 |
| Temas de FC |
R. Cancho Candela, M. Andrés de Álvaro
Unidad de Neurología pediátrica. Servicio de Pediatría. Hospital Universitario Río Hortega. Valladolid
| Resumen
La epilepsia es un trastorno cerebral en el que existen crisis epilépticas no provocadas; es decir, episodios de descarga neuronal que no son secundarios a una agresión inmediata al cerebro. Existen diferentes tipos de crisis según las características clínicas del modo en el que ocurren. Los síndromes epilépticos son enfermedades diferenciadas caracterizadas por tipos de epilepsia que cursan con crisis específicas y una historia natural definida (edad, pronóstico). Numerosas epilepsias muestran comorbilidades en el desarrollo del paciente infantil, con un elevado potencial de discapacidad. El reconocimiento precoz y específico de algunos tipos de crisis y de epilepsias por parte del pediatra, pueden mejorar sustancialmente el manejo y el pronóstico. |
| Abstract
Epilepsy is a brain disorder with unprovoked epileptic seizures, that is, neuronal discharge episodes that are not secondary to an immediate insult on the brain. There are different types of seizures according to their clinical char-acteristics. Epileptic Syndromes are differentiated diseases characterized by specific epilepsy types that present with specific crises, and a defined natural history (age, prognosis). Numerous epilepsies show comorbidities in the development of the infant patient, with a high potential for disability. The early and specific recognition of some types of seizures and epilepsies by the pediatrician can substantially improve management and prognosis. |
Palabras clave: Crisis epiléptica; Epilepsia; Síndrome epiléptico.
Key words: Epilepsy; Epileptic seizure; Epileptic syndrome.
Pediatr Integral 2020; XXIV (7): 375 – 382
Síndromes epilépticos según la edad
Epilepsia: clasificación, definiciones, diagnóstico
La epilepsia es un agrupamiento de diferentes síndromes y trastornos con base cerebral y con un síntoma común, la recurrencia de crisis epilépticas no provocadas.
La definición de epilepsia ha sido controvertida a lo largo del tiempo. Diversos conceptos relacionados con la epilepsia, como son su clasificación y los tipos de crisis, han mostrado cambios profundos en las últimas décadas. Se trata de una de las enfermedades infantiles crónicas más frecuente; a veces, se dice de modo coloquial, que: “la epilepsia es una enfermedad pediátrica que, a veces, se prolonga u ocurre en el adulto”. Esta afirmación excesiva encierra una verdad, y es que la prevalencia de epilepsia es, en torno a cuatro veces superior en la infancia, que en adultos (aproximadamente 1% de toda la población infantil)(1); además, la epilepsia pediátrica es de mayor riqueza semiológica y complejidad clínica, e incide en un cerebro en desarrollo, por lo que las implicaciones en relación con posibles secuelas son mayores.
Antes de profundizar en la descripción de los diversos síndromes epilépticos pediátricos, es obligado revisar algunas definiciones. La base de la clasificación y de las definiciones son las propuestas por la ILAE (International League Against Epilepsy)(2); existe consenso internacional respecto a su uso y su última revisión corresponde a 2017.
Denominamos crisis epiléptica (CE): a la aparición transitoria de signos y/o síntomas provocados por una actividad neuronal excesiva o sincrónica en el cerebro(3). Las CE pueden considerarse como: provocadas (o sintomáticas agudas, o reactivas), cuando se asocian a un factor agudo transitorio o reversible sistémico, o del SNC (p. ej.: fiebre, infección, tóxicos); no provocadas, si no es así; y reflejas, en caso de ser CE provocadas, en los que su recurrencia asocia una predisposición anómala en relación con el resto de población (estímulos sensoriales, lectura, etc.)(4).
En este sentido, denominamos epilepsia, a una enfermedad de la función cerebral, definida por una de estas tres condiciones:
1. Al menos, dos CE no provocadas (o reflejas) que ocurran en un plazo superior a 24 horas.
2. Una CE no provocada (o refleja) y un riesgo de presentar nuevas crisis similares al derivado, tras tener dos crisis no provocadas (de, al menos, un 60% de probabilidad de recurrencia en los próximos 10 años).
3. Diagnóstico de síndrome epiléptico concreto.
Denominamos síndrome epiléptico a un trastorno epiléptico que muestra unas características específicas en cuanto a sus síntomas y signos, incluyendo: historia natural, edad de inicio, tipo de crisis, etiología, hallazgos neurofisiológicos, anomalías neurológicas asociadas, respuesta al tratamiento y pronóstico. Es importante hacer notar que no todas las epilepsias pueden ser encuadradas dentro de un síndrome epiléptico definido; de manera expresa (y a pesar del título de nuestro artículo), la ILAE evita en su clasificación, la distribución de los tipos de epilepsia dentro (o en paralelo) de síndromes epilépticos.
Es también necesario, antes de iniciar el estudio de los síndromes epilépticos pediátricos, definir brevemente otros dos conceptos. Se entiende por encefalopatía epiléptica a una epilepsia tal que la propia actividad epiléptica en sí misma contribuye al deterioro progresivo de la función cerebral (con discapacidad cognitiva, motora, etc.) de forma adicional sobre la discapacidad esperada solo por la etiología subyacente y en la que la mejoría de la actividad epiléptica podría minimizar esta discapacidad. La epilepsia farmacorresistente es aquella en la que existe fallo de dos fármacos antiepilépticos (en monoterapia o combinación): correctamente indicados, bien tolerados, pautados a dosis apropiadas y durante el tiempo adecuado(5). La ILAE ha recomendado no usar, dentro de los diagnósticos de tipos de epilepsia, el término “benigno” y recomienda el uso de los términos: “autolimitado” o bien “fármacosensible”.
Como se ha indicado, las CE son manifestaciones clínicas de disfunción cerebral transitoria. Las diferentes formas clínicas de estas CE son las que se denominan como: “tipos de crisis”. La descripción clínica precisa de la forma de ocurrir una crisis determinada es lo que se denomina “semiología”. Los diferentes tipos de crisis han sido clasificados de formas muy diferentes a lo largo del tiempo. En la actualidad, la clasificación de consenso diferencia signos clínicos cerebrales de inicio focal o generalizado, y la existencia de conciencia preservada o no (Fig. 1).
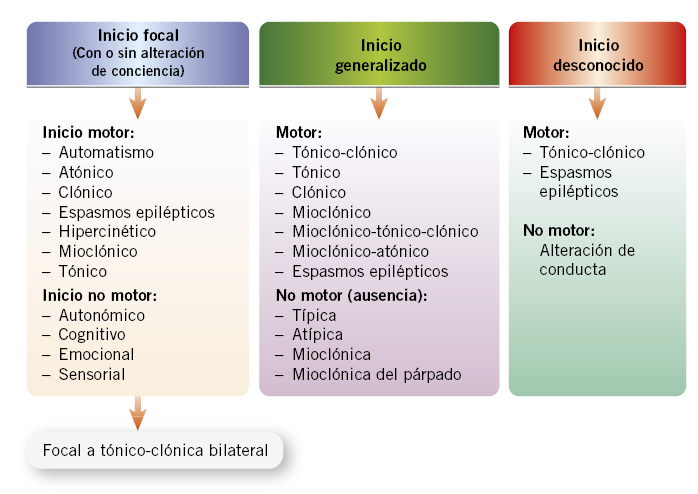
Figura 1. Clasificación de tipos de crisis según la ILAE (International League Against Epilepsy)(2).
Hablamos de signos de inicio focal, cuando existen signos y síntomas debidos a la activación inicial de un grupo de neuronas limitado a un hemisferio cerebral, que se denomina foco epileptógeno, con: semiología crítica motora, sensitiva, sensorial, psíquica o autonómica específicamente atribuida a esa zona neuronal. Pueden ocurrir con conciencia preservada o bien (en terminología previa “parcial simple”), o bien con conciencia alterada (término anterior “parcial compleja”), en las que existe percepción alterada (pero no inconsciencia). En las crisis generalizadas existe compromiso de ambos hemisferios cerebrales desde el inicio, e incluyen pérdida de conciencia, salvo en las crisis mioclónicas y atónicas, de gran brevedad. Debe remarcarse que el término convulsivo se reserva para crisis con componente motor y, por tanto, no debe emplearse para, por ejemplo, las ausencias típicas, que suelen carecer de fenómenos motores clínicamente relevantes.
Existe, por tanto, un escalonamiento en la orientación diagnóstica que debe orientar al profesional que se enfrenta al paciente con crisis que se sospecha sean CE(6) (Fig. 2).

Figura 2. Escalonamiento en la orientación diagnóstica de la epilepsia.
En primer lugar, debe determinar si los eventos paroxísticos del paciente no son en realidad trastornos de naturaleza no epiléptica, como: síncopes, parasomnias, trastornos del movimiento, etc. En segundo lugar, una vez determinadas las crisis como CE, debe intentarse su encuadre dentro de un tipo específico de epilepsia. En tercer lugar, y no siempre será posible, todo el cuadro clínico global (tipo de epilepsia, comorbilidades, edad, etc.) será incluido dentro de un síndrome epiléptico. Este proceso es, a veces, de gran simpleza diagnóstica y, en otras, de elevada complejidad, requiriéndose estudios neurofisiológicos, de neuroimagen, genéticos, etc. El tipo de epilepsia o síndrome epiléptico de un paciente puede, además, ser clasificado desde el punto de vista etiológico.
Existen diversas confusiones y errores habituales en el enfoque diagnóstico de la epilepsia. Expondremos algunos de los más usuales. Es frecuente la confusión entre tipo de CE y tipo de epilepsia o directamente síndrome. Ejemplos clásicos son: los espasmos epilépticos, típicos del síndrome de West (o previos a), pero no exclusivos, pudiendo aparecer en otros tipos de epilepsia de infancia temprana; o, por ejemplo, las CE tipo ausencias típicas, que son la CE habitualmente exclusivas de la epilepsia generalizada de tipo ausencias típicas, pero pueden aparecer en otras epilepsias como la epilepsia mioclónica juvenil. Debe hacerse, por tanto notar, que muchos tipos de epilepsia y síndromes epilépticos muestran varios tipos de CE. Otra confusión habitual es la reducción o pobreza descriptiva clínica que lleva a considerar todas las CE, en las que existen fenómenos motores preeminentes (crisis “hiperquinéticas”), como tónico-clónicas. De modo similar, es también habitual encontrar cómo se califican erróneamente de ausencias las CE focales, en las que existe alteración del nivel de conciencia con fenómenos motores no llamativos, e incluso contamina a los episodios no epilépticos de ensimismamiento, hoy en día, sobrediagnosticados de “ausencias”.
La descripción semiológica precisa es de elevado valor diagnóstico. Los fenómenos clínicamente “menores” que aparecen en las CE, son de un valor crítico en la orientación y manejo adecuado del cuadro. De este modo, la descripción precisa de: automatismos, fenómenos motores focales, movilidad ocular, descripción subjetiva de la crisis, etc., es mucho más importante que, por ejemplo, establecer la duración de una crisis tónico-clónica. Un ejemplo evidente es la orientación hacia neuroimagen, con mayor o menor celeridad en caso de focalidad en la expresión clínica; o como la clara impresión clínica de CE generalizadas, en las que el rendimiento de neuroimagen es escaso, debe hacer incidir en la mayor importancia de un estudio neurofisiológico adecuado.
Estudios complementarios para orientación diagnóstica
El visionado de vídeos domésticos se ha convertido en una importante herramienta para la orientación inicial diagnóstica.
Existen tres grupos de estudios complementarios principales en epilepsia, desde un punto de vista básico y orientativo, que son: la revisión de filmaciones domésticas de CE, los estudios neurofisiológicos (electroencefalograma: EEG) y los estudios de neuroimagen. Los estudios genéticos, inmunológicos, etc., pertenecen a una fase de profundización diagnóstica que no es pertinente que sea valorada en este manuscrito.
Hoy en día, existe una mayoría de familias que consultan por primera vez con sus pediatras de Atención Primaria o con un neuropediatra, aportando ya una filmación doméstica de episodios sospechosos de CE. Existen estudios que valoran, como más precisas, las filmaciones domésticas que, por ejemplo, el EEG, pero siempre que el evaluador sea una persona con cierta formación en epilepsia(7). En nuestra opinión, el valor clínico de los vídeos domésticos es muy elevado y, en la práctica clínica diaria, nos permite en consulta, hacer un primer despistaje de CE versus Trastornos Paroxísticos No Epilépticos (TPNE), que suele ser coincidente con el diagnóstico final. Por tanto, debe motivarse a las familias en la filmación de episodios sospechosos, sobre todo, en casos dudosos o no diagnosticados. En cierto modo, puede decirse que los vídeos son una herramienta muy valiosa para diagnóstico de CE y tipo, pero no tanto en el diagnóstico de tipo de epilepsia y síndrome.
El EEG es la herramienta fundamental en el manejo y, sobre todo, en el diagnóstico del tipo de epilepsia y síndrome. Sin embargo, la interpretación del EEG es un proceso complejo que requiere elevada especialización. Existen múltiples factores de confusión en este sentido. Algunos de ellos son(8):
- Aproximadamente el 4% de todos los niños no epilépticos muestran actividad paroxística en el EEG. Este porcentaje se eleva en caso de enfermedad neurológica sin epilepsia activa, como: migraña, daño cerebral, etc.
- En torno al 40% de los niños epilépticos tienen un EEG interictal basal normal (pero solo un 8% si se realizan activaciones adecuadas, como deprivación de sueño).
- La intensidad de las anomalías epileptógenas no siempre refleja la severidad o importancia del cuadro epiléptico.
- Las CE provocadas remedan, en ocasiones, patrones EEG que son básicamente similares a las CE de epilepsias específicas.
En cualquier caso, es probablemente adecuado que, en una primera consulta con Neuropediatría por sospecha de CE, pueda aportarse ya un primer estudio EEG, aunque sea basal, solicitado desde Atención Primaria siempre que esta actitud no demore en exceso la consulta con el especialista.
En relación al papel del EEG como decisor de otros estudios o de inicio de tratamiento, hay que reflejar que su uso debe ser específico e individualizado. Los algoritmos derivados de meta-análisis suelen ser erróneos en la atribución de riesgo de recurrencia, porque son incapaces de reflejar la multiplicidad de situaciones clínicas individuales, y reducen la situación de primera CE a, en realidad, primera CE tónico-clónica. Esta situación, clínicamente, no suele ser “real”, ya que se calcula que el 74% de los pacientes con “primera CE” ya han manifestado otras CE previas “menores”(9). El uso de EEG en estos algoritmos mejora la precisión diagnóstica, pero siempre teniendo en cuenta el adecuado uso de maniobras de activación (deprivación de sueño, hiperventilación y fotoestimulación). La situación prototípica, en este sentido, es la de primera CE tónico-clónica, en la que en el EEG aparecen descargas generalizadas con provocaciones, no acompañadas de crisis clínica. Esta situación orienta al riesgo de desarrollo de epilepsia generalizada idiopática, pero puede posiblemente asumirse no iniciar tratamiento por el momento.
Respecto a la neuroimagen, en nuestro ámbito habitual de trabajo, puede decirse que el estudio de elección en el proceso diagnóstico de epilepsia debe ser la resonancia magnética cerebral (RMC)(10). La mayoría de epilepsias requieren, para un proceso completo de estudio de la realización de RMC, en particular, las epilepsias focales (las asociadas a síntomas o trastornos con focalidad cerebral), las no clasificables o las de difícil control. Pueden existir controversias respecto a la obligada indicación en algunas epilepsias, aunque puede decirse que no es preciso si existe diagnóstico sindrómico claro no ligado a patología focal (como en la mayoría de las generalizadas idiopáticas, o en síndromes genéticos como el Dravet). En las epilepsias focales infantiles autolimitadas/benignas, es controvertido por la posible confusión de estos síndromes con epilepsias secundarias a lesiones cerebrales focales. Nuestra opinión es que, si existe acceso a RMC, se realice también en estos casos, sobre todo, si existe alguna atipicidad en la supuesta epilepsia benigna del paciente. En relación al uso de ecografía cerebral en lactante con CE o a la tomografía axial computarizada, debe reservarse su uso dentro del diagnóstico, a las situaciones de urgencia y primeras evaluaciones, para descartar cuadros agudos secundarios a patología: vascular, hemorrágica o infecciosa.
Síndromes epilépticos según edad de debut habitual
Se ha optado por realizar una descripción sintomática y electroclínica de los principales tipos y síndromes, minimizando los estudios genéticos y la terapéutica; para profundizar en estos ámbitos, se recomienda consultar con las referencias recomendadas(11-12).
Periodo neonatal
La semiología de las crisis neonatales es muy diferente a las de otras edades y son frecuentemente secundarias.
La mayoría de CE neonatales son provocadas y son secundarias a agresiones como: alteraciones metabólicas, hipoxia-isquemia o infección de SNC. Fuera de estas, existen algunos cuadros epilépticos específicos, frecuentemente relacionados con alteraciones genéticas. Una gran parte de CE neonatales muestran una semiología “sutil”, ya que presentan signos clínicos que frecuentemente no son identificados de forma inmediata como CE, en particular, en prematuros. Las más habituales involucran movimientos orofaciales, como: chupeteo, movimientos linguales y desviación ocular, siendo también posibles alteraciones autonómicas, apnea o movimientos de tipo pedaleo. La interpretación del EEG neonatal es particularmente compleja; es frecuente la existencia de patrones paroxísticos eléctricos sin aparente sintomatología clínica. Es por ello que, es de gran utilidad el uso de monitores de EEG simplificados de uso a pie de cama del paciente y que pueden facilitar el diagnóstico de CE y del patrón EEG. Se describen brevemente los principales síndromes epilépticos neonatales.
Epilepsia familiar neonatal benigna
Las comúnmente llamadas “crisis del quinto día” constituyen un cuadro heterogéneo, relacionado frecuentemente con mutaciones en KCNQ2 y KCNQ3, de herencia autosómico dominante, por lo que pueden existir antecedentes familiares. Su pico de inicio es entre el 4º y 6º día de vida, pero pueden debutar en las semanas siguientes. Las CE suelen ser: focales o multifocales, tónicas o clónicas, afectando a ambos hemicuerpos y, por lo general, asociadas a apnea, breves y frecuentes, ocurriendo principalmente durante la transición sueño-vigilia. El EEG interictal suele ser normal. La norma es la resolución espontánea en los primeros meses de vida, sin secuelas, pero con un riesgo de aproximadamente 10 a 15% de epilepsia futura.
Síndrome de Ohtahara (encefalopatía infantil precoz)
Se trata de una encefalopatía epiléptica. Las causas más frecuentes son estructurales (porencefalia, hemimegalencefalia y disgenesia) y mutaciones en genes específicos entre otros: KCNQ2, ARX, STXBP1, CDKL5, SCN2A y SCN3A. Las metabolopatías son causa menos habitual. Suele iniciarse en las primeras semanas de vida, en forma de CE, de tipo espasmos tónicos en el debut, a menudo en salvas. Pueden aparecer también CE de otros tipos, como focales motoras y mioclonías. El EEG muestra un característico brote-supresión continuo en todos los estadios sueño-vigilia que, con el paso de los meses, evoluciona hacia hipsarritmia. Los pacientes muestran deterioro neurológico global, falleciendo la mayoría en los primeros años de vida. Los supervivientes presentan encefalopatías epilépticas evolutivas. Se trata de un cuadro farmacorresistente.
Encefalopatía mioclónica temprana
Es un cuadro similar al síndrome de Ohtahara, pero con algunas especificidades. En este caso, hay una mayor asociación con metabolopatías (en particular, hiperglicinemia no cetósica) y, menos habitual, con malformaciones o mutaciones específicas. El debut suele ser anterior (en las primeras horas de vida), con CE predominantes en forma de mioclonías segmentarias, migratorias, que usualmente afectan a la musculatura distal de extremidades, pero con también crisis: clónicas focales, mioclónicas generalizadas o espasmos tónicos, más tardíos. El EEG es similar al Ohtahara, con patrón brote-supresión, pero el brote es más breve, de 1-3 segundos, con supresión más larga y aparece de forma predominante o exclusivamente en sueño. Puede no existir patrón EEG asociado a las crisis mioclónicas. El pronóstico vital y secuelar es similar al Ohtahara, pero debe ponerse énfasis en el despistaje de metabolopatías, ya que existe posibilidad de tratamiento específico en algunas de ellas (déficit de piridoxina, ciclos de urea, etc.). Es por ello, que debe insistirse en la diferenciación de este cuadro con el Ohtahara, a pesar de las similitudes clínicas y en EEG.
Lactancia
Los espasmos epilépticos son un tipo de crisis poco frecuente, pero que debe ser reconocido de manera precoz, para un adecuado manejo.
Síndrome de West
El síndrome de West (SW) debuta habitualmente entre los 3-7 meses de edad. Este síndrome no es específico de un grupo de etiologías, aunque existen algunas causas más proclives a producir el síndrome de West. Dado su debut en periodo de lactante, existe un grupo numeroso de pacientes en los que ya existe diagnóstico o, al menos, sospecha de enfermedad neurológica desde periodo neonatal; es el caso de los síndromes de West secundarios a daño cerebral por: encefalopatía hipoxico-isquémica perinatal, ictus perinatal, infección perinatal de SNC y la mayoría de malformaciones cerebrales(13). Existen, además, numerosas mutaciones patogénicas que pueden producir encefalopatía epiléptica, debutando como síndrome de West o bien de forma evolutiva, partiendo de otro tipo de encefalopatía precoz. Entre los numerosos genes implicados están: ARX, CDKL5, SPTAN1, STXBP1, SCN2A, etc. Entre las causas primarias genéticas, deben considerarse también las cromosomopatías (Down, del1p36, inv dupl 15, etc.). Debe hacerse una consideración especial hacia las enfermedades neurocutáneas, en particular, la esclerosis tuberosa (ET), dada su particular asociación con el síndrome de West. Dada la diversidad etiológica del síndrome de West y que el pronóstico se relaciona con la rapidez y eficacia del tratamiento, la identificación rápida de esclerosis tuberosa es una prioridad en cualquier paciente con síndrome de West de causa no obvia. La visualización de estigmas típicos de esclerosis tuberosa en el periodo de lactante (manchas hipomelanósicas y placas fibrosas), deben dirigir el esfuerzo diagnóstico hacia neuroimagen precoz y no hacia estudios metabólicos o genéticos no específicos; el diagnóstico de esclerosis tuberosa con síndrome de West indica el inicio de tratamiento con vigabatrina, que se ha mostrado muy superior a otros fármacos en este grupo de pacientes.
El síndrome de West muestra un tipo de CE específico, los espasmos epilépticos, que son típicos, pero no exclusivos de este síndrome. El diagnóstico de síndrome de West requiere de regresión en el desarrollo y patrón EEG específico denominado hipsarritmia, con un patrón desorganizado de ondas lentas de gran amplitud y, frecuentemente, combinado con puntas multifocales. Los espasmos son el tipo de CE paradigmático en relación con la discordancia entre su relativa sutilidad y la gravedad del cuadro clínico. Cuando el paciente no ha mostrado previamente enfermedad neurológica y no se vigila activamente al paciente, es frecuente que exista un decalaje de semanas o meses entre el debut de los espasmos y su diagnóstico. Los espasmos consisten en una contracción repentina en flexión, extensión o flexo-extensión de la musculatura proximal y del tronco (sobre todo, de miembros superiores, produciendo separación y elevación de ambos brazos con respecto al tronco), en general, simétricos y sincrónicos, con una duración habitual en torno a un segundo de duración, con clara tendencia a ocurrir agrupados en salvas o rachas, separados entre 5 a 20 segundos, principalmente en etapas de transición sueño-vigilia; suele existir una sutil supraelevación de la mirada durante el espasmo y llanto posterior. Es frecuente que, en una etapa inicial de algunas semanas, los espasmos ocurran con poca frecuencia y muy selectivamente antes de dormir o al despertar. Con posterioridad se incrementan y es evidente la detención o regresión en desarrollo.
El pronóstico del síndrome de West está ligado a la causa, pero en cualquier caso, es siempre mejor en casos de diagnóstico y tratamiento adecuado precoz. Al menos, tres cuartas partes de los pacientes presentan retraso significativo en el desarrollo y se ha reportado farmacorresistencia en síndrome de West en más de la mitad de los pacientes, con evolución hacia otros tipos de epilepsia y encefalopatía epiléptica. Desde el punto de vista diagnóstico, es clave cómo se ha indicado la identificación de los espasmos como tales. Asimismo, la realización de RMC precoz es también importante; en torno al 60-70% de pacientes tienen neuroimagen cerebral alterada. Puede existir gran complejidad en el proceso diagnóstico de síndrome de West de causa no evidente y requerir de múltiples estudios, en particular, genéticos y metabólicos. El tratamiento básico del síndrome de West se basa inicialmente en la diferenciación entre síndrome de West de causa estructural en los que suele existir mayor eficacia de vigabatrina versus resto de causas, en los que aparte de tratamientos específicos, suelen ser más eficaces los corticoides. Si existe ineficacia de estos dos tratamientos solos o combinados, deben probarse otros fármacos (zonisamida, valproico…), dieta cetogénica, etc.
Epilepsia familiar benigna de la infancia
Con anterioridad, la mayoría de estas epilepsias se englobaban como síndrome de Watanabe-Vigevano, pero se ha apreciado diversidad en cuanto a tipo de CE y en cuanto a pronóstico. Existen pacientes con mutaciones en PRRT2 (también relacionado con: coreoatetosis, convulsiones febriles, migraña hemipléjica y ataxia episódica), así como en SCN2A y SCN8A; las mutaciones patogénicas de estos dos genes pueden producir un espectro amplio de patología, que va desde cuadros relativamente benignos, a encefalopatías epilépticas muy severas. El caso típico es el de: un lactante previamente sano, entre los 3 y 8 meses de edad, que presenta CE agrupadas (varias en unas horas o días), de semiología variada, focales, con o sin generalización secundaria. Suele haber: cese de la actividad motora, ausencia de respuesta, desviación de la cabeza y/o ojos a un lado, mirada fija, parpadeo y clonias unilaterales o bilaterales. El EEG interictal suele ser normal. Tienen buena respuesta a fármacos. El pronóstico es bueno, con cese de CE antes de los dos años de edad, sin secuelas.
Epilepsia mioclónica de la infancia
Se trata de una epilepsia generalizada idiopática, con debut entre el segundo semestre de vida y los 3-4 años, en un paciente sin déficits neurológicos previos. Las CE suelen ser mioclónicas axiales, con afectación de EESS, generalmente sin afectación de conciencia y, a menudo, se desencadenan por estímulos sensoriales. El EEG interictal es generalmente normal y requieren a menudo de registro ictal o de EEG durante el sueño, visualizándose descargas generalizadas de punta-onda/polipunta onda y, a veces, fotosensibilidad. La respuesta a valproico o a levetiracetam suele ser la norma, pero existe un porcentaje apreciable (10-20%) de pacientes que presentan años después déficit cognitivo leve o trastornos de neuroconducta. Es también posible la recurrencia de crisis en infancia y adolescencia, con otros tipos de epilepsias generalizadas.
Epilepsia de la infancia con crisis focales migratorias
Se trata de una encefalopatía epiléptica que debuta en el primer semestre de vida. Suele tener origen genético (KCNT1, TBC1D24). Las CE se caracterizan por ser crisis focales múltiples de diversa semiología y localización, en particular motoras y autonómicas, con generalización en la mitad de los pacientes. Existe cierta complejidad en el EEG, apareciendo en el interictal puntas multifocales, pero en el ictal, se aprecian múltiples focos independientes, con actividad continua y migrante, conformando status epilépticos. La evolución es hacia una epilepsia farmacorresistente, con retraso global en desarrollo.
Síndrome de Dravet
El síndrome de Dravet (SD) o epilepsia mioclónica severa de la infancia es una encefalopatía epiléptica de inicio temprano, caracterizada por epilepsia refractaria asociada a crisis febriles (CF) atípicas y con una cronología característica. En aproximadamente el 80% de los pacientes, el síndrome es causado por mutaciones patogénicas en el gen SCN1A, pero pueden existir otros genes implicados. En realidad, existe un espectro clínico amplio asociado al gen SCN1A. Las mutaciones patogénicas de este gen pueden producir trastornos convulsivos, que van desde las CF aisladas a combinaciones de estas con CE afebriles, de tipo: mioclónico, focales, crisis tónico-clónicas (epilepsia con CF plus), etc. Las diferentes formas clínicas presentan implicación en el desarrollo, que varía, sobre todo, en función de la severidad del cuadro epiléptico. El debut del síndrome de Dravet (y de la mayoría de epilepsias relacionadas con SCN1A) se produce en un lactante en el primer año de vida, en relación con sus primeros procesos febriles, en forma de CF atípica (por edad, pero también usualmente por focalidad y duración). En la actualidad y en nuestro ámbito, es usual que el primer proceso febril de un lactante sea producto de una vacunación, en particular, tosferina y meningococo B, debido al potencial pirético de ambas. La discusión de la indicación de vacunación en pacientes con CF, sospecha de síndrome de Dravet, etc., rebasa los límites de este artículo, pero el lector debe recordar que la aparición de CF prolongada o/y asimétrica en primeras vacunas de un lactante, es de sospecha para desarrollo de epilepsias relacionadas con SCN1A. En estos primeros meses no existe regresión o detención en desarrollo, pero a partir del año de vida este empieza a ser evidente a la vez que debutan diversos tipos de CE afebriles; las más frecuentes son: tónico-clónicas o clónicas, generalizadas o focales con focalidad alternante, mioclónicas (segmentarias, multifocales o generalizadas, aisladas o en brotes) y crisis de ausencia (generalmente atípicas, con mioclono asociado frecuente). Entre los signos neurológicos suele ser frecuente la ataxia y los problemas cognitivos. El EEG interictal no suele ser específico y, a menudo, es normal a pesar de la elevada frecuencia de la crisis. En general, a partir de los 5-7 años de edad, existe una fase de estabilización, sin deterioro, pero con discapacidad intelectual ya establecida y con persistencia de CE, aunque menos frecuentes. El síndrome de Dravet muestra aproximadamente, un 7% de casos de muerte súbita inesperada. Es una epilepsia farmacorresistente, pero existe margen terapéutico para minimizar número de crisis y deterioro. Deben usarse combinaciones de: valproato, topiramato, benzodiacepinas, estiripentol (y, posiblemente, Cannabidiol en el futuro inmediato); es muy relevante el papel de la dieta cetogénica en este síndrome, de manera específica.
Infancia
Las epilepsias pediátricas más frecuentes debutan en este periodo: epilepsia focal rolándica y ausencias.
Epilepsias focales benignas (autolimitadas) de la infancia
Las epilepsias focales benignas de la infancia (EFBI) son un grupo de epilepsias caracterizadas por ser edad dependientes en debut y curso, con síntomas ictales que sugieren focalidad específica, relacionadas a menudo con el sueño; son idiopáticas, en el sentido de mostrar algunas variantes genéticas que favorecen su aparición, pero sin un factor etiológico único. En casos claros con escaso número de crisis sin generalización y no amenazantes, se intenta no administrar tratamiento; además de los riesgos directos del fármaco que se emplee, existe potencial favorecedor de transformación de EFBI en cuadros de mayor severidad con status eléctrico nocturno y regresión en desarrollo. En todos los casos, la deprivación de sueño o la transición vigilia-sueño son activadores claros en el EEG y debe indicarse esta maniobra si la sospecha es específica. En todos estos casos de EFBI, existe cierta controversia respecto a la realización de RMC, ya que pueden existir epilepsias secundarias a lesiones cerebrales ubicadas en áreas habituales de EFBI; en general, si existe: atipicidad, dudas, episodios frecuentes, etc., debe realizarse resonancia. Es interesante señalar que puede existir comorbilidad entre diferentes EFBI, de manera evolutiva. Las principales EFBI son las que se citan a continuación.
Epilepsia occipital de la infancia de inicio temprano (síndrome de Panayiotopoulos)
Se trata de una EFBI de debut antes de los 5 años, con crisis nocturnas de una semiología, a menudo, difícil de sospechar, ya que predominan: vómitos y signos vegetativos (palidez, síncopes, sialorrea…). Puede existir impresión de alteración del nivel de conciencia, así como desviación ocular y torsión cefálica, junto a más raros movimientos clónicos focales. Son crisis largas, en la mitad de ocasiones, rondan los 30 minutos, pero poco frecuentes; la mitad de los pacientes solo muestran 1 o 2 crisis en toda su vida. En el EEG con deprivación suelen hallarse las puntas occipitales características. En general, el cuadro remite 2-4 años después del debut.
Epilepsia occipital de la infancia de inicio tardío (síndrome de Gastaut)
En este caso, el debut es más tardano, en torno a los 7-9 años; existen síntomas visuales claros, como amaurosis y alteraciones de movilidad ocular. Frecuentemente, se acompaña de cefalea migrañosa, breve y, a menudo, clonias focales hemicorporales que pueden generalizar. Al contrario que en el anterior cuadro, suele haber muchas crisis y breves, lo que unido al potencial de generalización lleva a tratamiento más frecuentemente. El EEG es similar al Panayiotopoulos.
Epilepsia benigna de la infancia con puntas centrotemporales (epilepsia “rolándica”)
Se trata del síndrome epiléptico infantil más frecuente de todos. Aproximadamente, uno de cada cinco niños epilépticos tiene este diagnóstico. El debut es amplio, entre 3 años a preadolescencia, pero la mayor parte de pacientes debutan a los 6-8 años. La mayoría de crisis ocurren en sueño o en transiciones sueño-vigilia, sobre todo, cuando la noche previa ha existido una reducción de número de horas de sueño habitual, lo que aumenta la posibilidad de crisis en: viajes, eventos festivos o estresantes. La semiología es característica: espasmo hemifacial clónico, con sialorrea e imposibilidad para hablar, pero preservación de conciencia (al tocarle el paciente mira al observador y da la impresión de querer hablar, pero no puede hacerlo); pueden aparecer clonias de hemicuerpo homolateral y, no raramente, generalización. Cuando la rapidez del cuadro o la no observación no permiten objetivar el inicio focal, es importante el hallazgo de paresia facial y/o segmentaria postictal. A pesar de la supuesta benignidad del cuadro, existe evidencia respecto a que los pacientes con mayor persistencia de paroxismos nocturnos en EEG muestran déficits neuropsicológicos y alteraciones del comportamiento. En el EEG suele aparecer actividad de puntas en región centro-temporal uni o bilateral.
Síndrome de Landau Kleffner
Se trata de una encefalopatia epiléptica relacionada posiblemente con las EFBI, pero en el que el papel de mutaciones en gen GRIN2A son determinantes. Los pacientes tipo presentan, entre los 3 a 6 años, afasia receptiva y expresiva, partiendo de una situación de normalidad en desarrollo/lenguaje previa a debut. Puede existir también problemática conductual comórbida. Existe un amplio repertorio de crisis, en particular, focales motoras y generalizadas. El EEG en vigilia es similar a EFBI, pero es característica la actividad paroxística continua en sueño NREM, con status eléctrico.
Síndrome de Lennox-Gastaut
Se trata de una encefalopatía epiléptica de debut habitual entre los 2 y 6 años. Siempre existe una situación de patología neurológica o de desarrollo más o menos severa y, a menudo, se trata de una situación evolutiva de un paciente que previamente mostraba epilepsia de difícil control u otra encefalopatía epiléptica. El paciente afecto de este síndrome presenta diversos tipos de crisis sobre una base de regresión en desarrollo. Los tres tipos de crisis más típicos son: las tónicas, las atónicas con caída y las ausencias atípicas. Pueden también aparecer crisis generalizadas tónico-clónicas, mioclónicas, etc. Las crisis tónicas suelen ser: axiales, en extensión, acompañadas de apnea y revulsión ocular, y son más frecuentes en la transición vigilia-sueño. En el EEG suele existir punta-onda lenta interictal, con actividad paroxística rápida durante el sueño. Se trata de un síndrome caracterizado por la refractariedad y en el que el papel de los tratamientos no farmacológicos debe ser considerado, en particular, dieta cetogénica.
Epilepsia de ausencias infantiles
Es una epilepsia de tipo generalizado idiopático, que acontece en niños sin patología de desarrollo. Los antecedentes de epilepsia generalizada en la familia y/o crisis febriles no son infrecuentes. Es la segunda epilepsia más frecuente en la infancia junto a la EFBI rolándica. Ocurre en niños sin patología de desarrollo, con cierto predominio femenino. Suelen debutar en torno a los 6-7 años de edad, con crisis típicas de desconexión completa del medio de inicio y final brusco, habitualmente entre 5-10 segundos, sin atonía. Es frecuente que se acompañe de leve supraversión ocular que acompaña a mínimo parpadeo o cierre parcial ocular. La hiperventilación produce crisis en la mayoría de pacientes no tratados. Se trata de una epilepsia que tarda en ser diagnosticada, ya que puede pasar desapercibida con facilidad o no atribuirse patogenicidad a los episodios. El EEG interictal es normal, pero en un registro de cierta longitud, sobre todo, con hiperventilación, aparecen las características descargas de punta-onda generalizada, sincrónicas y simétricas a 2,5-3 Hz. Respecto al tratamiento, hoy en día, suele preferirse la etosuximida sobre el valproico, con uso más inhabitual de lamotrigina y levetiracetam. Suele haber buena respuesta y, en la mayoría de casos, tras retirada de fármacos a los 2-3 años de tratamiento, no existe recurrencia.
Epilepsia con ausencias mioclónicas
Es una epilepsia generalizada, con debut habitual en torno a los 6-8 años. Muestra crisis de ausencia, pero con mioclonías bilaterales y difusas, prolongadas (entre 10 segundos y el minuto), por lo que el paciente va elevando los brazos según acontecen las mioclonias, dando aspecto de crisis tónica que no es tal. Suelen existir también crisis tónico-clónicas. En el EEG hay descargas punta-onda generalizadas. El control de crisis es complicado, con un porcentaje elevado de regresión cognitiva significativa.
Epilepsia con crisis mioclono atónicas (síndrome de Doose)
Es una epilepsia infrecuente, con debut a los 3-4 años, en los que en general, desde una base de desarrollo normal, muestran crisis febriles de debut tardío, que son seguidas de crisis mioclónicas-atónicas, con mioclonía axorrizomélica, que es seguida de caída al suelo por pérdida de tono. Suele haber comorbilidad con crisis tónico-clónicas generalizadas y con ausencias. No existen genes asociados conocidos. En una mayoría de pacientes existe regresión, con deterioro cognitivo. El EEG es poco específico, con habituales brotes breves de punta-onda a 2-5 Hz y complejos de polipunta-onda. Es una epilepsia con tendencia a refractariedad.
Epilepsia frontal nocturna autosómica dominante
Se trata de una epilepsia de base genética, habitualmente de transmisión dominante, con cierta heterogeneidad intrafamiliar. El trastorno es de cierta complejidad; ya que, en muchas ocasiones, a la epilepsia le acompaña distonía episódica y coreoatetosis nocturna en sueño NREM. La edad de debut es amplia, siendo más frecuente en torno a los diez años. Las crisis epilépticas suelen ser casi exclusivamente: nocturnas, motoras, breves, de escasos segundos, que pueden ser de difícil diagnóstico; en las crisis más largas aparecen posturas tónicas/distónicas asimétricas, junto movimientos complejos (pedaleo, coreoatetosis…). Muy habitualmente, se acompañan de miedo o vivencia desagradable, lo que frecuentemente conduce al diagnóstico diferencial con parasomnias (pesadillas, terrores nocturnos). Es habitual, comorbilidad cognitiva leve o patología psiquiátrica.
Adolescencia
El síndrome epiléptico más común en el adulto, la epilepsia mioclónica juvenil, debuta en este periodo.
Epilepsia mioclónica juvenil
Si se consideran tanto infancia como periodo de adulto, se trata de la epilepsia más frecuente. La edad de debut es amplia, con pacientes desde los 5-6 años, pero el pico se produce a los 12-18 años. Hay predominio femenino. Es una epilepsia generalizada idiopática, en la que existen, sobre todo, crisis mioclónicas distales al despertar en extremidades superiores y crisis tónico-clónicas más adelante, junto a ausencias que son frecuentemente infradiagnosticadas. El EEG suele mostrar polipunta-onda generalizada y, comúnmente, respuesta fotoparoxística. La mejor respuesta se obtiene con valproato, pero dada la recomendación de evitación de dicho fármaco en mujeres, debe recurrirse a menudo a: levetiracetam, benzodiacepinas, etc. En la mayoría de pacientes debe mantenerse tratamiento de por vida.
Epilepsia de ausencias juvenil
Es una epilepsia generalizada, de debut al principio de la adolescencia, en forma de ausencias, apareciendo con posterioridad, crisis tónico-clónicas. En el EEG aparecen descargas generalizadas de punta-onda a 3-4 Hz. La respuesta a fármacos es peor que en ausencias típicas y suele requerirse tratamiento mantenido de por vida.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Aaberg KM, Gunnes N, Bakken IJ, Søraas CL, Berntsen A, Magnus P, et al. Incidence and Prevalence of Childhood Epilepsy: A Nationwide Cohort Study. Pediatrics. 2017; 139: e20163908.
2.*** ILAE: guidelines: definition and classification. En: https://www.ilae.org/guidelines/definition-and-classification. Acceso el 15 de julio de 2020.
3.*** Fisher R, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58: 522-30.
4. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014; 55: 475-82.
5. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010; 51: 1069-77.
6.** Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58: 512-21.
7. Dash D, Sharma A, Yuvraj K, Renjith A, Mehta S, Vasantha PM, et al. Can home video facilitate diagnosis of epilepsy type in a developing country? Epilepsy Res. 2016. 125: 19-23.
8.** Panayiotopoulos CP. General aspects on the diagnosis of epileptic seizures and epileptic syndromes. En: Panayiotopoulos CP Ed. A clinical guide to Epileptic Syndromes and their treatment. Oxford. Bladon Medical Publishing; 2002. p. 1-35.
9. Hauser WA Rich SS, Annegers JF, Anderson VE. Seizure recurrence after a 1st unprovoked seizure: an extended follow-up. Neurology. 1990; 40: 1163-70.
10. Bernasconi A, Cendes F, Theodore WH, Gill RS, Koepp MJ, Hogan RE, et al. Recommendations for the use of structural magnetic resonance imaging in the care of patients with epilepsy: A consensus report from the International League Against Epilepsy Neuroimaging Task Force. Epilepsia. 2019; 60: 1054-68.
11.*** Arzimanaglou A, Duchowny MS. Epilepsy and other seizure disorders. En: Arzimanaglou, O´Hare, Johsnton, Ouvrier Eds. Aicardi´s Diseases of the Nervous System in childhood. 4th ed. MacKeith Press. 2018. p. 845-998.
12.*** Panayiotopoulos CP. A clinical guide to Epileptic Syndromes and their treatment. Panayiotopoulos CP Ed. Oxford. Bladon Medical Publishing. 2002.
13. Osborne JP, Edwards SW, Dietrich-Alber F, Hancock E, Johnson AL, Kennedy CR, et al. The underlying etiology of infantile spasms (West syndrome): Information from the International Collaborative Infantile Spasms Study (ICISS). Epilepsia. 2019; 60: 1861-9.
Bibliografía recomendada
- ILAE: guidelines: definition and classification. En: https://www.ilae.org/guidelines/definition-and-classification. Acceso el 15 de julio de 2020.
La aproximación al estudio de la epilepsia requiere el conocimiento y lectura previas de las guías de clasificación y consenso internacionales. En la propia página web, se ofrece traducción al español de la mayoría de documentos más importantes, así como material para exposiciones y clases.
- Fisher R, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58: 522-30.
Aunque puede ser ardua la clasificación de los tipos de crisis, la profundización en el estudio de la epilepsia requiere del conocimiento de los tipos reconocidos como tales.
- Arzimanaglou A, Duchowny MS. Epilepsy and other seizure disorders. En: Arzimanaglou, O´Hare, Johsnton, Ouvrier Eds. Aicardi´s Diseases of the Nervous System in childhood. 4th ed. MacKeith Press. 2018. p. 845-998.
Revisión completa y actualizada de la epilepsia pediátrica dentro del que es probablemente el texto de Neurología pediátrica de referencia.
- Panayiotopoulos CP. A clinical guide to Epileptic Syndromes and their treatment. Panayiotopoulos CP Ed. Oxford. Bladon Medical Publishing. 2002.
Se trata de un libro excelente, accesible, en cuanto a la descripción clínica y la correlación EEG, orientado a la clínica, escrito por uno de los mayores expertos mundiales en Epileptología y recientemente fallecido. Dada la fecha de su edición, las importantes aportaciones de la genética no se encuentran por desgracia actualizadas.
| Caso clínico |
|
Un niño de siete años tras acudir a la Cabalgata de Reyes Magos, el 6 de enero se levanta a las 5:00 de la mañana para abrir sus regalos. Esa tarde tras la comida familiar, se queda dormido en el sillón. De modo súbito, parece despertarse, se incorpora y dirige la mirada a sus padres, mientras babea, sin poder hablar y con sacudidas en lado facial derecho y brazo del mismo lado. Unos segundos después, el paciente pierde la conciencia y comienza con sacudidas de ambos brazos y piernas durante un par de minutos, mostrándose a continuación con depresión postcrisis. Es transportado a Urgencias de su hospital de referencia, donde es evaluado. Al llegar, está despierto, con cierta agitación psicomotriz, cojea de forma ostensible y no es capaz de escribir su nombre con su mano dominante, la derecha.
|
