 |
| Temas de FC |
J. Campistol Plana
Catedrático de Pediatría Universidad de Barcelona. Servicio Neuropediatría. Hospital Sant Joan de Dèu. Universitat de Barcelona
| Resumen
Los Trastornos Paroxísticos no Epilépticos (TPNE) son episodios de aparición brusca y de breve duración que imitan a una crisis epiléptica, originados por una disfunción cerebral de origen heterogéneo y, a diferencia de la epilepsia, no obedecen a una descarga neuronal excesiva. Su incidencia es mucho más elevada que la epilepsia y son más comunes en los primeros años de vida. La inmadurez del sistema nervioso central en la infancia favorece que, en este período, las manifestaciones clínicas sean mucho más floridas y diferentes de otras edades. La función del pediatra es conocer estos trastornos e intentar identificarlos. El primer paso para un diagnóstico correcto, es establecer si este primer episodio corresponde a una crisis epiléptica o puede tratarse de un primer episodio de TPNE. Es importante seguir un protocolo de diagnóstico, analizando: antecedentes, factores desencadenantes y detalles de cada episodio, examen físico, así como conocer y tener experiencia en estos trastornos. Solamente debemos proceder a los exámenes complementarios básicos en caso de duda o para confirmación diagnóstica. |
| Abstract
The immaturity of the central nervous system in childhood contributes to clinical manifestations that differ greatly from those in other periods of life. Non-epileptic Paroxysmal Disorders (NEPD) are defined as episodes of sudden onset and of short duration that imitate an epileptic seizure, originated by brain dysfunction of heterogeneous origin and, unlike epilepsy, are not due to excessive neuronal discharge. Their incidence is much higher than epilepsy and they are more common in the first years of life. The role of the Pediatrician is to have knowledge of these disorders, and try to identify them. The first step for a correct diagnosis is to establish if this first episode corresponds to an epileptic seizure or if it may be a first episode of NEPD. It is important to follow a diagnostic protocol, analyzing the history, triggers, details of each episode, physical examination, and to have experience with these disorders. Only proceed to basic complementary tests in case of doubt or for diagnostic confirmation. |
Palabras clave: Trastornos paroxísticos no epilépticos; Epilepsia; Métodos diagnósticos; Genética; Tratamiento.
Key words: Non-epileptic paroxysmal disorders; Epilepsy; Diagnostic methods; Genetics; Treatment.
Pediatr Integral 2020; XXIV (7): 383 – 392
Trastornos paroxísticos no epilépticos en la infancia
Introducción
Los Trastornos Paroxísticos no Epilépticos (TPNE) se definen como: episodios que se van repitiendo con cierta frecuencia, más o menos estereotipados, y que remedan a una crisis convulsiva, pero que en realidad no son epilépticos(1). Su aparición es generalmente brusca y de breve duración, originados por una disfunción cerebral de origen diverso y, a diferencia de la epilepsia, no obedecen a una descarga neuronal excesiva(2).
Constituyen un grupo heterogéneo de manifestaciones muy polimorfas desde el punto de vista semiológico, en las que se producen cuadros clínicos diversos que pueden simular una crisis epiléptica y que están causados por procesos fisiológicos, psicológicos o de origen desconocido(3).
Una crisis epiléptica se define como: un evento clínico ya sea motor, comportamental, sensorial o sensitivo, como resultado directo de un cambio o una descarga en la actividad eléctrica cerebral. La diferencia es pues muy sutil y, en ocasiones, es difícil de demostrar; de ahí, la facilidad con que pueden confundirse ambos trastornos(4,5).
Incidencia
El 15% de niños menores de 15 años han padecido algún tipo de trastorno paroxístico. La incidencia de los TPNE en la infancia es 10 veces más elevada que la de los trastornos paroxísticos epilépticos (TPE) (10:1). Los tratados de epilepsia son muy numerosos, y muy escasos los que analizan los TPNE(2). El listado de los TPNE que pueden simular una crisis epiléptica es enorme (Tabla I).
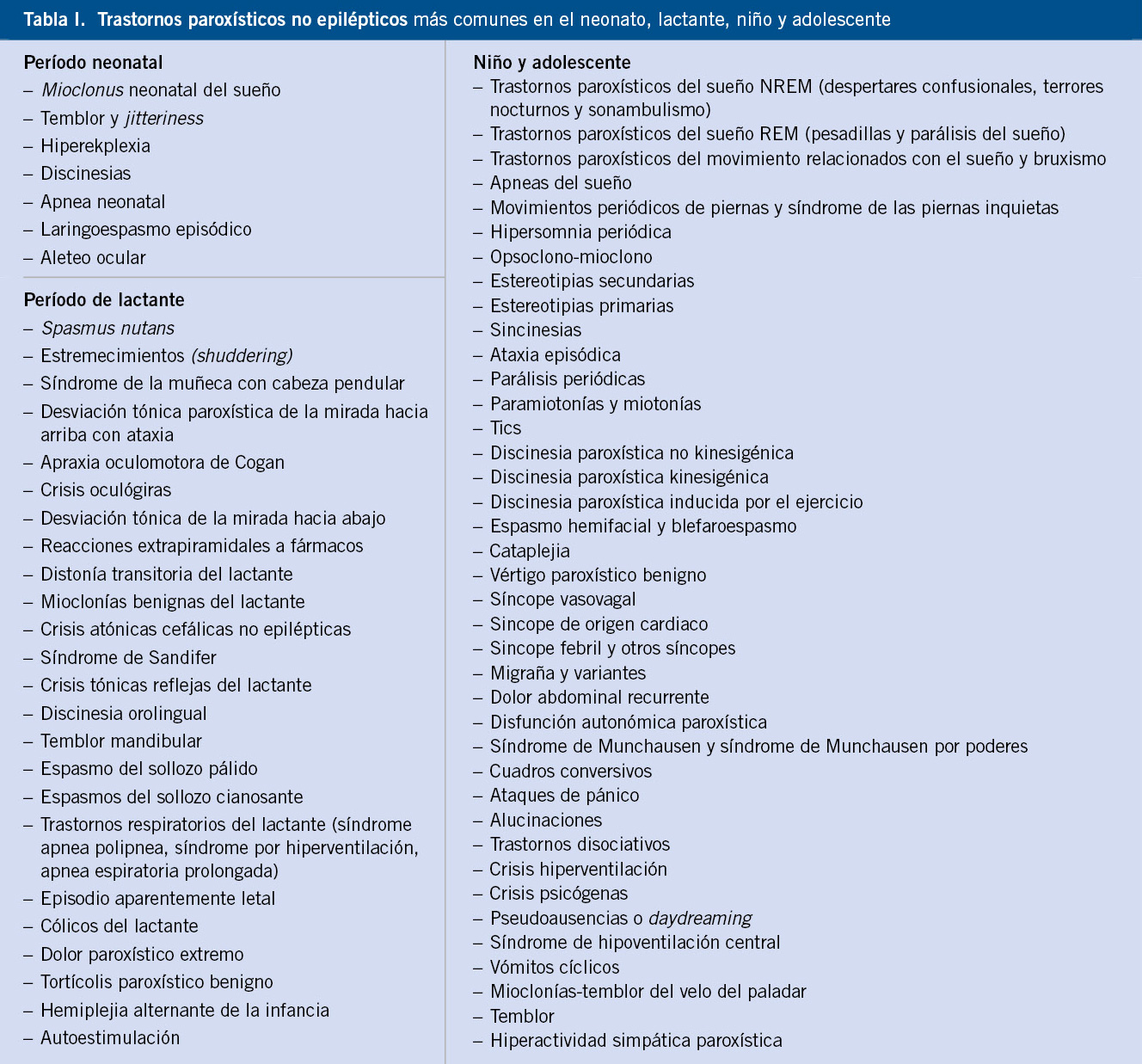
Muchos de estos trastornos son edad-dependientes y, en la mayoría de casos, suelen desaparecer sin dejar secuelas en los primeros años de vida(2,5).
Causas de error en el diagnóstico
Existen varios factores responsables de estos errores tan frecuentes en la práctica diaria y que están resumimos en la tabla II(2,5,6).

La inquietud que generan en el paciente y, muy especialmente, en los familiares que han presenciado el episodio, es enorme. Mayor angustia genera no diagnosticarlos o etiquetarlos erróneamente de epilepsia, cuando en realidad no lo son. Someterlos a medicación antiepiléptica cuando no la necesitan, a muchos exámenes complementarios que no precisan, o a las limitaciones propias de la epilepsia cuando no lo requieren. Todo ello forma parte de los errores habituales que se pueden cometer cuando no se analizan en profundidad o se desconocen los TPNE de la infancia. Paradójicamente, es menos común que un trastorno epiléptico se diagnostique erróneamente como trastorno paroxístico no epiléptico.
Pauta de actuación frente a un cuadro paroxístico
Es importante disponer de una pauta de estudio frente a un paciente con un cuadro paroxístico y que pasa por: un interrogatorio completo, una adecuada valoración de los antecedentes, la exploración física completa y los exámenes complementarios, dirigidos siempre en función del cuadro clínico (Tabla III)(2,3,5,6).

Determinar el origen epiléptico no siempre es tan sencillo y puede requerir, en caso de duda, una prudente espera, en ocasiones, de un registro vídeo-EEG, idealmente un registro vídeo-EEG durante el episodio o la solicitud de otras exploraciones complementarias(2,3,5).
Clasificación de los TPNE
Existen muchas clasificaciones de los trastornos paroxísticos no epilépticos, pero preferimos agruparlos por edades de presentación y, dentro de cada edad, relacionarlos: con el movimiento, con la hipoxia, con el dolor, con el sueño, con trastornos psicógenos y, para terminar, un amplio grupo de miscelánea (Tabla I). No podemos repasar en este capítulo todos y cada uno de los TPNE, existen algunas monografías al respecto, donde se detallan todo los TPNE(2). Nos limitaremos a revisar algunos de los TPNE más comunes en la práctica diaria, valorando sus características principales y diferenciales, fisiopatología y opciones terapéuticas.
TPNE en el período neonatal
Relacionados con el movimiento
Temblor y tremulaciones
El temblor es el TPNE más frecuente en el período neonatal. Consiste en movimientos rítmicos oscilatorios de igual amplitud. Se originan por inmadurez del sistema interneuronal inhibidor espinal. Pueden ser muy sutiles, de alta frecuencia (más de 6 Hz) y baja amplitud (menos de 3 cm), o pueden ser groseros, de baja frecuencia y elevada amplitud. Las tremulaciones (jitteriness) se manifiestan por un temblor recurrente.
El temblor puede afectar a una extremidad o al mentón, puede ser simétrico o asimétrico, y cede en general al sujetar la zona afecta. Aparece espontáneamente y suele aumentar con los estímulos, el llanto o el frío.
Más del 70% de los neonatos sanos presentan temblor de tipo sutil en los tres primeros días de vida, mientras que las tremulaciones afectan a casi la mitad de los neonatos. Ambas situaciones son más frecuentes en neonatos irritables y se consideran fisiológicos o benignos. Los temblores patológicos, habitualmente son movimientos más amplios y se observan en la encefalopatía hipóxico-isquémica, y pueden confundirse con crisis epilépticas. En este caso, junto con los temblores, se puede asociar: hiperreflexia, síndrome de hiperexcitabilidad neonatal y afectación del sensorio. También se observan en situaciones de: hipoglucemia, hipocalcemia, sepsis, hemorragia intracraneal, hipotermia, hipertiroidismo y en hijos de madres con abuso de sustancias o que han sido tratadas durante embarazo con inhibidores de la recaptación de serotonina. Los temblores pueden confundirse, con más frecuencia, con crisis epilépticas, especialmente en pacientes con problemas perinatales. En este caso, el EEG crítico y el vídeo-EEG ayudan al diagnóstico diferencial(1,2).
Respuesta exagerada al estímulo (reacción de sobresalto)
En el neonato existe una reacción de sobresalto normal ante un estímulo inesperado, que consiste en una mueca facial y parpadeo, seguido de flexión de tronco. La respuesta, cuando es exagerada, interfiere el estado basal del neonato y puede producir incluso apnea.
Este proceso es preciso diferenciarlo de la hiperekplexia, que se caracteriza por una reacción de sobresalto exagerada a los estímulos auditivos, táctiles e incluso visuales. Se asocia, además, rigidez muscular y mioclono nocturno.
El neonato presenta respuestas de sobresalto exageradas que se caracterizan por: espasmo tónico generalizado, con flexión tónica de extremidades y tronco, y cierre de puños. La apnea es posible durante el espasmo, debido a la rigidez de la pared torácica. El baño, el despertar o un estímulo auditivo o táctil, pueden desencadenar los episodios. Esta respuesta exagerada, se induce también fácilmente, golpeando el puente nasal o la glabela, lo que permite diferenciarlas de las crisis mioclónicas. El trastorno puede ser fácilmente confundido con: crisis mioclónicas, tetania neonatal o el síndrome del bebé-rígido congénito (stiff-baby). La hiperekplexia puede ser esporádica o transmitirse con un patrón autosómico dominante, con expresión variable. Se debe a mutaciones en el gen CLRA1, localizado en el cromosoma 5p33-35. Este defecto causa una dificultad en el paso del cloro a través de la subunidad α1 o β1 del receptor de la glicina estricnina-sensible. La maniobra de Vigevano es útil para abortar el episodio de sobresalto que dificulta la respiración, al forzar la flexión del neonato, presionando la cabeza hacia las rodillas.
Suelen responder bien al clonazepam (0,1-0,2 mg/kg/día). Superado el período neonatal, la enfermedad persiste varios años, con riesgo de muerte súbita por apnea central por afectación a nivel de tronco cerebral y/o por apnea durante el espasmo tónico.
La rigidez tiende a desaparecer antes de los 3 años, pero las caídas al suelo a consecuencia del espasmo y la respuesta exagerada al sobresalto, persisten mucho tiempo incluso en la edad adulta(2,3,5,6).
Movimientos coreico-distónicos de extremidades, boca y lengua
Pueden observarse durante el primer mes de vida en niños prematuros con displasia broncopulmonar grave. Estos episodios empeoran con la dificultad respiratoria y mejoran con el sueño. Se atribuyen a una hipoxia crónica de los ganglios basales. La evolución neurológica posterior no es muy favorable por la patología de base. Otros movimientos anormales del neonato son: el temblor, la agitación o las mioclonías.
Crisis sutiles sin correlato EEG
Se definen por la presencia de alteraciones en la conducta, de la actividad motora o de sus funciones autonómicas, sin que se asocien a anomalías epileptiformes en el EEG crítico. Estos episodios no se acompañan de movimientos tónicos, clónicos ni mioclónicos, pero sí de movimientos anormales de los ojos (mirada lateral mantenida, nistagmo) y movimientos oro-bucales (succión, masticación o protusión lingual). No responden a los anticonvulsivos y, en ocasiones, se asocian a verdaderas crisis, por lo que existe dificultad para discernir realmente cuando se trata de verdaderas crisis epilépticas. En estos casos, se requiere un estudio vídeo-EEG prolongado. Se cree que estos episodios representan un fenómeno de liberación del tronco cerebral(3).
Relacionados con el sueño
Mioclonus del sueño
El mioclono se define por un brusco y breve movimiento de un miembro causado por una contracción muscular. Puede ser focal, afectando a una parte del cuerpo o generalizado y, a diferencia del temblor, es: irregular, arrítmico y de mayor amplitud. Su origen puede situarse en: córtex, tronco o médula espinal(2). El mioclono epiléptico es infrecuente en el neonato, no así el mioclono patológico que se asocia a una complicación neurológica importante (hemorragia intraventricular, encefalopatía hipóxico-isquémica, encefalopatía metabólicas), como expresión de un fenómeno de liberación de tronco cerebral por pérdida de la inhibición cortical lesionada.
En el mioclono nocturno neonatal benigno, las contracciones mioclónicas solo se observan durante el sueño NREM. Es un trastorno relativamente frecuente y se confunde con crisis epilépticas y con estado epiléptico con mucha frecuencia, cuando se presentan de forma muy repetida. Las mioclonías son: breves, multifocales, repetitivas y arrítmicas, afectan a cualquier extremidad, especialmente a los miembros superiores, respetan la cara y ceden inmediatamente al despertar al neonato. En caso de duda sera necesario un vídeo-EEG de sueño
Se presentan en accesos y pueden persistir durante 20-30 minutos(5-7). Se inician en los primeros días de vida y suelen ceder espontáneamente antes de los 4 meses de vida. El uso de fenobarbital puede empeorarlas y la normalidad en el EEG crítico descarta un origen epiléptico. Probablemente, se producen por una inmadurez de las vías serotoninérgicas a nivel del tronco cerebral.
Relacionados con la hipoxia
Apneas
La apnea consiste en una interrupción del flujo aéreo, con una duración superior a los 20 segundos y con o sin disminución de la frecuencia cardíaca. También se consideran apneas, las detenciones del flujo aéreo más cortas, pero con repercusión cardiocirculatoria. Se distinguen tres tipos básicos de apnea, según su mecanismo de producción: centrales, obstructivas y mixtas. Se pueden dividir, por la etiología, en: primarias o idiopáticas, que tienen relación fundamental con la madurez; y secundarias o sintomáticas, que reflejan la presencia de una patología de base(1,2).
El episodio clínico de apnea incluye: cianosis, palidez e hipotonía; aunque puede aparecer de forma aislada y habitualmente, se acompaña de bradicardia y desaturación periférica de oxihemoglobina. El origen de los episodios de apnea se relaciona con varios factores: impulso respiratorio central deficiente, inmadurez de los quimiorreceptores centrales del neonato, alteraciones mecánicas de la vía aérea por obstrucción o colapso de esta, estimulación de reflejos vagales a través de maniobras terapéuticas, como la colocación de sonda nasogástrica, maniobras de aspiración nasotraqueales o el reflujo gastroesofágico.
Todo recién nacido por debajo de las 34 semanas con apneas o que acumule factores de riesgo, debe ser sometido a monitorización. Es importante, en caso de duda, la utilización de un monitor de función cerebral o un registro vídeo-EEG para establecer si las alteraciones del flujo y ritmo respiratorio detectadas están en relación con crisis epilépticas(3,6).
El primer objetivo del tratamiento es prevenir la aparición de nuevos episodios. La estimulación táctil suele ser suficiente para corregir la mayoría de los casos en los que la apnea se presenta como un episodio aislado. En los problemas posicionales de la vía aérea superior, se debe considerar la corrección postural. Cuando la apnea se acompaña de signos persistentes de cianosis o de bradicardia, se recurrirá a la administración de O2 con presión positiva. Si los episodios recurren varias veces al día, se recomienda iniciar tratamiento farmacológico con cafeína y, si persisten, se puede utilizar apoyo ventilatorio con CPAP.
Miscelánea
Laringoespasmo episódico
Es una contracción involuntaria de la musculatura laríngea, que produce una obstrucción parcial durante la inspiración. Esto puede causar la muerte en el niño o una hipoxia cerebral. En neonatos, el laringoespasmo ocurre generalmente tras una extubación o durante la intubación, pero también puede relacionarse con: hipocalcemia, reflujo gastroesofágico, crisis convulsiva, síndrome miasténico congénito o miotonía.
El cuadro clínico característico del laringoespasmo episódico neonatal se caracteriza por: episodios de rigidez generalizada con cianosis y bradicardia con o sin pérdida de conocimiento, y crisis de apnea durante la primera semana de vida. Pueden presentar episodios de estridor asociados y confundirse con laringomalacia. También se puede manifestar como: episodios de sollozo transitorios con rigidez muscular generalizada y palidez facial durante el llanto. La exploración entre los episodios puede ser normal, presentar un ligero estridor o una discreta rigidez.
El laringoespasmo episódico neonatal se ha relacionado con mutaciones del gen SCN4A en el cromosoma 17, que codifica la subunidad alfa del canal de sodio dependiente del voltaje del músculo esquelético Nav 1.4. Los pacientes tienen buena respuesta a los bloqueantes de canales de sodio: carbamacepina (10-40 mg/kg/día) o mexiletina (15 mg/kg/día)(2).
TPNE en el lactante
Relacionados con el movimiento
Mioclonías benignas del lactante
Suelen debutar entre los 3 y 9 meses de vida, aunque también se han descrito en el período neonatal.
El lactante, por lo demás normal, presenta, en general, al despertar y bruscamente, repetidas contracciones mioclónicas en flexión de cuello y extensión, y abducción de los miembros superiores en salvas y que recuerdan a los espasmos epilépticos del síndrome de West. Se presentan en vigilia y varias veces al día.
Pueden también simular una mioclonía generalizada. El vídeo-EEG en vigilia y sueño es normal, y no existe deterioro en el desarrollo a diferencia del síndrome de West. Ceden espontáneamente hacia los 9 meses y no precisan tratamiento. Aunque se han descrito con más frecuencia en niños sobrestimulados, se desconoce el origen del trastorno(1,3,7).
Crisis tónicas reflejas del lactante
Se presentan entre los 2 y 3 meses de edad con: episodios de contracción tónica y extensión de las 4 extremidades, apnea y cianosis, y congestión con rubicundez facial durante 3-10 segundos.
Se desencadena por estímulos táctiles o cambios posicionales, solo cuando el niño está en vigilia y muchas veces cuando está sujetado por un adulto en posición vertical o cuando es desplazado en dirección céfalo-caudal. Es más frecuente en lactantes irritables y, en ocasiones, aparece en salvas durante 2-3 días o, a veces, repite periódicamente. Ceden espontáneamente pocos meses después. El EEG ictal e interictal es normal. Se desconoce su origen(2).
Desviación tónica paroxística de la mirada hacia arriba con ataxia
El cuadro se inicia entre los 3 meses y los 2 años, y consiste en episodios prolongados (segundos, horas o días) de desviación continua o episódica de los ojos hacia arriba, con ataxia asociada.
Durante el episodio, el intento del niño para fijar la mirada induce un nistagmo repetido en dirección hacia abajo. Los síntomas desaparecen con el sueño y se incrementan con el cansancio o las infecciones. El cuadro clínico, en general, suele remitir espontáneamente en unos años y, casi la mitad de los casos, evolucionan con retraso del desarrollo y lenguaje. Se desconoce la causa, la neuroimagen y EEG son normales, existen cuadros familiares y otros relacionados con infecciones. Algunos casos mejoran con L-dopa(8).
Estereotipias
Se trata de conductas motoras repetitivas (orofaciales, cefálicas o de miembros superiores con movimientos de aleteo), que se presentan de forma rítmica y continuada en niños, por lo demás normales, especialmente cuando se sienten a gusto con el juego o en situaciones de excitación o estrés(1,2). Suelen comenzar al año, y remiten entre los 2 y 3 años. Son más acentuados y persistentes en pacientes con patología neurológica (discapacidad intelectual, TEA o en algunos síndromes genéticos como, por ejemplo, en el síndrome de Rett).
Spasmus nutans
Se presenta en lactantes y se caracteriza por episodios espontáneos de nistagmo asimétrico, asociado a inclinación y movimientos de negación de la cabeza o, a veces, de afirmación. La duración de los episodios es de escasos segundos, pero tiene tendencia a repetir. El proceso es autolimitado y desaparece entre los 3 y 6 años(3). El EEG es normal. Hay que descartar la posibilidad remota de lesiones quiasmáticas o diencefálicas.
Estremecimientos
Son eventos paroxísticos breves y frecuentes que se inician hacia los 4 meses de edad, pero que se extienden a lo largo de la infancia. Consisten en episodios de cese de la actividad sin pérdida de conciencia, con temblor brusco y rápido de corta duración, especialmente de la cabeza, con posturas tónicas en flexión o extensión de la cabeza y cuello con estremecimiento de tronco y cefálico.
Puede haber un factor precipitante y repetir varias veces al día, sin período post-crítico. El EEG ictal es normal. No requieren tratamiento(2,5).
Tortícolis paroxístico
Se inicia en el primer año de vida y remite espontáneamente antes de los 5 años. El niño presenta, de forma recurrente y subaguda (a menudo, desencadenado por cambios posicionales, una postura de inclinación cefálica no dolorosa, a veces, precedida de movimientos oculares anormales (Fig. 1).

Figura 1. Tortícolis paroxístico con inclinación cefálica y del tronco hacia la izquierda.
El paciente durante los episodios puede manifestar: malestar, irritabilidad, agitación, palidez, vómitos o incluso ataxia(3,4).
Los episodios, que suelen variar de un lado a otro en cada evento, pueden durar minutos, horas y, a veces, días, cediendo espontáneamente y repetir con una frecuencia variable, a veces, constante.
Entre los episodios, la exploración neurológica, neuroimagen y EEG son normales. Es más frecuente en niñas (3:1) y tiene un predominio matutino. Se desconoce la etiología, aunque se sospecha una canalopatía (CACNA1A y PRRT2) y relación con la migraña; ya que, en ocasiones, el cuadro puede evolucionar hacia vértigo paroxístico, ataxia episódica y cefalea migrañosa. No requiere tratamiento y, si son muy frecuentes, algunos responden a cinaricina o flunaricina.
Síndrome de Sandifer
Se presenta con posturas anómalas del cuello, tronco y extremidades, como consecuencia de un reflujo gastroesofágico, hernia hiatal o disfunción esofágica. En otras ocasiones, puede presentarse en forma de episodios súbitos de: tortícolis, rigidez generalizada y opistótonos, que pueden acompañarse de apnea y mirada fija con mioclonías de miembros. Suelen ocurrir pocos minutos después de comer y las regurgitaciones no siempre son evidentes; otra característica importante es la normalidad intercrítica. En casos dudosos, debe procederse a una completa exploración esofágica. Se presenta especialmente entre los 18-36 meses(2).
El cuadro es muy aparatoso, puede remedar una distonía o una crisis epiléptica. La presencia de regurgitaciones junto con un EEG crítico normal, descarta epilepsia y apoya el diagnóstico. El cuadro cede al tratar el reflujo gastroesofágico.
Episodios de autoestimulación
Se inician entre los 3 meses y 5 años. Se pueden expresar de formas diferentes: posturas distónicas, gruñidos, diaforesis, agitación, cianosis o palidez, mirada perdida, movimientos de labios y sonrisa o incluso miedo(9). El paciente puede estimular directamente sus genitales o mediante maniobras de frotamiento de los muslos.
Ciertas situaciones favorecen los episodios (aburrimiento, viaje sentado en la sillita, cansancio, para conciliar el sueño). El episodio cesa cuando se distrae al niño, aunque suele mostrarse contrariado y con tendencia a reiniciar la actividad. Pueden confundirse con facilidad con crisis focales complejas. Un registro del episodio puede ayudar al diagnóstico. El pronóstico es favorable y suelen desaparecer antes de los 5-6 años(2,5,9).
Hemiplejía alternante de la infancia
Este raro fenómeno debuta antes de los 18 meses. Consiste en episodios de hemiplejía flácida subaguda, asociado a síntomas autonómicos y nistagmo monocular ipsilateral a la hemiplejía. El episodio se suele iniciar con posturas tónicas o distónicas de miembros, con agitación y sensación de miedo. Se sigue de desviación de la cabeza hacia el hemicuerpo afectado y progresa hacia una hemiplejía completa con dificultad para la deglución y respiración.
La hemiplejía dura desde pocos minutos a varias horas, puede afectar a ambos lados de forma simultánea y remite espontáneamente para volver a repetir en el mismo o en otro hemicuerpo, al cabo de pocos días o semanas. El paciente muestra recuperación parcial o completa tras el sueño. El gen responsable es ATP1A3 en el 85% de los casos. Responden parcialmente a la flunaricina, que puede limitar los ataques, pero no evita el deterioro neurológico(4,10).
Relacionados con el sueño
Head banging
El episodio consiste en movimientos rítmicos de la cabeza, cuando el lactante inicia el sueño. La persistencia de estos movimientos durante el sueño se denomina jactatio capitis nocturno. Los episodios remiten espontáneamente a los 2-3 años, y se observan más frecuentemente y con más intensidad en niños con: retraso del desarrollo, deficiencia sensorial o trastorno dentro del espectro autista. El body rocking se inicia en el primer año con movimientos de balanceo, cuando el niño está en sedestación con movimientos rítmicos de tronco y cabeza, tanto en dirección lateral como postero-anterior. Aunque puede presentarse en niños normales, es más común en lactantes poco estimulados, con retraso cognitivo o déficit sensorial(2).
Relacionados con hipoxia
Espasmos del sollozo
Constituye uno de los TPNE más frecuentes en la edad pediátrica. Los episodios suelen aparecer entre los 6-18 meses y pueden persistir más tiempo o iniciarse antes.
Existen dos tipos: pálido y cianosante.
El espasmo del sollozo pálido es básicamente un síncope vasovagal por un mecanismo cardioinhibitorio neurológicamente mediado, que aparece desencadenado por situaciones de dolor, con frecuencia, por leves traumatismos craneoencefálicos especialmente en la región occipital, que conlleva un factor de sorpresa desagradable (espanto).
Se inicia con un llanto débil que se aborta con rapidez e incluso casi no llega a iniciarse, instaurándose rápidamente una intensa palidez de duración variable (5-30 segundos), seguido de hipertonía con rigidez axial y caída al suelo. Ocasionalmente, se asocian movimientos clónicos. El episodio puede confundirse fácilmente con una crisis epiléptica si no se detecta que fue precedido por traumatismo y si, además, se sigue de mioclonías. La recuperación tiene lugar en poco tiempo (segundos-minutos)(11).
La exploración intercrítica es normal, así como los exámenes complementarios, que en general no son necesarios. Los episodios remiten con la edad, aunque puede evolucionar hacia síncopes vasovagales. Algunos niños presentan anemia ferropénica asociada y de dudosa relación con el trastorno(1,2,5). No precisan tratamiento, pero se debe ofrecer a las familias, información del cuadro y de cómo proceder. Cuando se asocia a una notable variabilidad del ritmo cardíaco, se ha llegado a la implantación de un marcapasos. Existen otras posibilidades terapéuticas en casos extremos, como el empleo de un anticolinérgico, como el glicopirrolato, para disminuir la inhibición cardíaca junto con una preparación oral de teofilina por su efecto cronotrópico.
El espasmo del sollozo cianosante tiene su origen en una apnea espiratoria prolongada. El episodio se inicia por llanto o grito seguido de: apnea, pérdida de conciencia, cianosis y, en ocasiones, seguidos de posturas tónicas, movimientos convulsivos y relajación vesical. Es importante identificar el factor desencadenante responsable (traumatismo, enojo, frustración, ansiedad, etc.).
Es fundamental también conocer la situación clínica tras finalizar el episodio, dado que la recuperación es inmediata, sin estado post-crítico, aunque puede constatarse un estado de hipoactividad, que es tanto más elevado cuanto más alto es el nivel de sobreprotección que desarrollan unos padres asustados por la experiencia vivida. El examen intercrisis es siempre normal. No son necesarios los exámenes complementarios y, cuando se solicitan, pueden provocar mayores dudas e incrementar el riesgo de errores diagnósticos.
La evolución es favorable, no precisan tratamiento aparte de: identificar el trastorno y tranquilizar a la familia y el entorno. Se debe ofrecer una amplia información a los padres sobre la benignidad del proceso, instaurando un programa de modificación de pautas educacionales para disminuir, en la medida de lo posible, los componentes de ansiedad y de reacción neuropática del niño. En ocasiones excepcionales, si el espasmo es muy frecuente, puede mejorar con risperidona. Suelen desaparecer hacia los 4-6 años. La incidencia de epilepsia es la misma que la de la población general. Tampoco se ha hallado relación entre el espasmo del sollozo cianótico y el riesgo de muerte súbita. Los espasmos del sollozo cianosantes son mucho más frecuentes que los pálidos y existen formas mixtas. Es común encontrar familiares de cuadros similares.
Relacionados con el dolor
Dolor paroxístico extremo
Puede debutar ya en período neonatal o, especialmente, en el lactante con un dolor paroxístico extremo, especialmente a nivel rectal y en región perianal. Posteriormente, con la edad, el dolor puede persistir en esta zona o localizarse a nivel: ocular, submandibular o submaxilar. Otro hallazgo constante es la rubicundez facial que acompaña al dolor, especialmente en los pacientes más jóvenes(5,6,12). Durante los episodios, los pacientes manifiestan una extrema rigidez de extremidades y tronco, semejando una crisis tónica. Cuando el dolor es ocular, puede acompañarse de: midriasis, lagrimeo y rinorrea. La duración de los ataques es muy variable (de segundos a horas). El fenómeno autonómico que con frecuencia se acompaña de rigidez y espasmo tónico, se manifiesta por bradicardia y asistolia, conduciendo finalmente a un cuadro sincopal. En niños mayores y adultos, la identificación del trastorno es más sencilla y la localización cambiante del dolor, los antecedentes familiares y la normalidad intercrisis, ayudan al diagnóstico. Con la edad, el cuadro tiende a disminuir en severidad. El factor desencadenante más común es la defecación, la ingesta de comida o incluso la toma de medicación. Se trata de una enfermedad hereditaria autosómica dominante, debida a una mutación en el gen SCN9A responsable de una canalopatía con ganancia de función del Na v 1.7. Las opciones terapéuticas más efectivas son: identificar y evitar en lo posible los factores desencadenantes particulares en cada caso y el empleo de carbamazepina (10-15 mg/kg/d)(12).
TPNE en el niño y adolescente
Relacionados con el movimiento
Trastornos del movimiento
En ellos se incluyen: tics, corea, balismo, mioclono esencial, compulsiones, estereotipias, manierismos, coreoatetosis paroxística kinesigénica, coreoatetosis paroxística distónica y la distonía paroxística inducida por el ejercicio, que no trataremos por problemas de espacio(2,3).
Vértigo paroxístico benigno
Se caracteriza por la aparición, sin previo desencadenante, de episodios súbitos de vértigo sin aura, generalmente de breve duración (minutos u horas) que se resuelven espontáneamente. Es la causa de vértigo más frecuente en niños, cuando no se acompaña de pérdida de audición o patología ORL que lo justifique. Este cuadro se incluye dentro de los equivalentes migrañosos(2,13).
En niños pequeños, es difícil obtener una descripción adecuada, por lo que es preciso observar los signos que lo acompañan: inestabilidad, miedo a caminar, llanto, incapacidad para mantenerse de pie o búsqueda de apoyo o palidez. También puede acompañarse de vómitos. Durante el episodio, puede aparecer además nistagmo. El primer episodio suele ocurrir entre 1-4 años de edad. Puede ser confundido con epilepsia, debido a su carácter paroxístico, recurrente, asociado a sensación de miedo y manifestaciones autonómicas (sudoración, náuseas, vómitos), pero no se acompaña de pérdida de conciencia y no hay período post crítico. En caso de duda, el EEG puede permitir la diferenciación del vértigo de origen epiléptico y también es necesaria la valoración neurootológica en niños con vértigo. La neuroimagen es obligada, cuando los síntomas persisten o aparecen déficits neurológicos. El diagnóstico diferencial debe realizarse con otras formas de vértigo en la niñez como: el vértigo posicional paroxístico benigno, la ataxia episódica, la otitis media aguda y el traumatismo craneoencefálico. Los eventos son breves y generalmente no requieren tratamiento. Algunos responden al tratamiento profiláctico con ciproheptadina o flunaricina. Los episodios evolucionan hacia la desaparición 3-4 años después de su inicio, aunque una proporción de ellos progresa hacia migraña(2).
Reacciones extrapiramidales a fármacos
Los síndromes extrapiramidales de origen farmacológico se pueden manifestar por una amplia serie de alteraciones del tono y del movimiento muscular: acatisia, distonía, discinesia tardía, parkinsonismo y síndrome neuroléptico maligno. La semiología más frecuente es: distonía de cuello, musculatura facial y buco-lingual. Es importante la observación de los trastornos del movimiento y su coincidencia en el tiempo, con la administración de determinados fármacos(2,3,5,6).
Diversos fármacos pueden producir una reacción idiosincrásica aguda y transitoria que, en ocasiones, asemejan una crisis epiléptica. Los fármacos más implicados son: antieméticos (metoclopramida, cleboprida o domperidona) y antipsicóticos típicos (fenotiacinas y análogos, tioxantenos, butirofenonas, difenilbutilpiperidinas) y antipsicóticos atípicos, pero también pueden aparecer con psicoestimulantes (metilfenidato), antidepresivos tricíclicos, antiepilépticos (ácido valproico, fenitoína, carbamacepina, benzodiacepinas) y flunaricina. Es importante la historia clínica minuciosa y las determinaciones para identificar el producto ingerido. Responden al biperideno y a la retirada del desencadenante.
Relacionados con la hipoxia
Síncope
El síncope se define por una pérdida brusca de la conciencia y del tono postural, debido a hipoperfusión arterial cerebral con recuperación espontánea.
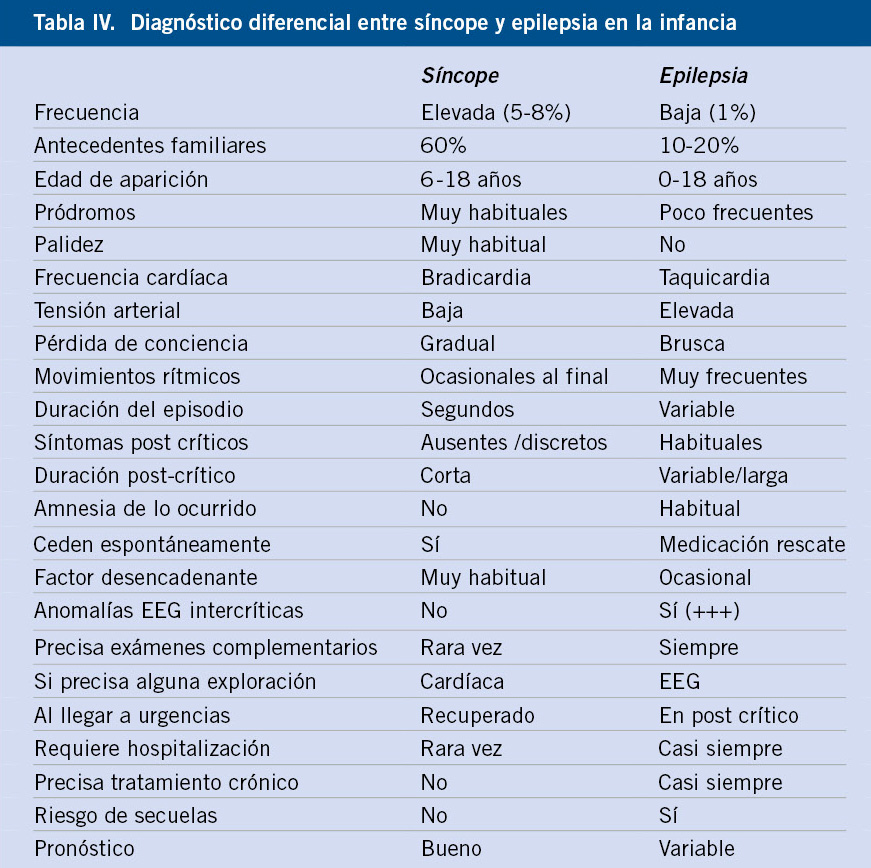
Los síncopes son los TPNE más frecuentes en este grupo de edad y que con mayor facilidad se confunden con crisis epilépticas generalizadas (Tabla IV).
El síncope se produce por un fallo hemodinámico que conduce a una hipoxia cerebral aguda y transitoria. Aparece cuando la falta de perfusión / oxigenación cerebral supera los 8-10 segundos. Con frecuencia, se asocia a: sudoración, palidez, debilidad y parestesias, así como a: cierta rigidez generalizada, opistótonos, trismus y, en ocasiones, sacudidas mioclónicas irregulares de los miembros (síncope convulsivo) (Fig. 2).

Figura 2. Síncope de origen cardíaco en un niño de 4 años por bloqueo aurículoventricular completo, evidenciado en el registro EEG.
Puede cursar, además, con enuresis o incluso mordedura de lengua en algún caso. La duración suele ser corta (10-30 segundos), aunque hay casos de mayor duración. Algunos pacientes presentan síntomas prodrómicos (presíncope) caracterizados por: mareo, parestesias en miembros inferiores, sensación de calor, náuseas, palidez y visión borrosa, que desaparecen con el decúbito. La recuperación es rápida sin síntomas de confusión postictal(1,3).
Hay varios tipos de síncope dependiendo de su mecanismo fisiopatológico. El más común es el síncope mediado neuralmente o no cardiogénico, y que incluye: síncope vasovagal, espasmos del sollozo pálidos y cianóticos. Otros síncopes pueden estar producidos por un incremento de la presión intratorácica, como resultado de: tos intensa (síncope tusígeno) o maniobras de Valsalva (micción o defecación), y síncope del seno carotídeo (hipersensibilidad del seno carotídeo) propio de adultos.
Síncope vasovagal. Resulta de una combinación de: excesivo tono vagal, respuesta anómala de catecolaminas al estrés y aumento del contenido venoso por bipedestación prolongada. Más común en escolares y adolescentes, pueden ser frecuentes y, en general, con carácter familiar. Para el diagnóstico diferencial con la epilepsia o con síncopes cardiogénicos, es muy importante identificar las circunstancias que rodeaban al paciente y los posibles factores desencadenantes (levantarse bruscamente después de un largo período de sedestación o decúbito, ayuno, calor ambiental excesivo, ambiente húmedo, no ventilado o con aglomeración de personas, ansiedad, uso de fármacos, traumatismo, visión de sangre o de escenas desagradables y, especialmente, por un estímulo doloroso brusco, como la venopunción) (Tabla IV). El EEG intercrítico es normal. El síncope vasovagal no suele ocurrir cuando el niño está sentado o tumbado, salvo en el caso de venopunción. Estos síncopes son, a veces, espectaculares, pero siempre benignos y, a veces, muy frecuentes. El test de la mesa basculante es un procedimiento para confirmar el diagnóstico de síncope vasovagal y es aconsejable su realización solo en pacientes con síncopes recurrentes (en los que no se haya establecido claramente la etiología vasovagal), en el estudio del síncope con ejercicio, en síncopes bruscos con traumatismo asociado y, en algunos casos para el diagnóstico diferencial entre síncope y epilepsia. El tratamiento pasa por las medidas de educación sanitaria, explicando la naturaleza benigna del trastorno, identificando los primeros síntomas y adoptando medidas profilácticas, valorar, en algunos casos, la ingesta de agua y sal, o incluso, en casos excepcionales, el empleo de fludrocortisona o midodrina (agente simpaticomimético)(2,5,6,14).
Síncope cardiogénico puede resultar tanto de una alteración del ritmo cardíaco (taquiarritmia, bradiarritmia, síndrome del QT largo, síndrome de Brugada), como de una alteración estructural cardíaca (cardiomiopatía, estenosis aórtica o mitral, tumor intracardíaco) (Fig. 2). Junto al cuadro clínico típico del síncope, suelen asociar: palpitaciones, dolor torácico, respiración entrecortada y/o fatiga.
El síncope cardíaco puede ocurrir en cualquier posición del paciente, incluida la posición de decúbito o el sueño, y especialmente durante períodos de: ejercicio intenso, emoción o excitación.
Es importante investigar los antecedentes familiares de: fallecimiento en personas jóvenes, de muerte súbita no aclarada o personales de cardiopatía o arritmias. La pérdida de conciencia y las convulsiones desencadenadas por miedo o sobresalto, particularmente, si las mismas ocurren durante el ejercicio, son muy sugestivas de un síndrome QT largo. Cuando este se acompaña de sordera congénita, forman parte de los síndromes de Jervell y Lange-Nielson (autosómica recesivo) o de Romano-Ward (autosómico dominante con penetrancia incompleta). En los casos de síncope infanto-juvenil, en los que claramente no se cumplan los criterios de ser mediados neuralmente o existan dudas, se debería efectuar un estudio cardiológico, dada la posibilidad de muerte súbita. El tratamiento depende de la patología de base y, en ocasiones, se debe recurrir a: tratamiento antiarrítmico, ablación quirúrgica por catéter, implantación de marcapasos o desfibrilador(2,3,6,14).
Existen otros síncopes que se deben conocer como: síncope febril (0-3 años en contexto febril), síncopes situacionales (tos, micción, dolor abdominal), síncopes por hipotensión ortostática (con cambios bruscos de postura) y el síncope por taquicardia postural ortostática (por disfunción autonómica y unos minutos después de adoptar la bipedestación). De nuevo insistir, en el valor de una buena historia clínica para identificar el factor desencadenante y no confundirlo con la epilepsia (Tabla IV).
Psicógenos
Pseudocrisis
Las crisis psicogénicas o pseudoepilépticas pueden confundirse con verdaderas crisis epilépticas. Pueden verse en la infancia a partir de los 5 años.
Suelen presentarse en forma de movimientos tónico-clónicos, generalmente: asíncronos, movimientos pélvicos o cefálicos de uno u otro lado, mirada fija o inconsciencia. Los episodios suelen ser muy aparatosos, acompañados de gritos y llanto, casi nunca se golpean y suelen presentarse en ambientes “especialmente seleccionados”(15).
En ocasiones, el evento puede ser provocado por inducción en el enfermo. En este caso, el diagnóstico diferencial es más sencillo. No ocurre lo mismo, cuando se trata de episodios puramente motores que podrían remedar crisis parciales complejas o crisis que se originan en el lóbulo frontal. Las pseudocrisis son frecuentes en pacientes con epilepsia y, por ello, es necesario llevar a cabo un estudio vídeo-EEG idealmente ictal e interictal, para discriminar si corresponde realmente a epilepsia o forma parte de la pseudocrisis. Para aumentar la dificultad en el diagnóstico, en algunos casos de crisis frontales, el EEG ictal puede ser normal. Hay datos que apoyan las pseudocrisis: no hay incontinencia urinaria, siempre con espectadores, sin estado postcrítico, sin traumatismos durante el episodio, nunca aparecen durante el sueño y reaccionan ante un estímulo doloroso. De todos modos, no resulta fácil el diagnóstico diferencial y, en muchos casos, se confunden las crisis epilépticas con pseudocrisis(3,16). Hasta el 70% de los casos referidos a unidades de epilepsia por sospecha de pseudocrisis, tenían una verdadera epilepsia. El abordaje terapéutico es complicado y debe ser multidisciplinar.
Relacionados con el sueño
Los TPNE relacionados con el sueño (terrores nocturnos, pesadillas, despertares nocturnos, sonambulismo, síndrome de las piernas inquietas, bruxismo, etc.) no se revisarán por problemas de espacio(1-3,5).
Miscelánea
Ensoñaciones
Consisten en episodios más o menos prolongados de desconexión o aislamiento del entorno, de contacto confuso con la realidad, con mirada fija o inatención cuando el paciente está en vigilia(2,17). Se presentan en la infancia-adolescencia. Los episodios tienen una duración inferior a cinco minutos, no se acompañan de ninguna sintomatología y se interrumpen voluntariamente, o cuando otra persona le aplica un estímulo como: tacto, cosquillas u oferta de algo que le pueda interesar.
Las pseudoausencias o “daydreams” suelen producirse por aburrimiento, especialmente durante la observación de la televisión o por inatención, y desinterés en el aula.
Habitualmente, pretenden la búsqueda de contenidos gratificantes, de modo que pueden crear cierta adicción, al desarrollar emociones hacia los personajes de sus fantasías.
Cuando existen dudas, se puede confirmar / descartar la epilepsia con ausencias, con el registro vídeo-EEG con hiperventilación (se inducen ausencias clínicas y aparecen alteraciones paroxísticas generalizadas de punta-onda a 3 Hz con la misma duración de la ausencia clínica).
Se debe identificar el trastorno y modificar los factores favorecedores: reducir el tiempo de observación de la televisión, e incluir al niño en actividades deportivas o lúdicas con las que se evite el aburrimiento. En niños con déficit de atención, trastorno generalizado del desarrollo o trastorno obsesivo-compulsivo, aplicar el tratamiento adecuado en estas problemáticas(3,5,17).
Conclusiones
Los TPNE tienen una incidencia mucho más elevada que la epilepsia y pueden aparecer a cualquier edad, pero son más frecuentes en los primeros años de vida. La función del pediatra de AP, es conocer estos trastornos e intentar identificarlos. El primer paso para un diagnóstico correcto es establecer si este episodio corresponde a una crisis epiléptica o puede tratarse de un primer episodio de TPNE. Es importante seguir un protocolo de diagnóstico, analizando: factores desencadenantes, pormenores de cada episodio, análisis detallado del episodio mediante un registro vídeo casero, examen físico, aplicar el sentido común y la experiencia en estos trastornos y solamente proceder a los exámenes complementarios básicos en caso de duda o para la confirmación diagnóstica.
Las opciones terapéuticas son escasas y la mayoría de TPNE tienen una evolución favorable. Si no responden a las medidas habituales o persisten dudas diagnósticas, hay que remitirlo al especialista.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1.** Kaplan PW, Fisher RS. Imitators of Epilepsy. Ed Demos Medical Publishing, 2 Ed. New York, 2005.
2.** Campistol J. Trastornos paroxísticos no epilépticos en la infancia. Barcelona, Viguera eds., 2014.
3.** Grattan-Smith P, Dale RC, Fernández-Álvarez E. Non-Epileptic Paroxysmal Movement Disorders en Aicardi J. Diseases of the nervous system in childhood. 4rd ed. Londres: Mac Keith Press, 2018.
4.* Martínez Bermejo A. Trastornos paroxísticos no epilépticos en los primeros años. En: Campistol J. Neurología para Pediatras. Madrid. Ed. Médica Panamericana; 2011. p. 191-202.
5.* Campistol J. Trastornos paroxísticos no epilépticos en la infancia. Pediatr Integral 2015; XIX(9): 622-31.
6.* Buonsenso D, Plosnic M, Bersani G, Monaco S, Ferrara P, Chiaretti A. Eventos paroxísticos no epilépticos en urgencias pediátricas. Eur Rev Med Pharmacol Sci. 2019; 23(5): 2188-193.
7. Ramelli GP, Sozzo AB, Vella S, Bianchetti MG. Benign neonatal sleep myoclonus: an under-recognized, non-epileptic condition. Acta Paediatr. 2005; 94: 962-3.
8. Campistol J, Prats JM, Garaizar C. Benign paroxysmal tonic upgaze of childhood with ataxia. A neuro-ophthalmological syndrome of familial origin? Dev Med Child Neurol. 1993; 35: 436-9.
9. Rodoo P, Hellberg D. Girls who masturbate in early infancy: diagnostics, natural course and a long-term follow-up. Acta Paediatr. 2013 102: 762-6.
10. Panagiotakaki E, Gobbi G, Neville B, Friedrich E, Campistol C, Nevsimalova S, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain. 2010; 133: 3598-610.
11. Carman KB, Ekici A, Yimenicioglu S, Arslantas D, Yakut A. Breath holding spells: point prevalence and associated factors among Turkish children. Pediatr Int. 2013; 55: 328-31.
12. Fertleman CR, Ferrie CD, Aicardi J, Bednarek NAF, Olofsson E, Emslie FV, et al. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology. 2007; 69: 586-95.
13. Krams B, Echenne B, Leydet J, Rivière F, Roubertie A. Benign paroxysmal vertigo of childhood: long term outcome. Cephalalgia. 2011; 31: 439-43.
14. Fenichel GM. Altered states of consciousness. In: Clinical Pediatric Neurology: a signs and symptoms approach. 6th ed. Philadelphia: Saunders & Elsevier; 2009. p. 71-2.
15. La France WC, Baker GA, Duncan R, Goldstein LH, Reuber M. Minimum requeriments for the diagnosis of psychogenic nonepileptic seizures: a staged approach. A report from the International League Againts Epilepsy. Nonepileptic seizures Task Force. Epilepsia. 2013; 54: 2005-18.
16. Salpekar JA, Plioplys S, Siddarth P, Bursch B, Shaw RJ, Asato MR, et al. Pediatric psychogenic nonepileptic seizures: a study of assessment tools. Epilepsy & Behavior. 2010; 17: 50-5.
17.* Obeid M, Mikati M. Expanding spectrum of paroxysmal events in children: potential mimickers of epilepsy. Pediatr Neurol. 2007; 37: 309-16.
Bibliografía recomendada
– Kaplan PW, Fisher RS. Imitators of Epilepsy. Ed Demos Medical Publishing, 2 Ed. New York, 2005.
Este libro es interesante, porque analiza los TPNE y las causas de error para confundirlos con epilepsia. Analiza, por edades de presentación, los TPNE y, en cada uno, reporta un caso clínico relacionado. Es interesante porque, además, analiza trastornos metabólicos, neuroendocrinos e, incluso, los accidentes cerebrovasculares y psiquiátricos que se confunden con más facilidad con epilepsia.
– Campistol J. Trastornos paroxísticos no epilépticos en la infancia. Barcelona, Viguera eds., 2014.
Monografía, patrocinada por la Sociedad Española de Neurología Pediátrica, donde se revisan hasta 74 trastornos paroxísticos no epilépticos de la infancia. Se analizan en todos y cada uno de ellos: su forma de presentación, los signos clave para identificarlos, el diagnóstico diferencial, las opciones terapéuticas y la evolución. Existen muy pocas monografías que analicen uno por uno todos estos trastornos y puede ser una herramienta muy útil para el pediatra y, en especial, en Atención Primaria.
– Grattan-Smith P, Dale RC, Fernández-Alvarez E. Non-Epileptic Paroxysmal Movement Disorders In: Arzimanoglou A, O’Hare A, Johnston M, Ouvrier R, eds. Aicardi’s Diseases of the Nervous System in Childhood, 4th ed. 2018 London: Mac Keith Press.
Excelente tratado de Neuropediatría, recientemente actualizado por el profesor Alexis Arzimanoglou, ya en su cuarta edición, este libro pretende respetar un enfoque clínico centrado en el paciente. Escrito para neurólogos infantiles, proporciona información actualizada sobre los TPNE y, en especial, sobre los trastornos del movimiento que están ampliamente desarrollados, tanto desde el punto de vista de la estrategia diagnóstica como terapéutica.
– Martínez Bermejo A. Trastornos paroxísticos no epilépticos en los primeros años. En: Campistol J, Neurología para Pediatras. Madrid. Ed. Médica Panamericana; 2011. p. 191-202.
Comprende este capítulo una interesante revisión, por un autor con mucha experiencia en los TPNE en la infancia, con especial énfasis en los que se manifiestan en los primeros años de vida.
| Caso clínico |
|
Francisco es un lactante sin antecedentes personales, que inicia a los 4 meses, diariamente y sin factor precipitante, eventos paroxísticos breves y frecuentes con interrupción de la actividad, sin pérdida aparente de conciencia, con temblor brusco y rápido de corta duración, especialmente de la cabeza y tronco superior, y con posturas tónicas en flexión o extensión.
|
