|
| Temas de FC |
J. Hinojosa Mena-Bernal1,2, B. Pascual1
1Servicio de Neurocirugía Pediátrica. Hospital Universitario 12 de Octubre. Madrid. 2Unidad de Neurocirugía Pediátrica. Hospital Universitario Quirón. Madrid
| Resumen
Los trastornos en la forma y el tamaño del cráneo son la causa más frecuente de derivación de pacientes desde el Pediatra de Atención Primaria a una Unidad de Neurocirugía Infantil. El diagnóstico diferencial es fundamental para reconocer la necesidad de seguimiento en una consulta especializada, informar a las familias sobre el pronóstico de estos pacientes y enfocar el tipo de tratamiento y el momento más adecuado para el mismo. Un diagnóstico acertado y precoz permitirá llevar a cabo una actitud terapéutica conservadora, pero eficaz en aquellos niños con deformidades craneales secundarias a moldeamientos externos (plagiocefalia posicional) y reconocer, sin embargo, a los pacientes afectados por una craneosinostosis verdadera, que requerirán una corrección quirúrgica temprana y un seguimiento estricto para descartar el riesgo de hipertensión intracraneal y otras alteraciones funcionales. |
| Abstract
Anomalies in the size and the shape of the skull are the commonest reason for consultation to a Paediatric Neurosurgical Unit from Primary Care paediatricians. An adequate differential diagnosis is essential to recognize the need to proceed for a specialized consultation, to inform families about prognosis and to offer the right type and moment for the treatment. Early and accurate diagnosis will lead to a conservative but highly effective management in children with cranial deformities secondary to external molding (positional plagiocephaly) and to the recognition of patients affected by a true craniosynostosis that will require a prompt surgical correction and strict follow up to avoid the incidence of intracranial hypertension and other functional abnormalities. The most frequent differential diagnosis in front of a cranial deformity is positional plagiocephaly, but there are at least three other common conditions that could be misleading for a primary care paediatrician: metopic synostosis (and all the spectrum), the dolicocephalic head shape of preterm infants and its differential diagnosis with scaphocephaly; and differentiation between positional plagiocephaly (due to external molding) and synostotic plagiocephaly, may it be anterior (coronal synostosis) or posterior (lambdoid synostosis). Positional plagiocephaly has an excellent response to posturing and physiotherapy in the first five months of life. After this period, cranial orthesis (helmets) could help to correct more severe flattening. Early recognition of these deformities from parents and caregivers would avoid excessive frequentation to referring units and the necessity for the use of cranial orthesis. On the other hand, early diagnosis of craniosynostosis will render the best results after referral and early precocious treatment. |
Palabras clave: Plagiocefalia; Craneosinostosis; Deformidad craneal; Plagiocefalia postural; Escafocefalia; Trigonocefalia; Ortesis craneal
Key words: Plagiocephaly; Craniosynostosis; Skull deformity; Positional plagiocephaly; Scaphocephaly; Trigonocephaly; Cranial orthesis
Pediatr Integral 2015; XIX (9): 591-599
Trastornos del tamaño y la forma del cráneo
Trastornos en la forma del cráneo
La deformidad craneal es la consulta procedente de Atención Primaria más frecuente en la especialidad de Neurocirugía Pediátrica. El diagnóstico diferencial permite distinguir las deformidades posicionales –leves y con excelente pronóstico estético y funcional– de las craneosinostosis verdaderas ocurridas por el cierre prematuro de una o más suturas del cráneo y que tienen un tratamiento y pronóstico completamente diferentes. Las deformidades posicionales pueden producirse en el periodo intrauterino, y encontrarse presentes en el momento del parto o aparecer más tardíamente, en las primeras semanas de vida. Entre las primeras, se encuentran la dolicocefalia del recién nacido pretérmino o algunas plagiocefalias posturales anteriores. Entre las postnatales, mucho más frecuentes, destaca la plagiocefalia occipital, de alta incidencia desde el inicio de la campaña “back to sleep” para la prevención de la muerte súbita del lactante iniciada por la sociedad americana de Pediatría.
Existen fundamentalmente tres tipos de deformidades craneales que pueden crear dudas diagnósticas en el clínico:
1. El cierre precoz de la sutura metópica (en todo su espectro).
2. La dolicocefalia del prematuro por su confusión con la verdadera escafocefalia.
3. La plagiocefalia, sea esta anterior (o frontal), posterior (u occipital) o mixta. Veámoslas por separado.
Dolicocefalia del prematuro vs. escafocefalia (sinostosis sagital precoz)
El diagnóstico diferencial entre una dolicocefalia postural (deformidad cosmética autolimitada) y la escafocefalia (craneosinostosis que precisa tratamiento quirúrgico) se basa en el cierre prematuro de la sutura sagital en esta última.
La dolicocefalia como deformidad postural, sin presencia de escafocefalia (sinostosis sagital precoz), aparece normalmente en recién nacidos pretérmino de bajo peso. Estos neonatos tienen cabezas alargadas y estrechas debido a la macrocefalia relativa que presentan y a la ausencia de tono en la musculatura cervical, lo que provoca una posición de reposo de la cabeza que se apoya lateralmente. El peso de la cabeza sobre un hueso muy fino y débil produce este aspecto característico. La sutura sagital no se encuentra fusionada y la deformidad se corrige espontáneamente en torno a los 3 o 4 meses de edad, siempre que el desarrollo psicomotor sea normal, al mejorar también el control postural de la cabeza. En casos de enfermedades asociadas prolongadas, si el paciente se encuentra con restricción al movimiento de la cabeza en la cuna, o si se acompaña de retraso motor, la deformidad puede mantenerse aún más tiempo y ocasionalmente alcanzar la edad adulta. Cuando la deformidad es muy marcada –y no se corrige espontáneamente– puede indicarse la utilización de una ortesis craneal por razones cosméticas.
A diferencia de la dolicocefalia postural, la escafocefalia se produce en recién nacidos a término, donde el aspecto alargado y estrecho de la cabeza se debe a la fusión precoz de la sutura sagital. Aunque el aspecto puede recordar a la dolicocefalia del prematuro, la escafocefalia se acompaña habitualmente de abombamientos frontales y occipitales compensadores y no mejora con el paso del tiempo. El diagnóstico es fundamentalmente clínico, aunque suele confirmarse con pruebas de imagen (Rx o TC craneales). El tratamiento, en este caso, es quirúrgico, en general con excelentes resultados (Fig. 1).
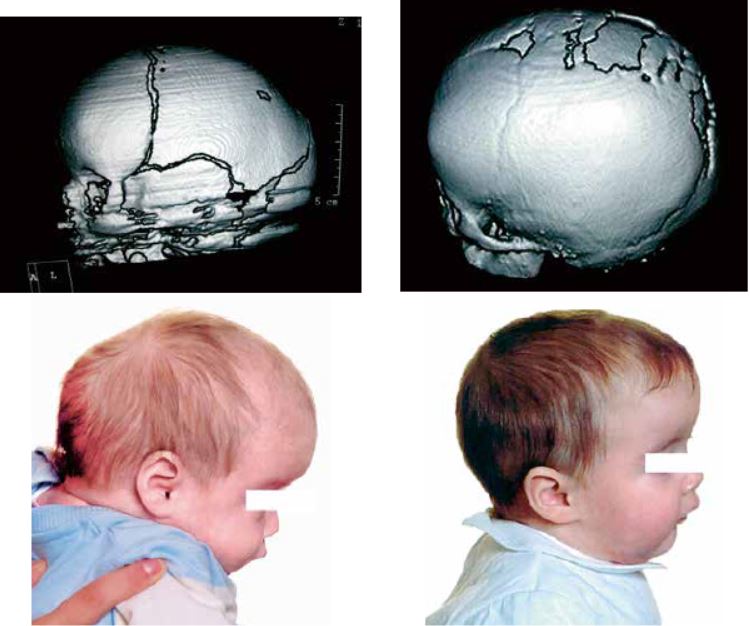
Figura 1. Escafocefalia. TC craneal antes y después de la cirugía. Resultado obtenido tras cirugía endoscópica.
Sinostosis metópica precoz
La trigonocefalia es el cierre precoz de la sutura metópica. Tiene una amplia variabilidad fenotípica y la necesidad de tratamiento quirúrgico debe ser establecida por un especialista en cirugía craneofacial.
La sutura metópica o frontal es la primera sutura craneal en desaparecer. Su cierre se inicia en torno al segundo año de vida(10), pero este momento es variable y en algunos lactantes sin patología puede encontrarse cerrada antes de los 10 meses de edad.
Ocasionalmente, un lactante puede desarrollar una cresta metópica visible o palpable en la región frontal, aun permaneciendo la morfología craneal normal (Fig. 2).

Figura 2. Cresta metópica (sin trigonocefalia).
En esta variante de la normalidad, la sutura puede parecer fusionada en las pruebas de imagen (Rx o TC), pero el estudio histopatológico demostraría que no existe una sinostosis verdadera. A veces, existe un antecedente familiar, y uno de los padres (generalmente la madre) presenta la misma cresta. Al igual que sus progenitores, estos niños suelen encontrarse en percentiles bajos en la curva de crecimiento del perímetro cefálico sin que esto suponga una patología. Puesto que la deformidad craneal que produce es mínima, el tratamiento es conservador. Muy ocasionalmente, por razones estéticas, puede realizarse una cirugía menor de remodelación frontal (fresado de la cresta y remodelación). Las cirugías de reconstrucción craneal no están indicadas en estos casos.
En el otro extremo del espectro, se encuentran los niños con una clara sinostosis metópica y una trigonocefalia significativa. Esta condición se halla presente en el momento del nacimiento y se caracteriza por una acusada retrusión fronto-orbitaria, aplanamiento temporal y de ambos frontales e hipotelorismo (Fig. 3). Estos pacientes son candidatos a procedimientos de reconstrucción craneal y orbitaria debido a la severidad de la deformidad y a la posibilidad de desarrollar un incremento de la presión intracraneal y trastornos del desarrollo cognitivo. Los resultados quirúrgicos son muy satisfactorios.
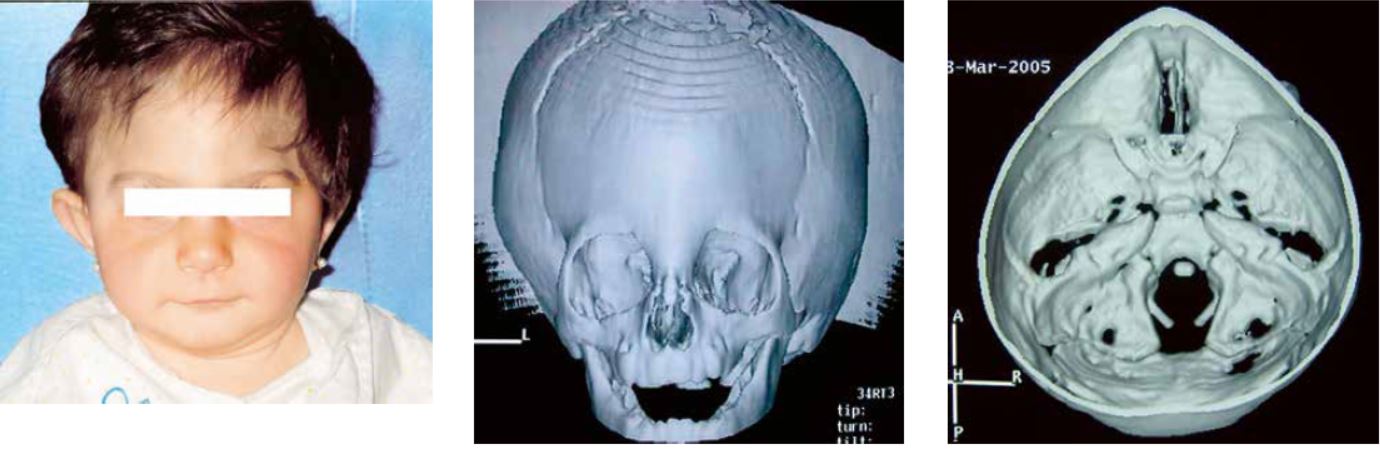
Figura 3. Trigonocefalia con la característica frente en quilla, retrusión de ambos hemifrontales y arcada supraorbitaria, indentación pterional e hipotelorismo. El TC craneal muestra cierre de la sutura metópica y el estrechamiento de la fosa craneal anterior.
Entre estos dos extremos, se encuentran los niños con variantes menores de esta patología, con una cresta frontal que se asocia a formas más leves de trigonocefalia. La indicación de tratamiento quirúrgico en estos casos es más delicada, y depende en cierto modo del deseo de los padres de conseguir un aspecto estético más favorable para sus hijos. Existen trabajos que han demostrado un mayor riesgo de deterioro cognitivo en estos niños, pero aún no está claro hasta qué punto la corrección quirúrgica de la deformidad beneficia a estos pacientes. Todos los casos de trigonocefalia, sean o no intervenidos, precisan de un seguimiento regular para descartar problemas de hipertensión intracraneal tardía o problemas de desarrollo.
Finalmente, los niños con microcefalia verdadera presentan habitualmente cierre de la sutura metópica por falta de crecimiento del encéfalo. La falta de empuje del telencéfalo produce un cierre precoz de todas las suturas craneales. El perímetro cefálico se estanca, a diferencia de la cresta metópica simple, y se acompaña de retraso psicomotor evidente. No existe, indudablemente, en estos casos, ninguna indicación de cirugía.
Las plagiocefalias
La plagiocefalia anterior –secundaria al cierre precoz de una de las suturas coronales– necesita tratamiento quirúrgico, a diferencia de la plagiocefalia occipital o postural, mucho más frecuente y que tiene únicamente repercusión cosmética. El diagnóstico diferencial se realiza mediante la anamnesis y exploración clínica. En caso de duda, un TC craneal puede confirmar la permeabilidad o cierre de la sutura implicada.
La plagiocefalia (del gr. plagio, oblicuo y kephalos, cabeza) es un concepto que engloba a varios tipos de deformidad craneal caracterizados por la asimetría u oblicuidad de la cabeza. Aunque existen diferentes clasificaciones, lo más sencillo es agruparlas en posicionales y sinostóticas. Las plagiocefalias posicionales generalmente afectan a la región posterior del cráneo (huesos occipitales) y muy rara vez se producen plagiocefalias anteriores posicionales. Las plagiocefalias debidas a una craneosinostosis (cierre prematuro de una sutura) pueden, sin embargo, afectar a las suturas coronales –y entonces las denominamos plagiocefalias anteriores o coronales– o a la sutura lambdoidea –craneosinostosis lambdoideas–, si bien, estas últimas son mucho menos frecuentes. Ambas producen una deformidad inicial o primaria (occipital, en el caso de las lambdoideas, y anterior o fronto-orbitaria, en el caso de las coronales) y una serie de deformidades compensadoras o secundarias; por lo que habitualmente observamos todo el cráneo deformado. Se describen a continuación las distintas formas de plagiocefalia insistiendo en su diagnóstico diferencial.
Plagiocefalia anterior
La plagiocefalia anterior afecta aproximadamente a 1 de cada 3.000 recién nacidos. Se trata de una craneosinostosis compleja que incluye el cierre unilateral de una sutura coronal y de otras suturas relacionadas de la base del cráneo: frontoesfenoidal, frontoetmoidal y esfenozigomática. La deformidad afecta a toda la región craneofacial y se produce un aplanamiento y retrusión del hueso frontal del mismo lado del cierre de la sutura, mientras que el frontal contralateral se abomba compensatoriamente. La órbita afectada se encuentra elevada, retruída y rotada (“órbita en arlequín”). Existe un grado variable de distopia vertical (un globo ocular más alto que el otro). El eje nasal es oblicuo, con la raíz nasal desviada hacia el lado de la sutura afecta y la punta hacia el lado sano (Fig. 4). El zigoma y el maxilar superior pueden presentar grados variables de hipoplasia. El peñasco temporal ipsilateral se encuentra mal posicionado, por lo que el pabellón auricular se halla adelantado y descendido.
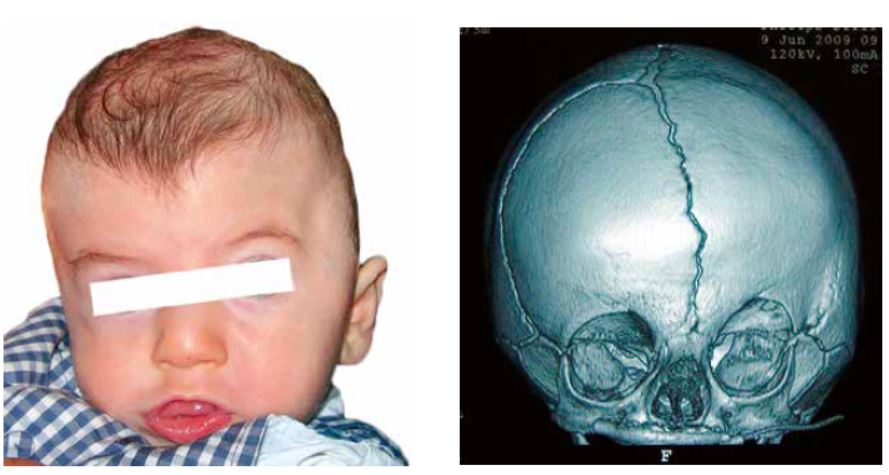
Figura 4. Plagiocefalia anterior por sinostosis de la sutura coronal anterior izquierda. Muestra los estigmas típicos, con aplanamiento del lado afecto (izquierdo) y elevación de la órbita y reborde supraorbitario izquierdos y abombamiento frontal compensador derecho con descenso del reborde supraorbitario y menor volumen de la órbita derecha. La raíz nasal se encuentra desviada hacia la sutura afectada y la punta se dirige hacia el lado sano.
Más allá de la deformidad estética (en ocasiones, severa), la distorsión anatómica de la órbita afectada produce un estrabismo que suele requerir tratamiento quirúrgico. La mala posición orbitaria, así como la alteración de los diámetros de la misma, da lugar a un estrabismo por acortamiento de la musculatura ocular extrínseca y sus tendones de reflexión. La alineación anormal del eje visual, como consecuencia de lo anterior, puede producir anomalías de la visión binocular y, en último término, un ojo vago.
El tratamiento de la plagiocefalia anterior secundaria a una craneosinostosis coronal es quirúrgico y consiste en la remodelación fronto-orbitaria uni o bilateral.
Craneosinostosis lambdoidea (plagiocefalia posterior sinostótica)
La sinostosis lambdoidea aislada (sin afectación de otras suturas) es poco frecuente y las diferentes series de la literatura estiman una incidencia mucho menor del 1% de todas las plagiocefalias posteriores. El diagnóstico diferencial con la plagiocefalia occipital posicional es sencillo atendiendo a criterios puramente clínicos (Fig. 5).
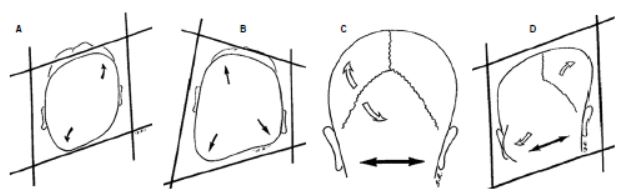
Figura 5. Diagnóstico diferencial de las plagiocefalias. A. Plagiocefalia occipital posicional. Aplanamiento occipital derecho con abombamiento frontal del mismo lado compensador y occipital contralateral. El pabellón auricular se adelanta en el mismo lado del aplanamiento. La cabeza toma la forma de un paralelogramo (modificado de Huang et al.[10]). B. Plagiocefalia por sinostosis lambdoidea. Aplanamiento occipital derecho. El pabellón auricular se desplaza hacia detrás en el mismo lado de la sinostosis. Existe un abombamiento parieto-occipital posterior contralateral, pero en la región frontal el abombamiento compensador es del lado contrario a la deformidad occipital. La cabeza toma forma de trapezoide. C. Plagiocefalia occipital posicional. En la visión posterior el crecimiento del hueso se produce de forma perpendicular a la sutura. Puesto que esta no está fusionada, la base de cráneo (línea bimastoidea) es horizontal y no se observa deformidad craneal. D. Plagiocefalia por sinostosis lambdoidea. El crecimiento se produce de forma paralela a la sutura fusionada y, por tanto, se produce un abombamiento parietal contralateral y occipitomastoideo del mismo lado, con una línea bimastoidea oblicua, descendida en el lado fusionado. La cabeza adopta desde detrás un aspecto de paralelogramo.
Desde una visión cenital, en la plagiocefalia posicional se observa el aplanamiento de la región posterior del cráneo correspondiente a la deformidad inicial (o primaria) producida por el apoyo continuado del lactante sobre esta zona. Se asocia habitualmente a un abombamiento compensador del occipital contralateral y del hueso frontal del mismo lado del aplanamiento occipital. El pabellón auricular está desplazado hacia delante. La cabeza vista desde arriba tiene aspecto de paralelogramo (Fig. 5A). Si se mira desde atrás, el perfil coronal de la cabeza es simétrico, con una línea bimastoidea horizontal (Fig. 5C).
Por el contrario, en la sinostosis lambdoidea, el aplanamiento posterior se asocia con un abombamiento parietal contralateral. En la región anterior existe un mínimo abombamiento frontal, pero si está presente es generalmente contralateral. El pabellón auricular del lado de la sinostosis está normal o, en todo caso, desplazado posteriormente. Suele observarse un abombamiento occipitomastoideo. Vista desde arriba, la cabeza presenta un aspecto de trapezoide (Fig. 5B). En una visión posterior de la cabeza, el abombamiento compensador occipitomastoideo del mismo lado de la sinostosis, y el parietal contralateral produce que la línea bimastoidea de la base de cráneo sea oblicua, desplazada hacia abajo en el lado de la sutura cerrada (Fig. 6).

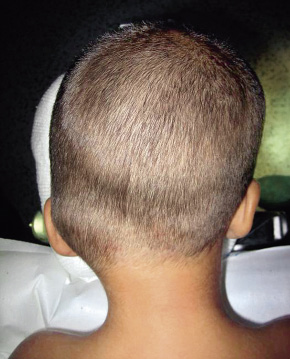
Figura 6. Visión posterior de plagiocefalia occipital por sinostosis lambdoidea. Obsérvese la fusión de la sutura lambdoidea izquierda y las deformidades características descritas en la figura 5D.
De este modo, el pabellón auricular del lado afectado se desplaza inferiormente y la cabeza vista por detrás presenta el aspecto de un paralelogramo (Fig. 5D).
Plagiocefalia occipital posicional
La plagiocefalia posicional se conoce también como: plagiocefalia por moldeamiento, plagiocefalia occipital, plagiocefalia deformativa, plagiocefalia sin craneosinostosis, plagiocefalia postural o plagiocefalia funcional.
Durante el año 1992, se desarrolló por parte de la Asociación Americana de Pediatría (AAP) una campaña de divulgación conocida como “back to sleep” para disminuir el número de casos de “muerte súbita del lactante”. Esta campaña recomendaba que los niños fueran colocados boca arriba para dormir. Diferentes publicaciones destacan un efecto beneficioso de la misma, que consiguió disminuir la mortalidad hasta en un 40%; por lo que, actualmente, no parece aconsejable cambiar una estrategia que ha tenido tanto éxito(5,6). Como consecuencia de esta campaña, el incremento de deformidades posicionales occipitales se ha multiplicado y, en la actualidad, es la causa más frecuente de asistencia en una consulta de neurocirugía pediátrica en el mundo occidental(7). Es difícil establecer la incidencia real de la plagiocefalia posicional en nuestro medio, pero algunos trabajos recientes indican que si los criterios diagnósticos empleados no son correctos, la cifra puede llegar hasta un 48% de niños sanos por debajo del año de edad(7,14). Por el contrario, la incidencia de la craneosinostosis occipital en la literatura es mucho menor, con una cifra en torno a 3 de cada 100.000 recién nacidos (0,003%)(15).
Atendiendo a la etiología de la deformación, la plagiocefalia posicional es de “carácter externo”, porque está producida por fuerzas mecánicas extrínsecas que actúan sobre la sutura lambdoidea o la región posterior del cráneo, bien durante la etapa intrauterina o más frecuentemente en el periodo postnatal(4,8-13). Por el contrario, la plagiocefalia por craneosinostosis es debida a factores intrínsecos que afectan a las propias suturas craneales (sinostosis lambdoidea precoz). Diversos factores pueden actuar sobre la cabeza fetal produciendo un fenómeno de moldeamiento craneal: oligohidramnios, bandas amnióticas, embarazos múltiples, encajamiento fetal prematuro, anomalías uterinas (útero bicorne o tabicamientos), macrocefalia, grandes fetos, partos con fórceps o ventosas, etc.
Después del nacimiento, son muchas las causas que pueden ocasionar una deformidad por moldeamiento(8,11,12): la posición de bienestar elegida por el lactante, el apoyo continuo de la cabeza por descuido de los cuidadores en la misma posición (de un lado o en supino estricto), la utilización abusiva de los modernos “carritos” y sillas con el niño apoyando siempre la cabeza de la misma forma(8), la presencia de tortícolis secundaria a hematomas del músculo esternocleidomastoideo o malformaciones de la unión occipito-cervical, estrabismos y numerosas otras circunstancias, incluida la hipotonía y el retraso psicomotor, que favorecen la aparición de una plagiocefalia al disminuir la motilidad espontánea del niño(7).
Un mecanismo frecuente en la clínica es de carácter mixto: niños que ya nacen con asimetría craneal, producida por moldeamiento intrauterino o durante el parto, y que durante las primeras semanas de vida empeoran por asociarse cualquiera de los factores previamente citados, especialmente porque la cabeza siempre tiende a apoyarse permanentemente sobre el lado previamente aplanado, agravándose la situación todavía más cuando existe tortícolis.
La fisiopatología de estas deformaciones no está todavía completamente explicada, especialmente en relación con las diferencias anatómicas existentes entre las craneosinostosis y las plagiocefalias no craneosinostóticas en la unión craneocervical. Algunos autores han propuesto incluso un origen fisiopatológico común para ambas entidades. Según Dias et al.(9), las fuerzas mecánicas que provocan la deformación primitiva de la región occipital podrían originar, si su actuación es muy persistente, cambios patológicos en las suturas lambdoideas y la base craneal al final de proceso, transformándose en auténticas craneosinostosis. En relación con ello, se han implicado también factores, como la inmovilización y la compresión mecánica de otras suturas similares, para explicar algunas formas de craneosinostosis. Tal es el caso en la escafocefalia (sutura sagital) y la trigonocefalia (sutura metópica)(7).
El diagnóstico es fundamentalmente clínico, por lo que en la mayoría de los niños no son necesarios los estudios radiológicos (Fig. 7).
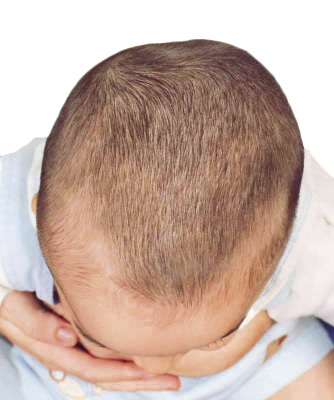
Figura 7. Plagiocefalia posicional derecha. Abombamiento compensador frontal derecho y occipital izquierdo. Adelantamiento del pabellón auricular derecho.
La morfología craneal y los antecedentes clínicos recogidos en la anamnesis permiten un diagnóstico de certeza en la práctica totalidad de los casos. La Rx simple puede dar lugar a confusión, ya que no es sencillo distinguir: “fusiones puntuales” de la sutura, puentes óseos, estenosis o esclerosis de los bordes suturales, que permitan diferenciar una sinostosis lambdoidea de una plagiocefalia postural(12). En caso de duda, la técnica de elección es el TAC craneal con reconstrucción tridimensional.
La controversia existente sobre si la plagiocefalia postural tiene implicaciones en el desarrollo cognitivo del niño no debería empañar la afirmación de que esta deformidad es un problema exclusivamente cosmético. La mayor parte de los estudios que han afrontado esta cuestión son de tipo retrospectivo y algunos de los que afirmaban encontrar una relación entre plagiocefalia y deterioro cognitivo habían incluido además plagiocefalias asociadas a craneosinostosis, valorando los resultados a través de encuestas telefónicas y sin un grupo control(16,21). Las guías de práctica clínica de la NICE (National Institute for Clinical Excellence), la Cochrane y varios estudios prospectivos y de revisión sistemáticos(17-19) no encuentran evidencia para sugerir que plagiocefalia sin craneosinostosis se asocie a retraso mental o psicomotor en la edad adulta, y concluyen que, la mayoría de los casos se resuelven a los dos años de edad sin secuelas cognitivo-motoras. Del mismo modo, no se ha encontrado evidencia que relacione la plagiocefalia con el estrabismo, excepto en la plagiocefalia frontal y la asociada a craneosinostosis(20).
El tratamiento de la plagiocefalia posicional incluye una serie de medidas que en opinión de muchos autores, deben ser escalonadas: reposicionamiento, rehabilitación, técnicas de ortesis craneal y en último –¡y excepcional!– lugar, la reconstrucción quirúrgica(7,11-13).
Durante todo el proceso, la información proporcionada por el pediatra y/o el rehabilitador a las familias debe de ser lo más clara posible, instruyéndoles especialmente en las medidas encaminadas a lograr una correcta re-educación postural del niño.
Así, por ejemplo: los cambios de posición de la cabeza mientras el niño duerme, con ayuda de cojines o la inclinación del colchón; el aprovechamiento del tiempo en que el niño esté despierto para que practique movimientos de cabeza y cuello y ejercicios sobre superficies duras; los juegos en decúbito prono y posición de gateo a partir de los 4,5-5 meses (tummy-time); y, por supuesto, el tratamiento del tortícolis con la fisioterapia adecuada, son todas ellas medidas fundamentales con las que es posible la corrección de la gran mayoría de los casos durante los primeros meses de vida. Son los propios padres quienes deben de realizar estos ejercicios al niño, para lo que deben ser instruidos adecuadamente por el pediatra o rehabilitador.
El tratamiento con ortesis craneal (“casco”), aunque haya sido cuestionado por algunos autores(13), parece adecuado en determinados pacientes, especialmente en los casos refractarios a las medidas de educación postural y fisioterapia (Fig. 8).
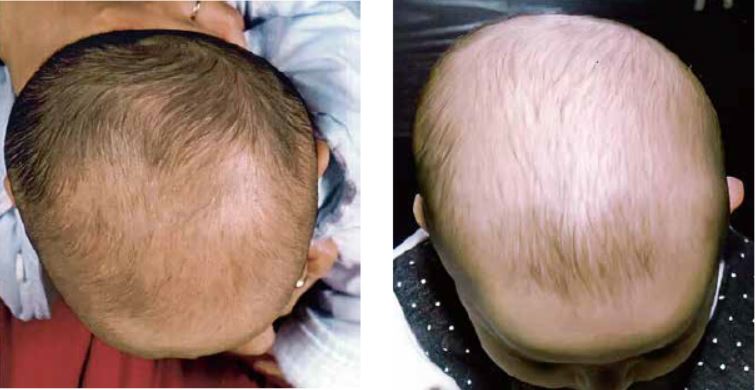
Figura 8. A. Plagiocefalia posicional occipital izquierda antes de tratamiento ortopédico (casco). B. Resultado tras tres meses de tratamiento con ortesis craneal (casco).
Existen diferentes tipos de “bandas o cascos” con principios terapéuticos y resultados similares.
La respuesta más idónea con este tipo de tratamiento se obtiene a partir de los 4,5 o 5 meses de edad y hasta un límite de 12 meses, más allá de los cuales, el cráneo deja de ser susceptible a moldeamientos externos. En todo caso y dado que este tratamiento genera unos costes evidentes, bien para las familias o –en el caso de que exista financiación pública– para la Administración, conviene sistematizar lo mejor posible su empleo. En este sentido, se han publicado y remitido a las diferentes Administraciones del Estado español diversos ejemplos de protocolo asistencial que inciden en la prevención y educación postural hasta los 5 meses de edad, para recomendar el tratamiento con casco en aquellos casos más severos que no respondan a la terapia conservadora(7).
Finalmente, el tratamiento quirúrgico debe reservarse para los pacientes afectados por una craneosinostosis verdadera (sinostosis lambdoidea), o bien para aquellos raros casos de deformidad severa persistente, en los que los tratamientos conservadores (fisioterapia y educación postural, casco) no hayan tenido el efecto deseado(8,12,13).
Trastornos en el tamaño del cráneo
Hidrocefalia externa benigna
La hidrocefalia externa benigna del lactante, caracterizada por la macrocefalia y una serie de hallazgos típicos en los estudios de neuroimagen, se considera una entidad nosológica autolimitada que, rara vez, precisa tratamiento quirúrgico.
La hidrocefalia es una condición relativamente común en Pediatría, que afecta a 1 de cada 1.000 recién nacidos vivos aproximadamente(22). Se define como un acúmulo anormal de LCR en las cavidades ventriculares o en el espacio subaracnoideo que conduce a un aumento de la presión intracraneal.
La hidrocefalia externa benigna es un subtipo caracterizado por el rápido incremento del perímetro cefálico durante el periodo de lactancia (generalmente a partir del 5º al 8º mes de vida) acompañado de un llamativo aumento en el tamaño del espacio subaracnoideo en pruebas de neuroimagen, fundamentalmente a nivel frontal, con un tamaño ventricular normal o mínimamente dilatado. Se han empleado muchos otros términos para definir esta condición, como: acúmulo extra-axial benigno de la infancia, efusiones subdurales, higromas subdurales o hidrocefalia obstructiva extraventricular, que hacen referencia a entidades en las que a pesar de la similitud en las pruebas de imagen subyace un mecanismo fisiopatológico diferente(23). Nosotros nos referiremos a esta condición como hidrocefalia extra-axial benigna de la infancia (HEBI).
En la actualidad, definimos la HEBI como: una condición en la que se produce un rápido aumento del perímetro cefálico del niño en la lactancia, con un marcado incremento de los espacios subaracnoideos y sin (o con mínima) dilatación del sistema ventricular. Algunos autores añaden en la definición que debe estar ausente todo signo o síntoma derivados de un aumento de la presión intracraneal, como pudiera ser el abombamiento de las fontanelas o la presencia de radiolucencia periventricular por exudado transependimario en las pruebas de imagen.
Es la causa más frecuente de macrocefalia en Pediatría, y por alguna razón desconocida es más prevalente en varones. En la mayor parte de los casos es de causa idiopática, y debe distinguirse de aquellos casos producidos después de una hemorragia intraventricular, meningitis, enfermedad metabólica, tratamiento con esteroides, quimioterapia, traumatismos o fenómenos congestivos venosos por enfermedad cardiaca o torácica. En más de un 50% de los pacientes, puede reconocerse la presencia macrocefalia en alguno de los progenitores, lo que hace sospechar la existencia de una herencia mendeliana de tipo autosómico dominante, o más probablemente un modelo de herencia multifactorial, en el que sobre un gen autosómico dominante inciden distintos factores en el periodo fetal y/o neonatal. Algunos autores han propuesto que el mecanismo fisiopatológico fundamental sería una inmadurez en el desarrollo de los villi aracnoideos, que serían incapaces de reabsorber todo el LCR producido en condiciones normales. La maduración de los villi aracnoideos en torno a los 18 meses de edad explicaría la condición autolimitada de esta entidad.
Clínicamente, el hallazgo más característico es el rápido incremento del perímetro cefálico (PC), generalmente a partir del 6º mes de vida, similar al que se produce en la hidrocefalia. El aumento del PC suele ser brusco y progresivo, manteniéndose por encima del p98, aunque paralelo a la curva (Fig. 9). Suele observarse un abombamiento de los huesos frontales, aunque la fontanela se encuentra sin tensión, y no existen otros signos o síntomas de aumento de la presión intracraneal. El fenómeno de “ojos en puesta de sol” (signo de Parinaud) está sistemáticamente ausente.
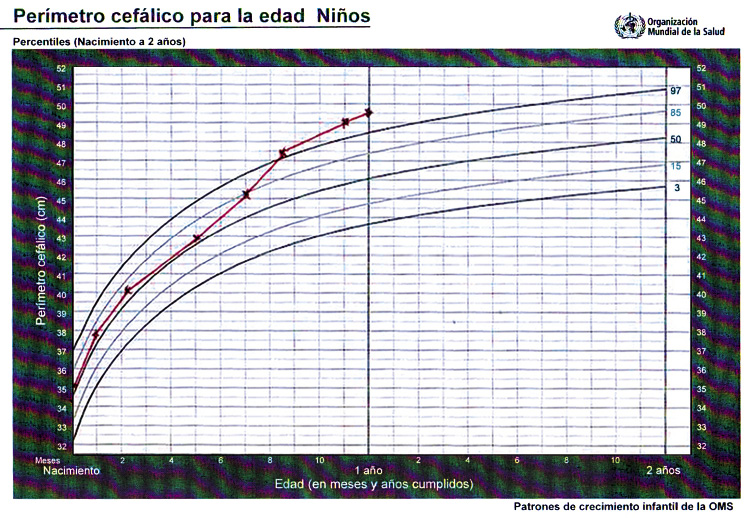
Figura 9.
Desde el punto de vista neurorradiológico, el signo más específico de la HEBI es el aumento del espacio subaracnoideo sobre los lóbulos frontales en ausencia (o con mínima presencia) de dilatación ventricular (Fig. 10).
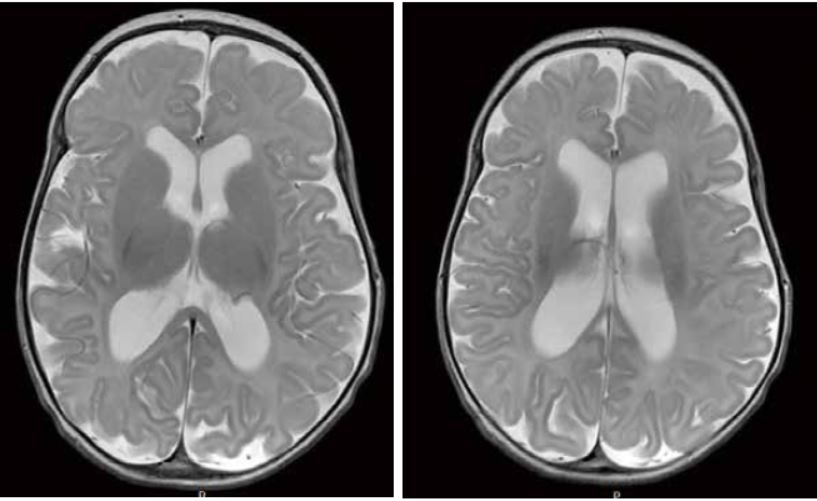
Figura 10.
Es frecuente encontrar también un ensanchamiento de la cisura interhemisférica. Una ecografía transfontanelar suele ser suficiente para el diagnóstico de la hidrocefalia externa benigna, evaluando la distancia entre corteza cerebral y tabla interna, que en condiciones normales se halla por debajo de 3 a 5 mm, y que en la HEBI se sitúa por encima de 5-8 mm. Es también útil para valorar el tamaño ventricular y realizar el seguimiento del paciente. Habitualmente, el estudio se completa con TC o preferiblemente RM, que permite hacer el diagnóstico diferencial con otras entidades que cursan con aumento del espacio extra-axial (atrofias cerebrales, síndrome de Sotos, enfermedades metabólicas, higromas subdurales posthemorrágicos o postinfecciosos, o trauma).
La historia natural de esta condición es generalmente benigna, dado su carácter autolimitado. Puede (suele) existir un retraso puntual del desarrollo cognitivo motor y verbal, aunque la mayor parte de los pacientes alcanzan un desarrollo normal en torno a los 2 o 3 años de edad. El porcentaje de pacientes que acaba con macrocefalia en la edad adulta varía entre un 10% y un 87%, dependiendo de los trabajos consultados(23).
Excepcionalmente, precisa tratamiento quirúrgico, en forma de derivación del LCR desde el espacio subdural o intraventricular a la cavidad peritoneal, y se reserva tan solo para los casos en los que el acúmulo de LCR es progresivo y no autolimitado, o aparecen signos o síntomas de aumento de la presión intracraneal.
Conclusiones
Un diagnóstico diferencial acertado ante la deformidad craneal de un recién nacido o lactante es fundamental para distinguir las deformidades posturales –una condición leve y con mínimas repercusiones–, de las verdaderas craneosinostosis producidas por el cierre precoz de una o más suturas craneales, que requieren las más de las veces, un tratamiento quirúrgico precoz y, en ocasiones, complejo. La sinostosis metópica se presenta con una amplia variabilidad fenotípica y, aunque solo las formas más severas requieren corrección quirúrgica, todos los pacientes precisan de un seguimiento estricto para descartar problemas de desarrollo cognitivo. La dolicocefalia del prematuro tiene un manejo conservador a diferencia de la verdadera sinostosis sagital (escafocefalia), cuyo tratamiento es quirúrgico. La plagiocefalia occipital se debe raramente a una sinostosis lambdoidea verdadera, y la mayor parte de los casos son consecuencia de un moldeamiento de origen externo. Un manejo conservador apropiado que incida fundamentalmente en la reeducación postural obtiene generalmente resultados muy satisfactorios en estos niños. El empleo de cascos se reserva para las deformidades posicionales más severas.
Cualquier paciente con un diagnóstico de sospecha de craneosinostosis debe remitirse precozmente a un servicio de Neurocirugía Pediátrica con experiencia en el manejo de la patología craneofacial.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1. Ayer A, Campbell A, Appleboom G, Hwang BY, McDowell M, Piazza M, et al. The sociopolitical history and physiological underpinnings of skull deformation. Neurosurg Focus. 2010; 29(6): E1.
2. Nichter LSPJ, Persing JA, Horowitz JH, Morgan RF, Nichter MA, Edgerton MT. External cranioplasty: historical perspectives. Plast Reconstr Surg. 1986; 77: 325-32.
3. Schjiman E. Artificial cranial deformation in newborns in the pre-Columbian Andes. Child´s Nerv Syst. 2005; 21: 945-50.
4.*** Persing J, James H, Swanson J, Kattwinkel J. Prevention and Management of Positional Skull Deformities in Infants. Pediatrics. 2003; 112; 199-202.
5. American Academy of Pediatrics, Task force on Positioning and Sudden Infant Death Syndrome. Positionings and SIDS. Pediatrics. 1992; 89: 1120-6.
6. American Academy of Pediatrics, Task Force on Infant Sleep Positional Sudden Infant Death Syndrome. Changing concepts of sudden infant death syndrome: implications for infant sleeping environment and sleep position. Pediatrics. 2000; 105: 650-6.
7.** Esparza J, Hinojosa J, Muñoz MJ, Romance A, García Recuero I, Muñoz A. Diagnóstico y Tratamiento de la Plagiocefalia Posicional. Protocolo para un Sistema Público de Salud. Neurocirugía. 2007; 18: 457-67.
8.** Chadduck WM, Kast JH, Donahue DJ: The enigma of lambdoid positional molding. Pediatr Neurosurg. 1997; 26: 304-11.
9.* Dias M, Klein D: Occipital plagiocephaly: deformation or lambdoid synostosis? A unifying theory regarding pathogenesis. Pediatric Neurosurgery. 1996; 24: 69-73.
10. Huang MH, Mouradian WE, Cohen SR, Gruss JS. T The Differential Diagnosis of Abnormal Head Shapes: Separating Craniosynostosis from Positional Deformities and Normal Variants. Cleft-Palate Craniofacial J. 1998; 35: 204-11.
11. Mottolese C, Szathmari A, Ricci AC. Plagiocephalies positionnelles: place de l’orthese crânienne. Neurochirurgie. 2006; 52: 184-94.
12.** Persing J, James H, Swanson et al.: Prevention and management of positional skull deformities in infants. Pediatrics. 2003; 112: 199-202.
13.** Pollack IF, Losken WH, Fasik P: Diagnosis and management of posterior plagiocephaly. Pediatrics. 1997; 99: 180-5.
14. Kane AA, Mitchell LE, Craven KP: Observations on a recent increase of plagiocephaly whitout synostosis. Pediatrics. 1996; 97: 877-85.
15. Rekate HL: Occipital plagiocephaly: a critical review of the literature. J Neurosurg. 1998; 89: 24-30.
16. Miller RI1, Clarren SK. Long-term developmental outcomes in patients with deformational plagiocephaly. Pediatrics. 2000; 105(2): E26.
17. Moulding helmets/cranial banding for plagiocephaly. NICE (National Institute for Clinical Excellence). http://www.nice.org.uk/page.aspx?o=263598.
18. Bialocerkowski AE, Vladusic SL, Howell SM. Conservative interventions for posicional plagiocephaly: a systematic review. Dev Med Child Neurol. 2005; 47(8): 563-70.
19. Hutchison BL, Hutchison LA, Thompson JM, Mitchell EA. Plagiocephaly and brachycephaly in the first two years of life: a prospective cohort study. Pediatrics. 2004; 114(4): 970-80.
20. Gupta PC, Foster J, Crowe S, Papay FA, Luciano M, Traboulsi EI. Ophthalmologic findings in patients with nonsyndromic plagiocephaly. J Craniofac Surg. 2003; 14: 529-32.
21. Collett B, Breiger D, King D, Cunningham M, Speltz M. Neurodevelopmental implications of “deformational” plagiocephaly. J Dev Behav Pediatr. 2005; 26(5): 379-89.
22.*** Zahl SM, Egge A, Helseth E, Wester K. Benign external hydrocephalus: a review, with emphasis on management. Neurosurg Rev. 2011; 34: 417-32.
23. Hellbusch L. Benign extracerebral fluid collections in infancy: clinical presentation and long-term follow-up. J Neurosurg (2 Suppl Pediatrics). 2007; 107: 119-25.
| Caso clínico |
|
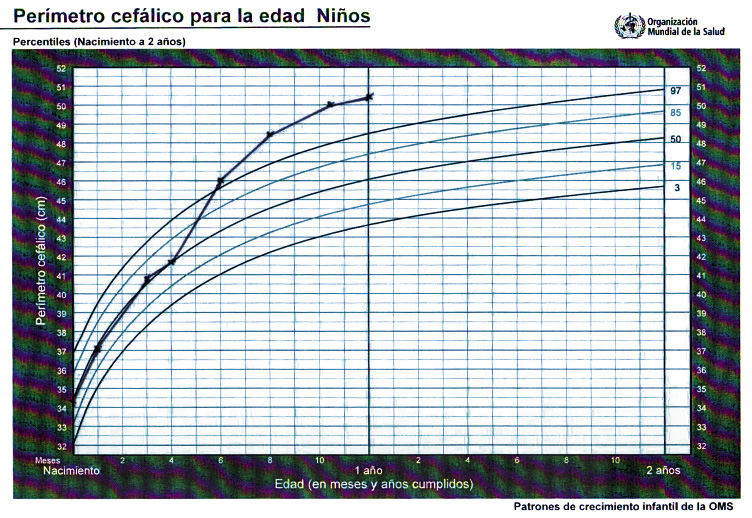
Milton M. es un varón de 12 meses al que seguimos en consulta de Pediatría del Niño Sano. Se trata de un un recién nacido en la semana 37 de gestación, con un peso al nacer de 3,150 kg y un perímetro cefálico de 34,3 cm (p25-50). No se refieren otros antecedentes obstétricos o perinatales de interés. Presenta un calendario vacunal correcto y acude a sus consultas rutinarias sin incidencias. A los padres les ha alertado un crecimiento excesivo del perímetro cefálico en las últimas semanas, aunque el comportamiento, alimentación y sueño del niño parecen normales. En la exploración, encontramos un niño con excelente estado general, correcto estado nutricional y exploración neurológica normal, con adquisición de hitos psicomotrices adecuados a su edad, aunque llama la atención una discreta hipotonía axial. El sostén cefálico y la sedestación son algo inestables, aunque no impresiona de retraso neuro-cognitivo. Interacciona alegremente con el explorador, presenta un seguimiento visual activo, manipula objetos cambiándolos de mano y palmotea y balbucea con frecuencia. La cabeza es de morfología normal, aunque con aspecto macrocéfalo y ligeramente braquicéfalo por un aplanamiento posicional posterior. No se palpan crestas óseas prominentes y el perímetro cefálico es 50,2 cm (> p98). El índice cefálico es 85. La fontanela anterior está abierta, amplia (2 x 2 cm), aunque sin tensión, y se deprime con el niño en sedestación. No hay dilatación de venas epicraneales. Los pares craneales explorados son normales, incluyendo los movimientos oculares. No se objetiva fenómeno de Parinaud. El perímetro cefálico de la madre es 56,5 cm (p50-75) y el del padre es 61,3 cm (>p98). La madre del niño asegura que su suegro es “cabezón”. El marido replica que la que es “cabezona de verdad” es su suegra.
|