando duerme, tiene siempre el cráneo lateralizado hacia la derecha.
|