|
| Temas de FC |
M.L. Cilleruelo Pascual![]() , A. García Díaz
, A. García Díaz
Servicio de Gastroenterología, Hepatología y Nutrición Pediátrica. Hospital Universitario Puerta de Hierro Majadahonda. Madrid
| Resumen
Valoración y manejo de la diarrea crónica en función de su mecanismo fisiopatológico, síntomas, exploraciones complementarias y tratamiento. La diarrea se clasifica en diarrea osmótica y diarrea secretora. También puede dividirse en diarrea por maldigestión, cuando no se producen las enzimas digestivas que hidrolizan las proteínas, grasas y carbohidratos, y diarrea por malabsorción, cuando existe una alteración del epitelio intestinal que impide el transporte de los nutrientes ya digeridos. Deben considerarse los signos y síntomas de alarma para efectuar una aproximación diagnóstica inicial de diarrea de origen orgánico o funcional. En la historia clínica se definirán las características de las heces, acuosas, con sangre o con grasa, lo que permitirá orientar el posible mecanismo fisiopatológico. En la exploración física se valorará especialmente la repercusión nutricional. El diagnóstico diferencial es muy amplio y las exploraciones complementarias numerosas, pero deben efectuarse en función de los hallazgos clínicos. Con las exploraciones de primer nivel se puede llegar al diagnóstico de las causas más frecuentes. Aparte del soporte nutricional, se efectuará el tratamiento específico de la enfermedad de base, que con frecuencia, pero no siempre, será exclusivamente nutricional. El correcto y temprano diagnóstico y tratamiento de estos pacientes minimizará la morbilidad y las consecuencias nutricionales de la diarrea crónica. |
| Abstract
This article focuses on the assessment and management of chronic diarrhea according to its pathophysiological mechanism, symptoms, complementary examinations and treatment. Diarrhea is classified as osmotic diarrhea and secretory diarrhea. It can also be divided into diarrhea due to maldigestion, when the digestive enzymes that hydrolyze proteins, fats and carbohydrates are not produced, and diarrhea due to malabsorption, when there is an alteration of the intestinal epithelium that prevents the transport of already digested nutrients. The warning signs and symptoms must be considered to make an initial diagnostic approach to diarrhea of organic or functional origin. The clinical history should define the characteristics of the stools, watery, bloody or greasy, which will help to orientate the possible pathophysiological mechanism. In the physical examination severity of malnutrition is specifically assessed. The differential diagnosis is broad and the complementary examinations numerous, but they must be carried out based on the clinical findings. First level investigations can lead to the diagnosis of the most common causes. In addition to nutritional support, specific treatment of the underlying disease is provided, which will often, but not always, be exclusively nutritional. Correct and early diagnosis and treatment of these patients will minimize the morbidity and nutritional consequences of chronic diarrhea. |
Palabras clave: Diarrea crónica; Diarrea funcional; Diarrea orgánica; Malabsorción; Maldigestión; Exploraciones complementarias; Tratamiento.
Key words: Chronic diarrhea; Functional diarrhea; Organic diarrhea; Malabsorption; Maldigestion; Complementary tests; Management.
Pediatr Integral 2024; XXVIII (8): 493 – 502
OBJETIVOS
• Comprender los mecanismos fisiopatológicos de la diarrea crónica para efectuar una primera aproximación diagnóstica.
• Conocer las diferentes etiologías de la diarrea crónica para poder establecer el diagnóstico diferencial.
• Efectuar una correcta historia clínica, concediendo la mayor importancia a las características de las heces y a la exploración física del paciente.
• Seleccionar las diferentes exploraciones complementarias en función de los hallazgos clínicos.
• Conocer el tratamiento, especialmente de las diarreas crónicas, cuyo manejo es fundamentalmente dietético.
|
|
|


Diarrea crónica
Introducción
La diarrea crónica dura más de 14 días y tiene un comienzo insidioso. El diagnóstico diferencial es complejo por la multiplicidad de causas.
La diarrea no es fácil de definir en la edad pediátrica, ya que la frecuencia y consistencia de las heces va a ir variando con la edad y con la introducción de la alimentación. Lo esperable es que el número de deposiciones disminuya y su consistencia aumente con el paso del tiempo. La OMS define la diarrea como el paso de 3 o más deposiciones al día, blandas o líquidas, o con una frecuencia superior al que era el ritmo previo del paciente(1).
La diarrea prolongada es aquella que persiste más de 14 días. Suele tener un comienzo agudo y es más frecuente en el niño menor de 5 años. La diarrea crónica es la que dura más de 14 días, el inicio es insidioso y la progresión lenta(2).
La diarrea crónica puede tener múltiples etiologías y diferentes mecanismos fisiopatológicos, por lo que el diagnóstico diferencial no es sencillo. Es necesario efectuar una historia clínica detallada y una exploración física enfocada al estado nutricional y a los potenciales déficits nutricionales específicos, para seleccionar las exploraciones complementarias dirigidas a conocer su causa.
Fisiopatología
La diarrea crónica se clasifica en diarrea osmótica y diarrea secretora. Asimismo, puede dividirse en diarrea por malabsorción y por maldigestión.
El intestino maneja una gran cantidad de líquidos que proceden de la ingesta y de las secreciones de saliva, jugo gástrico, biliar, pancreático e intestinal. Existen numerosos procesos de transporte de líquidos, electrolitos y solutos que tienen lugar fundamentalmente en el intestino delgado. El resto de los fluidos se reabsorben en el intestino grueso hasta formar el bolo fecal. Cualquier disbalance en estos procesos dará lugar a una diarrea.
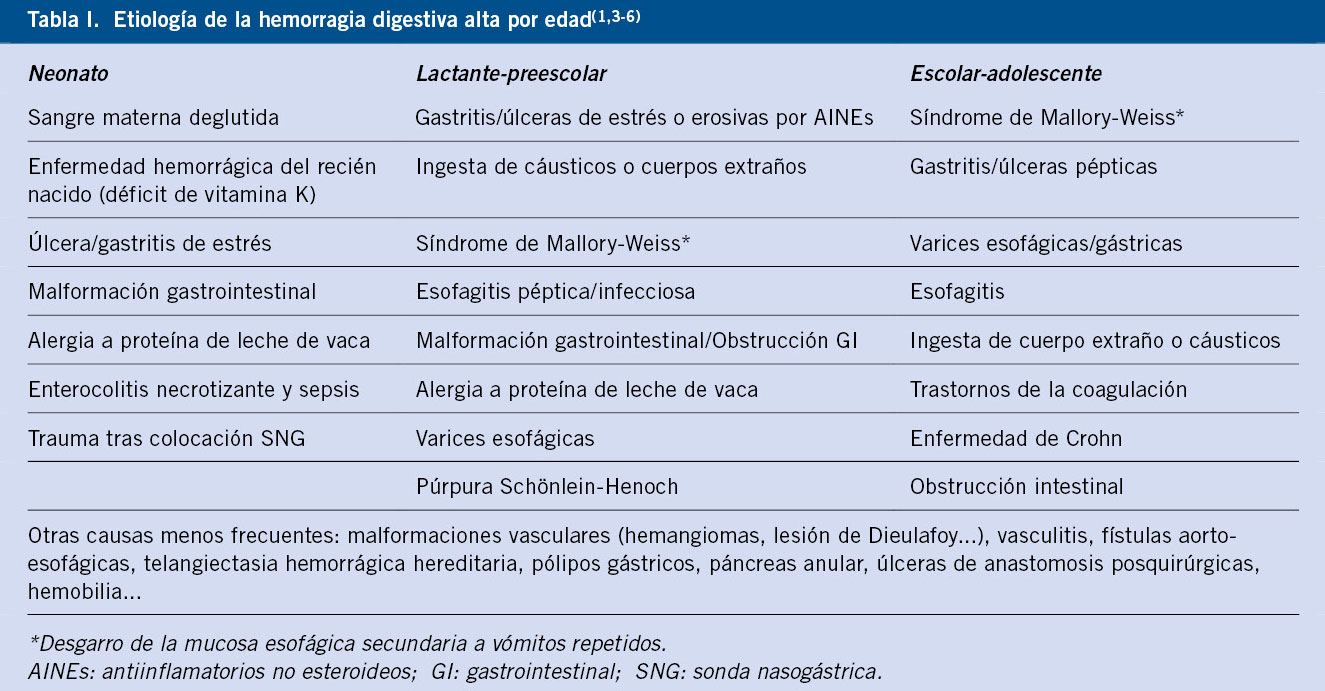
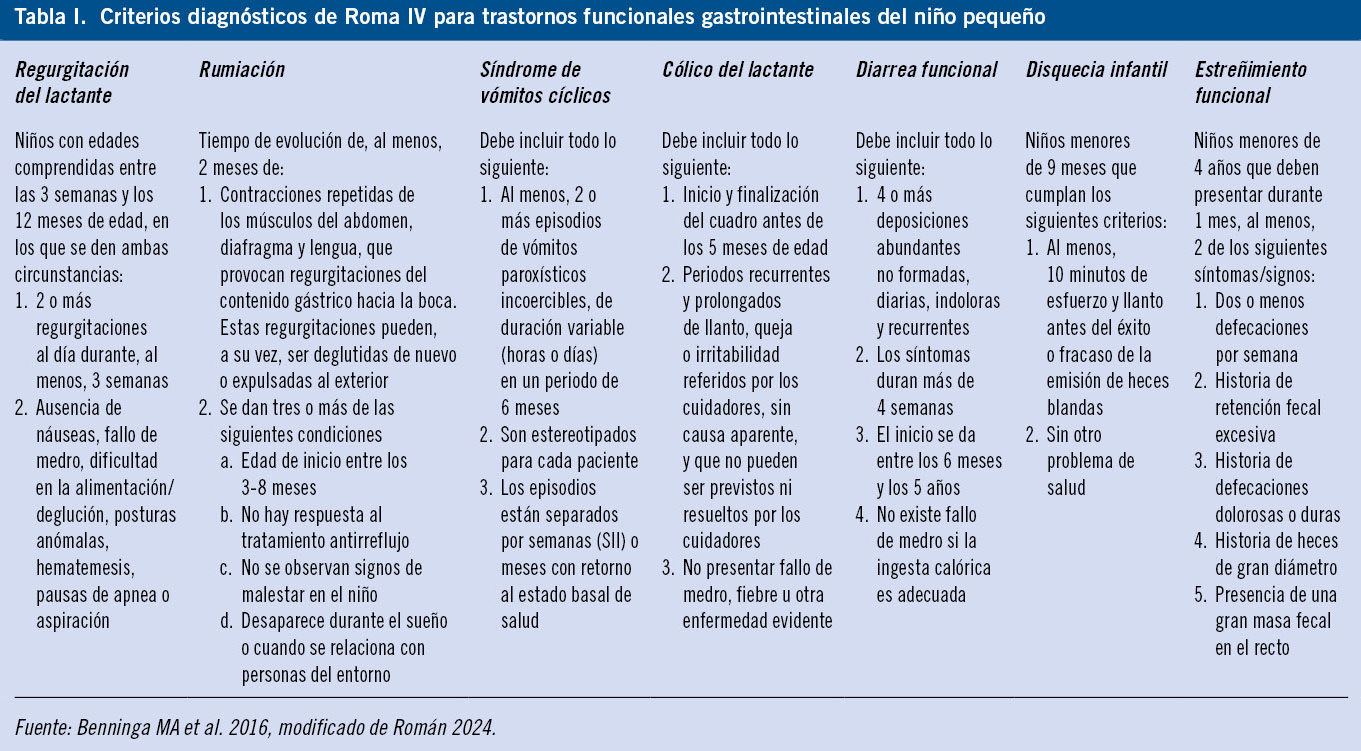
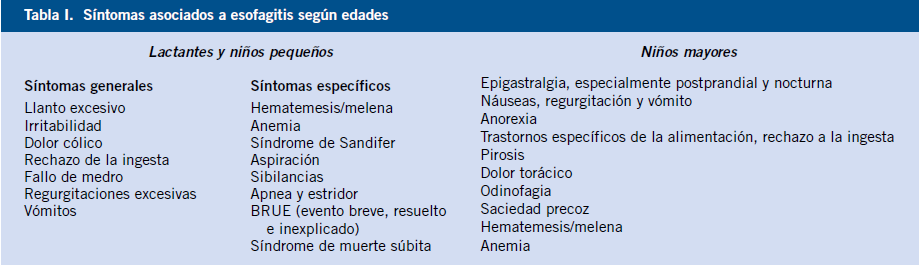
El mecanismo más importante de regulación del transporte de fluidos es mediante el transporte activo de iones (sodio, cloro y bicarbonato) a través del enterocito. El movimiento del sodio favorece la absorción y el del cloro la secreción de líquidos. También es necesaria la integridad de la barrera epitelial para evitar que los electrolitos y, por tanto, el líquido, sean transportados pasivamente tras la absorción o secreción. Por último, es necesaria una motilidad intestinal normal para que se pueda producir un adecuado contacto entre los nutrientes y el epitelio intestinal. El proceso de digestión se inicia con la acción de las enzimas luminales y se completa con la actuación de las enzimas del borde en cepillo en la mucosa intestinal (Tabla I).

La diarrea crónica se clasifica, según su mecanismo fisiopatológico, en diarrea osmótica y diarrea secretora.
• Diarrea osmótica: es aquella que resulta de la malabsorción de solutos. Como en todas las diarreas, se producen fuerzas osmóticas; hay autores que prefieren el término de diarrea inducida por alimentos(3). El aumento de la osmolaridad en la luz intestinal produce movimientos a favor del gradiente de agua y electrolitos. Este tipo de diarrea mejora característicamente con el ayuno(3).
• Diarrea secretora: en estas diarreas se produce un aumento de la secreción de líquidos arrastrados por aniones (cloro o bicarbonato) o potasio o por la pérdida del mecanismo de absorción de líquidos a través del sodio. Por esto, algunos autores prefieren utilizar el término de diarrea relacionada con el transporte de electrolitos(3).
La diarrea crónica también puede clasificarse en función del proceso digestivo por el que se produce la alteración en:
• Maldigestión: cuando fracasa la producción de las enzimas digestivas, tanto las intraluminales como las del borde en cepillo, que hidrolizan las proteínas, grasas y carbohidratos. Fundamentalmente están implicadas las enzimas pancreáticas y, clínicamente, predomina la esteatorrea.
• Malabsorción: cuando existe una alteración del epitelio intestinal que impide el transporte de los nutrientes ya digeridos. La fermentación en el colon de los alimentos no absorbidos produce ácidos grasos de cadena corta, que ejercen un efecto catártico en el intestino delgado, dando lugar a heces muy voluminosas.
Etiología
Debe establecerse la sospecha de diarrea de causa funcional u orgánica y, posteriormente, se valorarán las posibles causas de diarrea orgánica, teniendo en cuenta la edad de inicio y los síntomas de alarma.
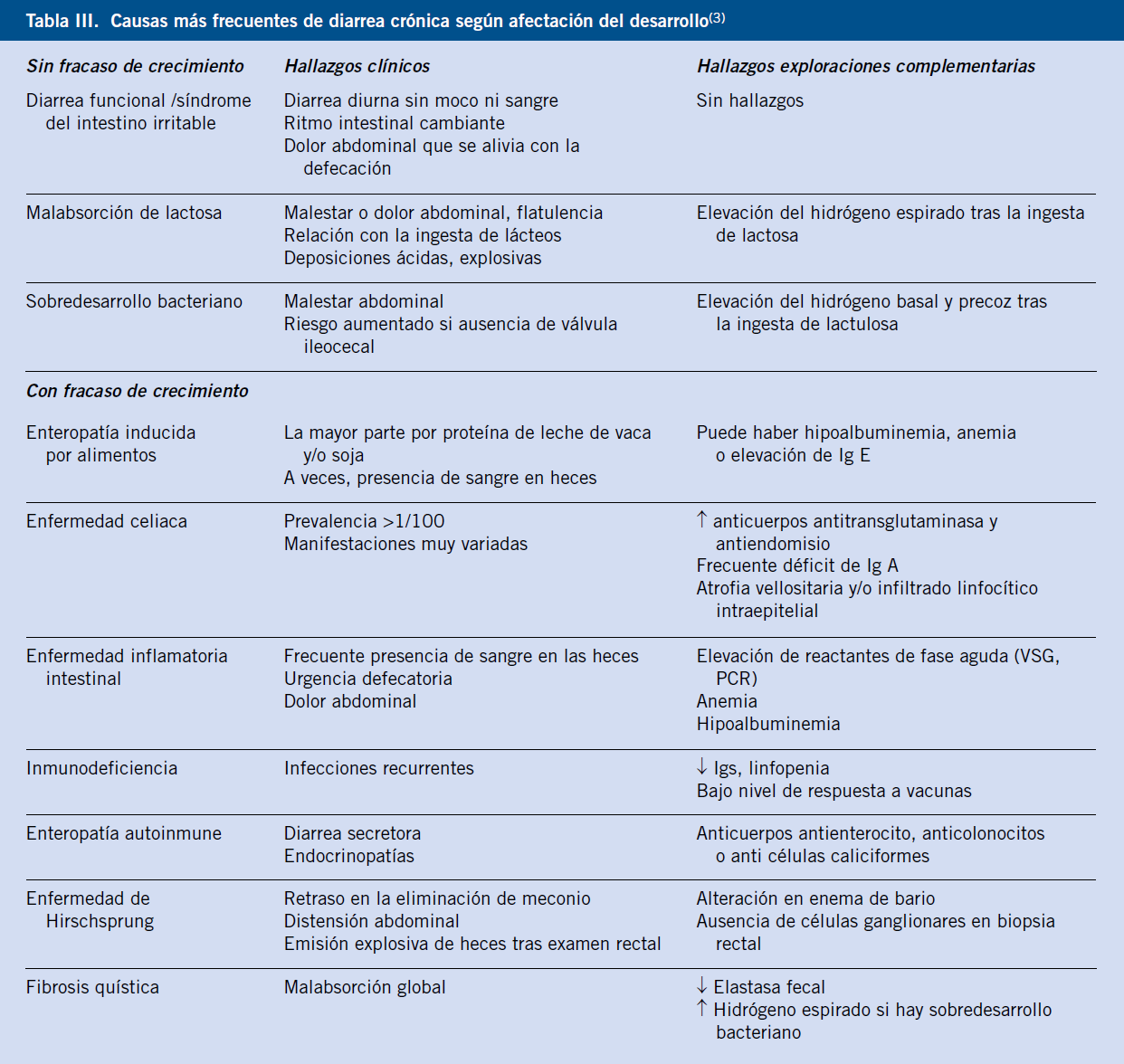
Las causas de diarrea crónica son muy numerosas. Es importante tener en cuenta la edad del paciente para considerar, de forma lógica, las distintas posibilidades etiológicas. En la tabla II se muestran las principales causas de diarrea crónica por grupos de edad.

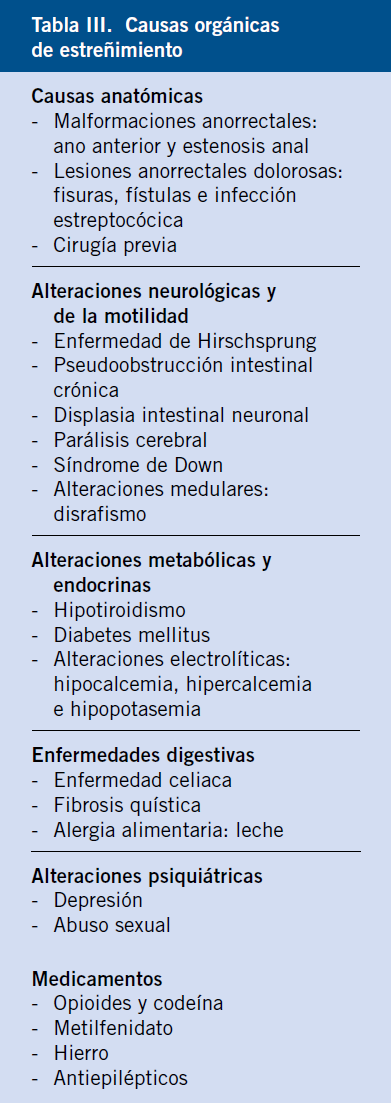
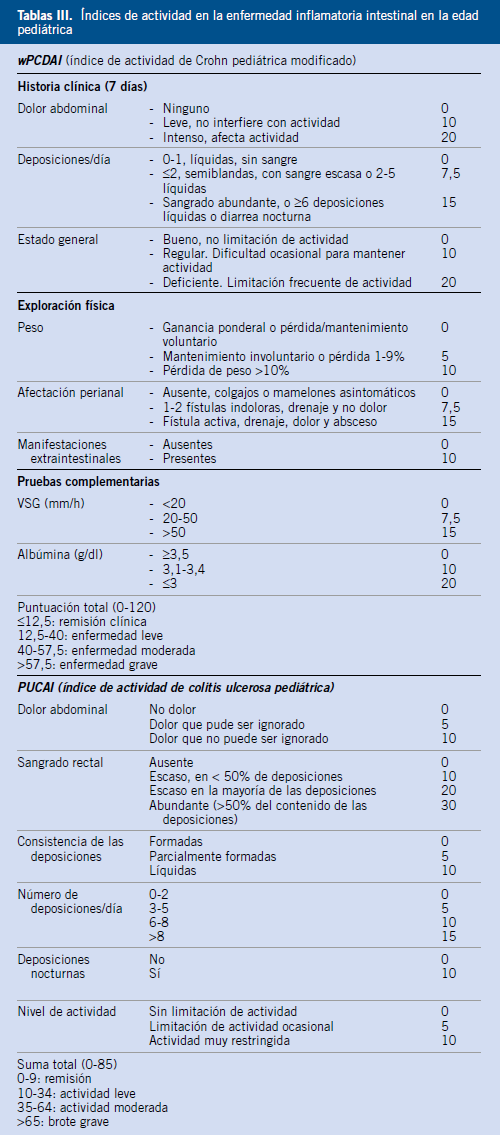
La diarrea crónica puede ser de origen funcional u orgánico. Deben buscarse los signos y síntomas de alarma (Tabla III), cuya ausencia orienta al diagnóstico de diarrea funcional, con la consiguiente limitación de las exploraciones complementarias a realizar.

Diarrea funcional
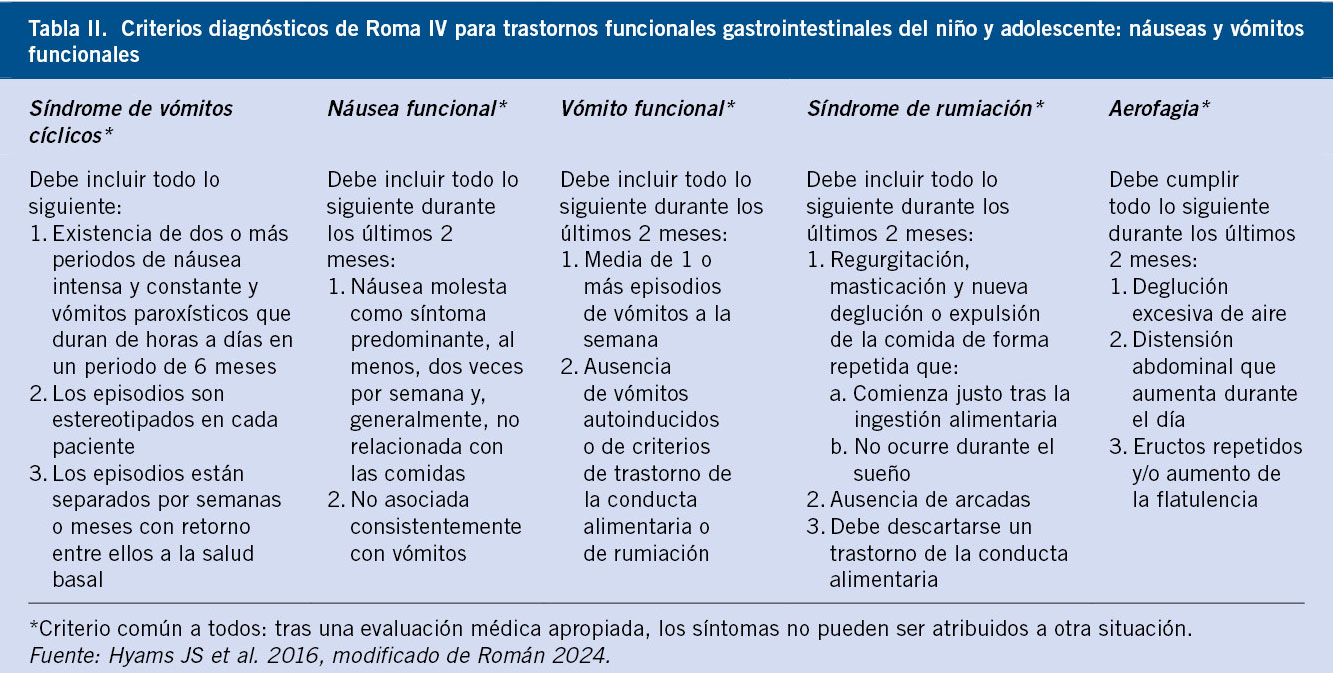
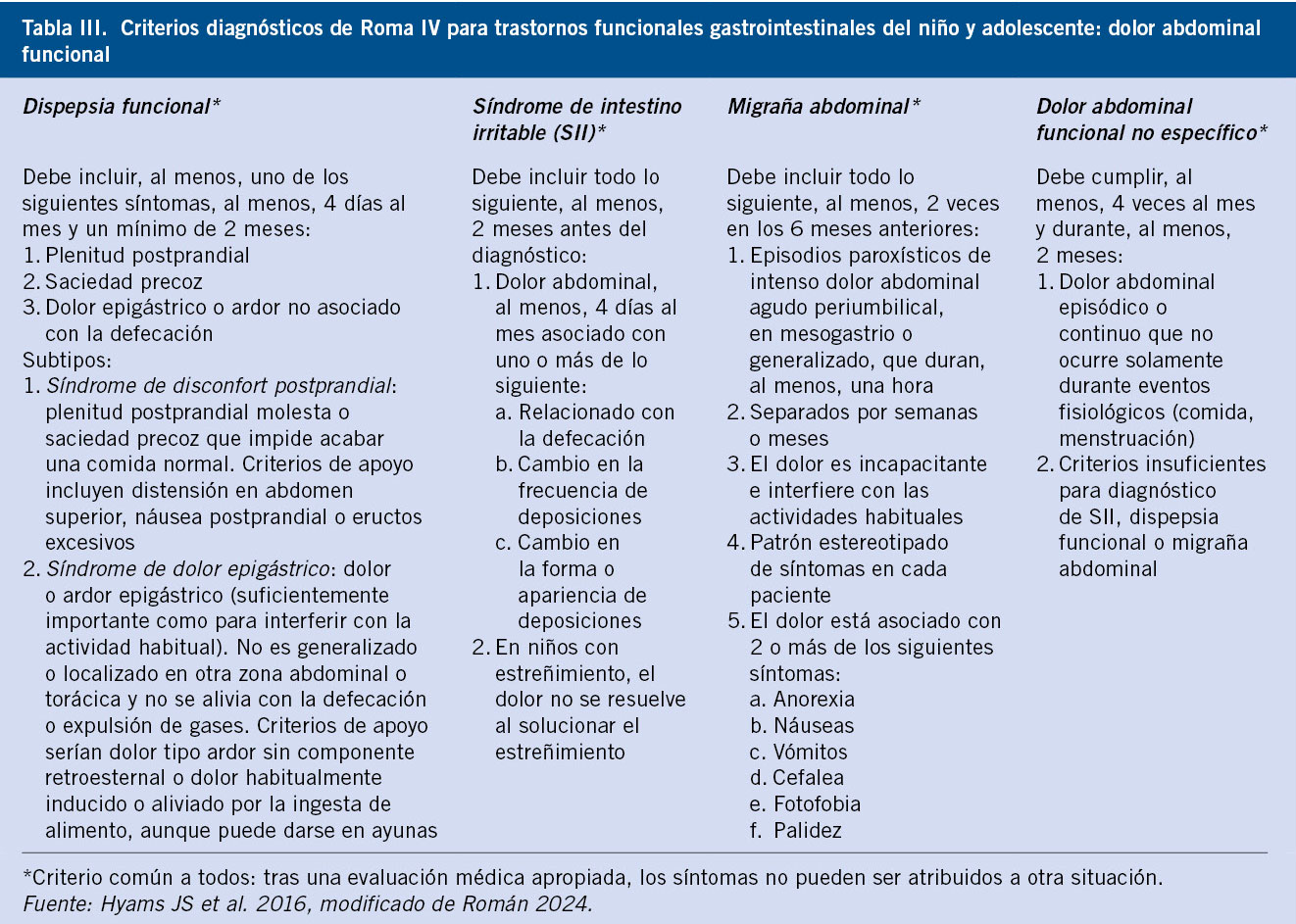
• Diarrea funcional del lactante: se caracteriza por el paso de, al menos, 4 deposiciones al día, abundantes, no formadas, con alimentos reconocibles y de color claro, sin dolor, pérdida de apetito ni alteración en la ganancia de peso. Se inicia a los 6 meses y suele resolverse a los 5 años, pero puede persistir a lo largo de la infancia y adolescencia(4,5).
• Síndrome de intestino irritable: se caracteriza por, al menos, 4 días al mes durante 2 meses de dolor abdominal asociado a uno o más de los siguientes síntomas: dolor con la defecación, cambio en la frecuencia y cambio en la apariencia de las deposiciones, en este caso con predominio de diarrea(6).
Diarrea orgánica
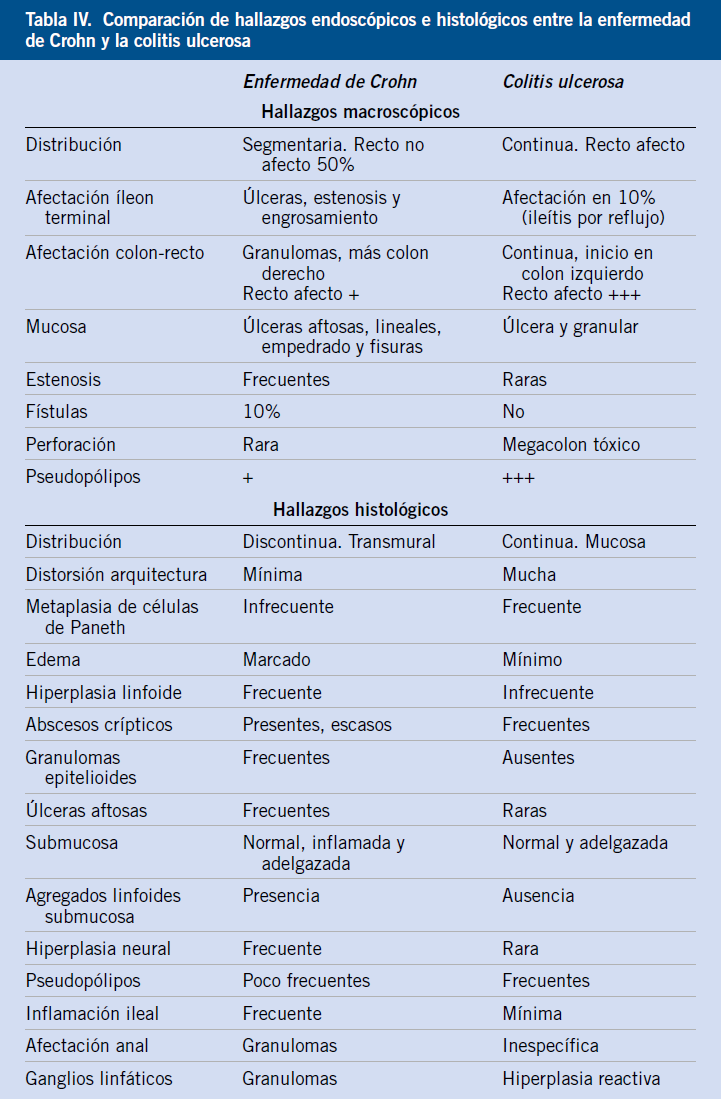
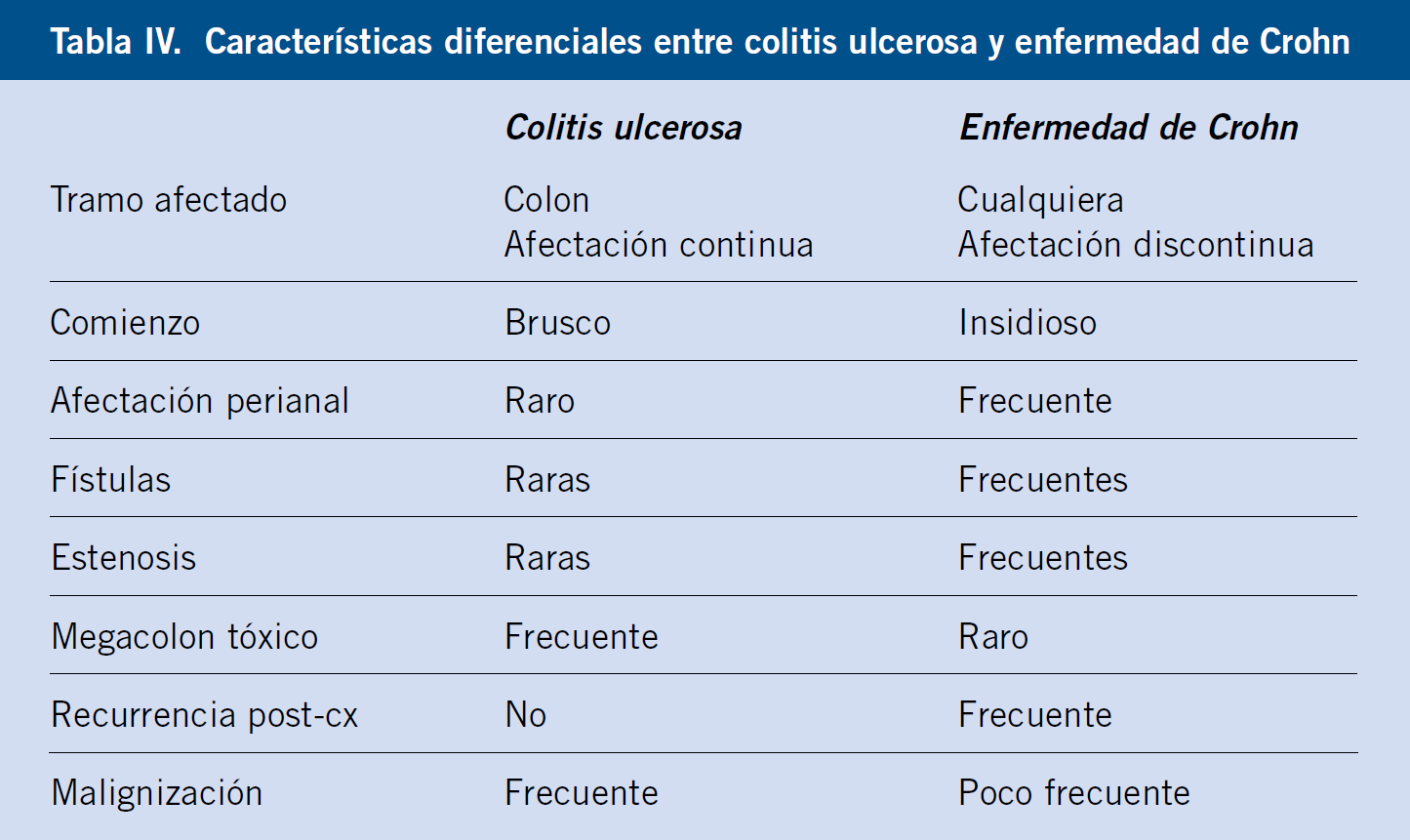
Las características clínicas más importantes de cada enfermedad se muestran en la tabla IV.


Diarreas y enteropatías congénitas
Son trastornos monogénicos, algunos de ellos extremadamente infrecuentes, con herencia autosómica recesiva, que se manifiestan en los días inmediatos al nacimiento e incluso intraútero, aunque hay excepciones, como es el caso del déficit de sacarasa-isomaltasa que se inicia con la introducción de la alimentación complementaria (frutas, zumos y alimentos con almidón)(7). Fundamentalmente, producen diarrea líquida, pero también pueden causar diarrea con sangre (enfermedad inflamatoria intestinal de comienzo muy precoz) o esteatorrea (déficit primario de sales biliares y alteraciones en el transporte o metabolismo de la grasa). Las diarreas congénitas se pueden dividir en 5 grupos en relación a su mecanismo fisiopatológico:
1. Alteraciones de los transportadores epiteliales(3,8,9).
2. Alteraciones de las enzimas epiteliales y el metabolismo(3,7,10,11).
3. Alteraciones estructurales y funcionales de la absorción(3,8).
4. Neuroendocrinopatías entéricas(3).
5. Enteropatías asociadas con alteraciones de la inmunorregulación(3,12) (Tabla IV).
El trastorno más frecuente de este grupo es el déficit primario de lactasa, hipolactasia de tipo adulto o no persistencia de la lactasa. Se debe a la disminución de la función de la lactasa-floricina hidrolasa, enzima presente en la zona más apical de las vellosidades intestinales. Es un trastorno autosómico recesivo que puede aparecer en la raza caucásica a partir de los 5 años. Los síntomas se inician con la ingesta de leche o productos lácteos, cuando el porcentaje de actividad de la enzima es inferior al 50 % y son muy variados en intensidad, ya que la adaptación de la flora intestinal contribuye a la tolerancia. La deficiencia secundaria de lactasa es transitoria y adquirida tras lesión de la mucosa intestinal.
Además de las diarreas congénitas, los recién nacidos pueden presentar diarreas por malformaciones congénitas digestivas, como en el intestino corto (<75 cm de longitud) y en la enterocolitis como complicación en la enfermedad de Hirschsprung. Otras diarreas crónicas son adquiridas y más frecuentes en prematuros, como los defectos anatómicos intestinales (atresias) o la enterocolitis necrotizante(3).
Respuesta inmune anómala
Este grupo comprende enfermedades mucho más frecuentes en la práctica clínica, como la enfermedad celiaca, la alergia a las proteínas de la leche de vaca (APLV) no IgE mediada(13) y la enfermedad inflamatoria intestinal (EII). Los trastornos gastrointestinales eosinofílicos primarios producen una gran variedad de síntomas, entre los que se incluye la diarrea crónica, y van adquiriendo protagonismo en las unidades especializadas. Las dos entidades que producen diarrea son la gastritis eosinofílica y la colitis eosinofílica(14). Por último, en la colitis microscópica, que engloba la colitis linfocítica y la colitis colágena, la mucosa tiene un aspecto normal y son los hallazgos histológicos específicos los que permiten el diagnóstico, al ser ambas enfermedades clínicamente indistinguibles(15).
Insuficiencia pancreática exocrina
En la insuficiencia pancreática exocrina se produce una disminución de la secreción de enzimas pancreáticas, bicarbonato o ambos, dando lugar a una maldigestión de los nutrientes. Las principales enzimas secretadas son la amilasa para la digestión de carbohidratos, la lipasa para la digestión de la grasa y las proteasas (tripsinógeno y quimotripsina) para la digestión de las proteínas. La insuficiencia de amilasa no origina síntomas, ya que se compensa por la secreción de amilasa salivar y de las glándulas del intestino delgado. Cuando la secreción de tripsina es menor del 5-10 % de lo normal, se produce un exceso de pérdida de nitrógeno en las heces. Por tanto, la principal consecuencia de la insuficiencia pancreática es la malabsorción de grasas.
Los síntomas más característicos son la diarrea crónica, la esteatorrea y la pérdida de peso asociadas a distensión abdominal, que aparecen cuando la función del páncreas es inferior al 10 %. Asimismo, se produce un déficit de las vitaminas liposolubles (A, D, E, K) y oligoelementos, como el magnesio y zinc(16).
Diarrea inducida por fármacos
• Diarrea asociada a antibióticos: aparece hasta 2 semanas tras el inicio de antibióticos, como cefalosporinas, ampicilina y amoxicilina-clavulánico.
• Colitis pseudomembranosa: causada por el Clostridium difficile en niños tratados con antibióticos (clindamicina, penicilina, fluoroquinolonas y cefalosporinas). También, puede producirse en niños con enfermedad inflamatoria intestinal o con inmunodeficiencias(17).
• Diarreas no asociadas a antibióticos: el abuso de laxantes (polietilenglicol, hidróxido de magnesio), sorbitol y edulcorantes artificiales y el tratamiento con eritromicina, que acelera el tránsito intestinal, no alteran la mucosa intestinal. Sin embargo, los antiinflamatorios no esteroideos y los inhibidores de la bomba de protones (IBP) pueden producir enteritis(17).
Miscelánea
El síndrome de sobrecrecimiento bacteriano (SIBO) se caracteriza por un crecimiento excesivo de microorganismos en el intestino delgado, bien de tipo colónico (coliformes) o de bacterias orofaríngeas y respiratorias. En un estudio reciente, los dos factores más frecuentemente asociados a su desarrollo fueron el antecedente de infección gastrointestinal en el último año y el uso de IBP en el último mes. Solo un 7,4 % tenía antecedentes de cirugía intestinal previa y hasta en un 22,2 % el cuadro recurrió tras el tratamiento(18).
Diagnóstico
El diagnóstico de la diarrea crónica se basa en una anamnesis y exploración física completas y en la realización de pruebas complementarias y herramientas diagnósticas de primer y/o de segundo nivel.
Historia clínica y exploración física
Al valorar al paciente con diarrea crónica, es muy importante evaluar la edad de comienzo y su posible relación con la introducción de nuevos alimentos, tratamientos farmacológicos previos o en curso, las características de las heces y los antecedentes familiares de enfermedades digestivas.
Las características de las deposiciones nos pueden informar sobre los mecanismos responsables de la diarrea: intolerancia a hidratos de carbono (deposiciones líquidas, explosivas y ácidas, pudiendo asociar eritema perianal); maldigestión de grasas (deposiciones pegajosas, brillantes, pálidas y que flotan); origen autoinflamatorio (deposiciones con moco y/o sangre, frecuentes, pequeñas, pudiendo asociar tenesmo y/o hábito nocturno) y origen funcional (deposiciones blandas, cuya consistencia disminuye a lo largo del día y que pueden contener restos de alimentos, en ocasiones, alternando con deposiciones normales o duras).
La exploración física y la valoración antropométrica son fundamentales para evaluar la posible repercusión nutricional de la diarrea y para detectar signos y síntomas de alarma. Se debe registrar la curva de peso y talla del paciente, ya que el descenso de esta puede ser un dato de patología orgánica.
En los trastornos funcionales, una clínica compatible y la ausencia de datos de alarma son suficientes para establecer el diagnóstico; mientras que, si se sospecha patología orgánica, habitualmente son necesarias pruebas complementarias orientadas en función de la edad del paciente, el cuadro clínico y los hallazgos a la exploración.
Pruebas complementarias de primer nivel
Son suficientes para establecer el diagnóstico de las causas más frecuentes de diarrea crónica en Atención Primaria.
• Análisis sanguíneo: hemograma, bioquímica con glucemia, urea, creatinina, proteínas totales, albúmina, colesterol, triglicéridos, iones, calcio, fósforo, fosfatasa alcalina y ferritina. Además, se recomienda solicitar: inmunoglobulinas, TSH, serología de enfermedad celiaca y reactantes de fase aguda, como VSG y proteína C-reactiva. Estos parámetros nos permitirán el diagnóstico de enfermedades, como la enfermedad celiaca y el hipertiroidismo, además de evaluar la posible malabsorción de nutrientes o la repercusión bioquímica. Así, en procesos inflamatorios podemos encontrar anemia (de origen multifactorial), trombocitosis, hipoalbuminemia y elevación de los reactantes de fase aguda. La anemia también puede estar presente en casos de malabsorción de hierro, ácido fólico y/o vitamina B12, y los niveles bajos de proteínas totales y albúmina en suero pueden orientar hacia una enteropatía pierde proteínas(8).
• Examen microbiológico de heces:
– Coprocultivo y antígenos virales: aunque estos patógenos producen habitualmente cuadros de diarrea aguda, pueden cronificarse, principalmente en niños con inmunodeficiencias.
– Parásitos en heces: resulta fundamental su determinación, ya que la infección por Giardia lamblia es la causa infecciosa más frecuente de diarrea crónica en los países desarrollados. Debido a la eliminación intermitente de los quistes del parásito, la recogida de heces de tres días diferentes aumenta la sensibilidad del estudio.
– Toxina de Clostridium difficile: indicado si la diarrea se acompaña de sangre en las heces, principalmente en pacientes inmunodeprimidos, con enfermedad inflamatoria intestinal o que hayan recibido tratamiento antibiótico reciente.
• Prueba terapéutica de exclusión-provocación: se puede considerar una prueba empírica de exclusión y provocación posterior, si se sospecha alergia no IgE mediada o intolerancia digestiva, como puede ocurrir con las proteínas de leche de vaca (en lactantes) o con algunos hidratos de carbono (lactosa o fructosa).
Pruebas complementarias de segundo nivel
En el caso de que tras una valoración clínica completa y un estudio de primer nivel no se obtenga el diagnóstico de la diarrea crónica, se deberán realizar estudios específicos por parte del pediatra gastroenterólogo. La elección de las pruebas a realizar vendrá orientada por la sospecha diagnóstica en función de los síntomas y las características de la diarrea.
• Analítica sanguínea orientada según sospecha clínica:
– Existen anticuerpos que pueden ser positivos en EII, como el anti-Saccharomyces cerevisiae (ASCA), más frecuente en enfermedad de Chron y los anticuerpos anti-citoplasma de neutrófilos (p-ANCA), que puede estar elevado en colitis ulcerosa(2).
– Los niveles bajos de vitaminas liposolubles (A, E y D) se detectan en caso de insuficiencia pancreática.
– Niveles bajos de zinc, en el contexto de diarrea crónica asociada a acrodermatitis, pueden orientar a la acrodermatitis enteropática.
• Estudios en heces:
– Test de Van de Kamer: consiste en la determinación de grasa en heces en 72 horas. Los recién nacidos y los lactantes excretan entre el 15 y el 20 % de la grasa de la dieta en las heces. El coeficiente de absorción de grasas aumenta con la edad y alcanza valores adultos de >95 % a partir del año de vida. Los valores normales son <3 g/24 horas en niños y <6 g/24 horas en adultos. Es recomendable hacer una encuesta dietética de tres días con la determinación del coeficiente de absorción grasa (grasa ingerida/grasa excretada) para evitar falsos negativos por baja ingesta de grasas. Aunque es el gold standard, han aparecido otras técnicas, como el análisis por reflexión en el infrarrojo cercano (NIRA), que facilita el manejo de la muestra y, además, aporta datos sobre la absorción de azúcares (valores normales <2,5 %), almidón (valores normales <1 %) y contenido de agua (valores normales <85 %).
– Osmolalidad de las heces: los electrolitos de las heces se utilizan para calcular el gap osmótico fecal. En la diarrea secretora, el gap osmótico en las heces es <50 mOsm/kg, mientras que en la diarrea osmótica es >125 mOsm/kg(2).
– Sustancias reductoras (>1 %) y pH en heces (<5,3) indican malabsorción de carbohidratos. Hay que considerar que la sacarosa no es un azúcar reductor.
– Elastasa-1 fecal: es una enzima pancreática resistente a la degradación por la flora intestinal, por lo que cuando el valor es bajo (<200 μcg/g de heces) sugiere insuficiencia pancreática exocrina(8).
– α1-antitripsina fecal: proteína sérica sintetizada en el hígado, resistente a la proteólisis, que normalmente está presente en bajas concentraciones en las heces. Valores ≥2 mg/g de heces en una muestra pueden indicar pérdida de proteínas.
– Calprotectina fecal: proteína citosólica de los neutrófilos que se eleva en cuadros de inflamación intestinal, siendo normales valores <50 μg/g. A pesar de su alta sensibilidad, este parámetro puede estar elevado en infecciones gastrointestinales, pólipos juveniles, uso de antiinflamatorios no esteroideos o hemorragia gastrointestinal. Además, los valores de calprotectina tienen una correlación negativa significativa con la edad, observándose niveles muy altos, de hasta 1.500 μg/g, en lactantes sanos y una amplia variabilidad hasta los 4 años, por lo que hay que tener precaución con su interpretación en niños menores de esta edad(19).
– Sangre oculta en heces: marcador inespecífico que indica pérdida gastrointestinal de sangre.
• Test de aliento: las pruebas de aliento de lactosa y fructosa consisten en administrar estos carbohidratos marcados, midiendo posteriormente el H2 exhalado. En cuadros de malabsorción de carbohidratos, estos no se absorben, por lo que son fermentados en el colon, produciendo H2 y metano, que pasan a la sangre y se eliminan durante la exhalación. En el SIBO, las bacterias del intestino delgado fermentan la glucosa antes de su absorción, detectándose un aumento temprano del hidrógeno en el aliento. Ante una sospecha de intolerancia a hidratos de carbono, estas pruebas solo están indicadas en casos de no respuesta clínica concluyente a la exclusión-provocación.
• Endoscopia y estudio histológico: esta prueba es fundamental en enfermedades, como la enfermedad inflamatoria intestinal, la enfermedad celiaca, las enteropatías congénitas, la colitis microscópica, los trastornos gastrointestinales eosinofílicos primarios, la abetalipoproteinemia y la linfangiectasia intestinal, así como para el estudio de disacaridasas en mucosa intestinal.
• Estudios genéticos: la mayoría de las diarreas congénitas, la fibrosis quística (FQ), la EII monogénica y los trastornos de inmunodeficiencia tienen una base genética. El análisis genético ayuda a realizar un diagnóstico definitivo y a orientar el manejo.
• Test del sudor: estimulación del sudor mediante iontoforesis con pilocarpina. Se realiza un primer test mediante conductividad y si los valores son >50 mmol/l, se medirá la concentración de cloro en sudor. Dos determinaciones de cloro de >60 mmol/L confirman el diagnóstico de FQ. Se consideran dudosos valores entre 40-60 mmol/L.
• Pruebas de imagen: la ecografía abdominal y la enterorresonancia tienen un importante papel en el estudio de la EII. La albúmina marcada con Tc99 permite visualizar la fuga de linfa en la linfangiectasia intestinal.
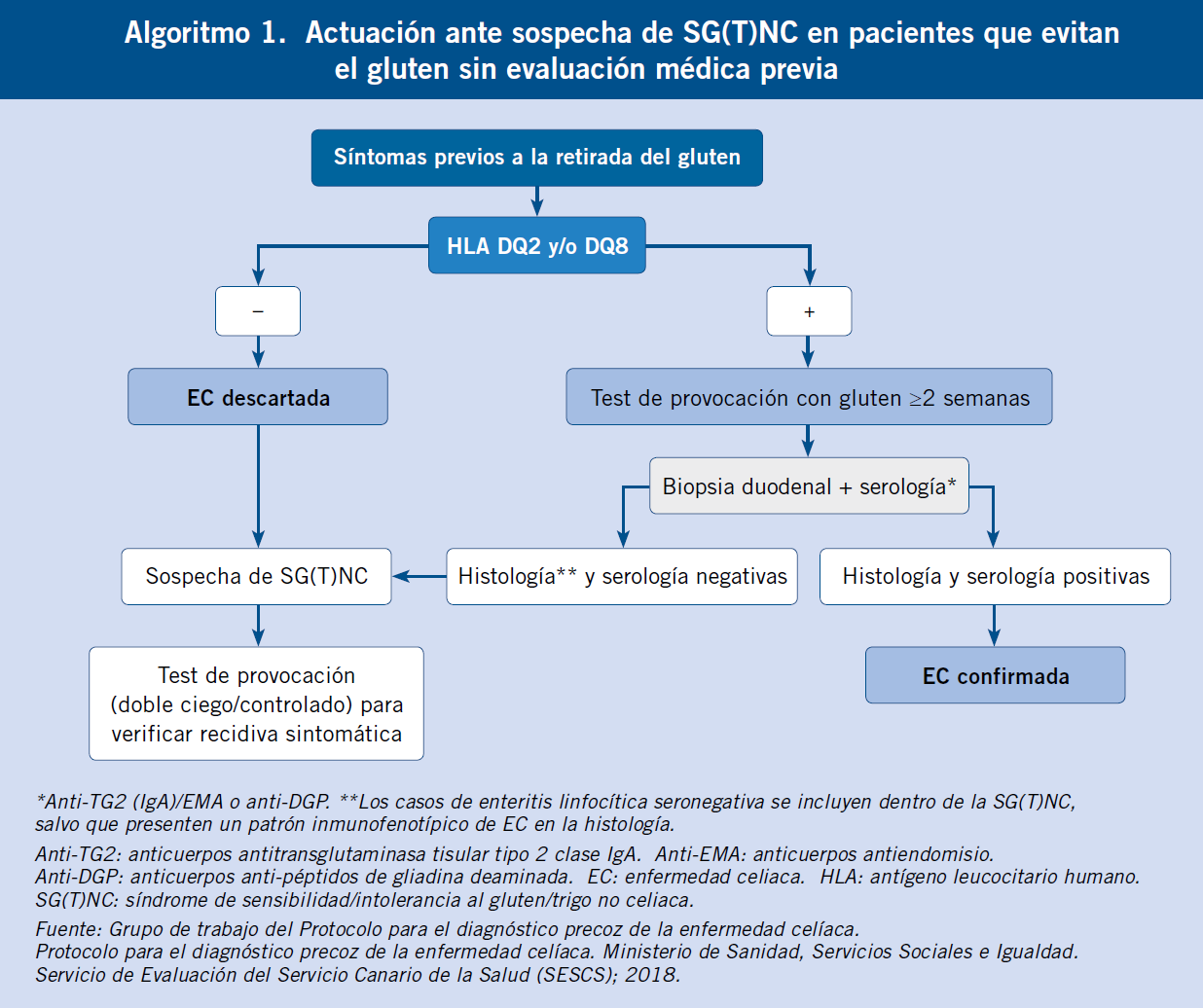
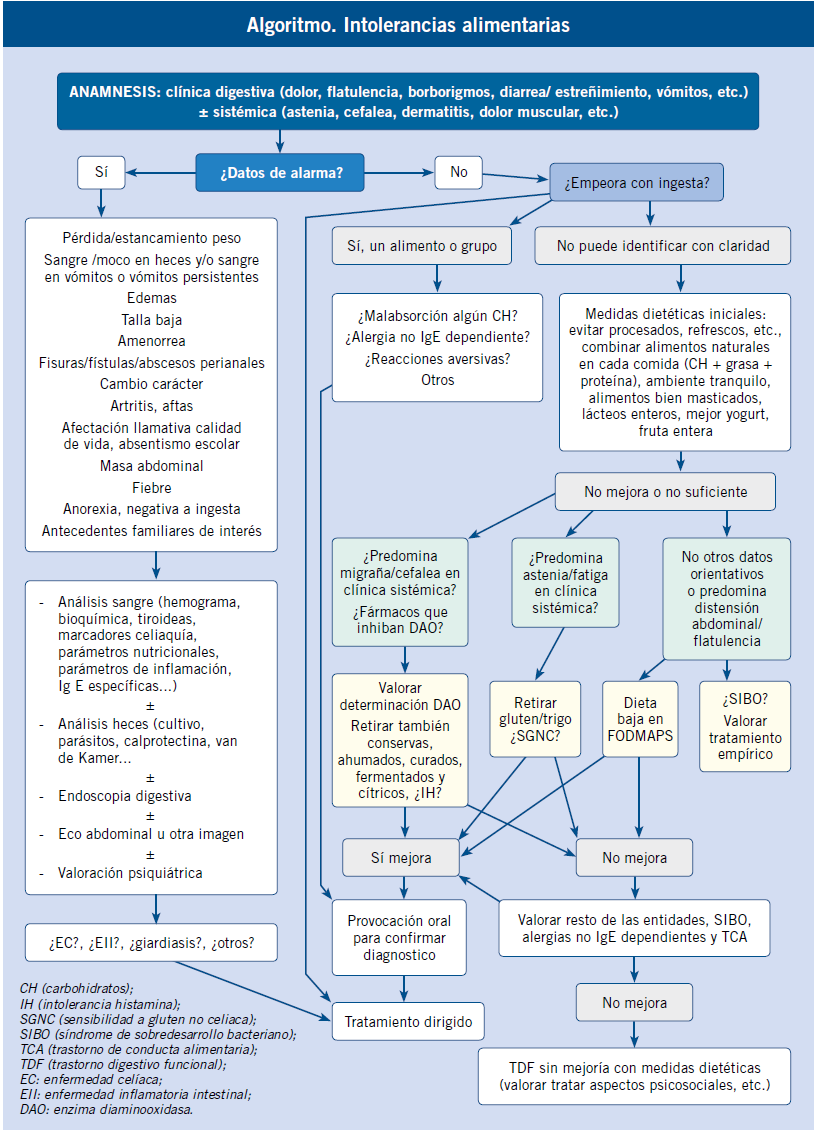
Al final del artículo se muestra el algoritmo diagnóstico de la diarrea crónica, con las exploraciones de primero y segundo nivel de los diferentes tipos fisiopatológicos de diarrea y por etiología.
Tratamiento
El tratamiento de la diarrea crónica se basa en el manejo general de la misma, asegurando un adecuado soporte nutricional, y en el tratamiento específico de la causa subyacente.
Tratamiento general
El tratamiento de la diarrea crónica requiere un diagnóstico preciso para un manejo adecuado y específico de la causa subyacente, siendo la rehabilitación nutricional el aspecto más importante en el tratamiento de estos pacientes. El aporte adecuado de nutrientes facilitará la recuperación de la mucosa intestinal, siendo la vía oral la de elección. En ocasiones, pueden ser necesarios suplementos nutricionales para cubrir los requerimientos del paciente. En casos de mala tolerancia oral o compromiso en la seguridad o la eficacia deglutoria, la alimentación puede administrarse a través de sonda nasogástrica o transpilórica a débito continuo o intermitente. La vía parenteral se reservará para aquellos pacientes con mala tolerancia enteral, desnutrición grave y diarrea intratable. El empleo de fármacos inhibidores del peristaltismo intestinal no está recomendado en población pediátrica. Los probióticos solo estarían indicados en la prevención de la diarrea asociada a antibióticos y no hay suficiente evidencia en la prevención o tratamiento del SIBO.
Tratamiento específico
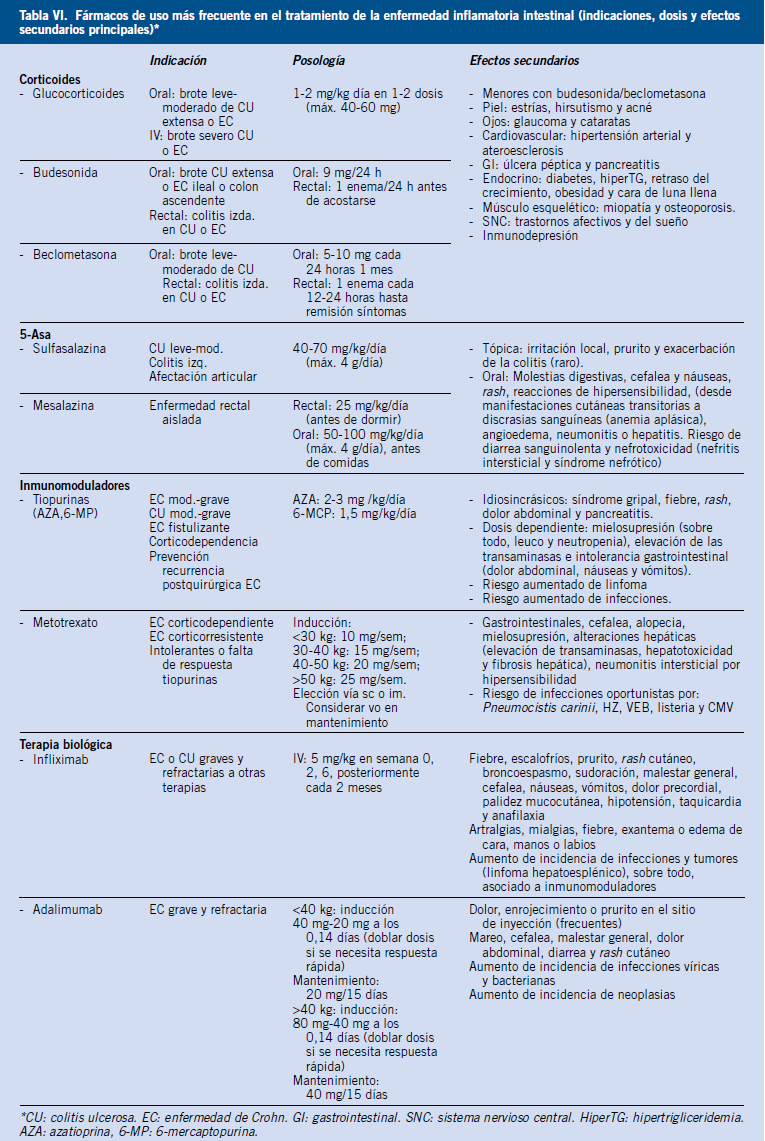
• Tratamiento dietético: será el de elección en patologías en las que existe una malabsorción por deficiencia de enzimas o proteínas transportadoras, como sucede en la malabsorción de glucosa-galactosa, intolerancia a la lactosa, a la fructosa o en el déficit de sacarasa-isomaltasa, basándose el manejo de estos cuadros clínicos en la retirada parcial o total del hidrato de carbono implicado(20). También es el tratamiento de elección en diarreas crónicas por alergias no IgE mediadas, como en la APLV(13). La dieta estricta sin gluten es el tratamiento de la enfermedad celiaca. En la linfangiectasia intestinal, el manejo nutricional es fundamental, indicándose una dieta pobre en grasas, con triglicéridos de cadena media (MCT) y rica en proteínas(20). En las alteraciones en el transporte y metabolismo de las grasas, como la abetalipoproteinemia, se restringirán las grasas de la dieta y se aportarán ácidos grasos esenciales(20). En la enfermedad de Crohn, el tratamiento dietético tiene un papel importante como alternativa a los corticoides. En la fibrosis quística se precisa suplementación con vitaminas liposolubles. En las diarreas que cursan con enteropatía pierde-proteínas, se precisa dieta sin o baja en grasa, con aceite MCT, rica en proteínas y suplementación de vitaminas liposolubles y ácidos grasos esenciales(20).
En la diarrea funcional, una dieta baja en oligosacáridos, disacáridos, monosacáridos y polioles fermentables (FODMAPS) puede ser útil en el síndrome de intestino irritable, aunque debe indicarse con precaución y realizarse un seguimiento estrecho por el riesgo de repercusiones nutricionales(20).
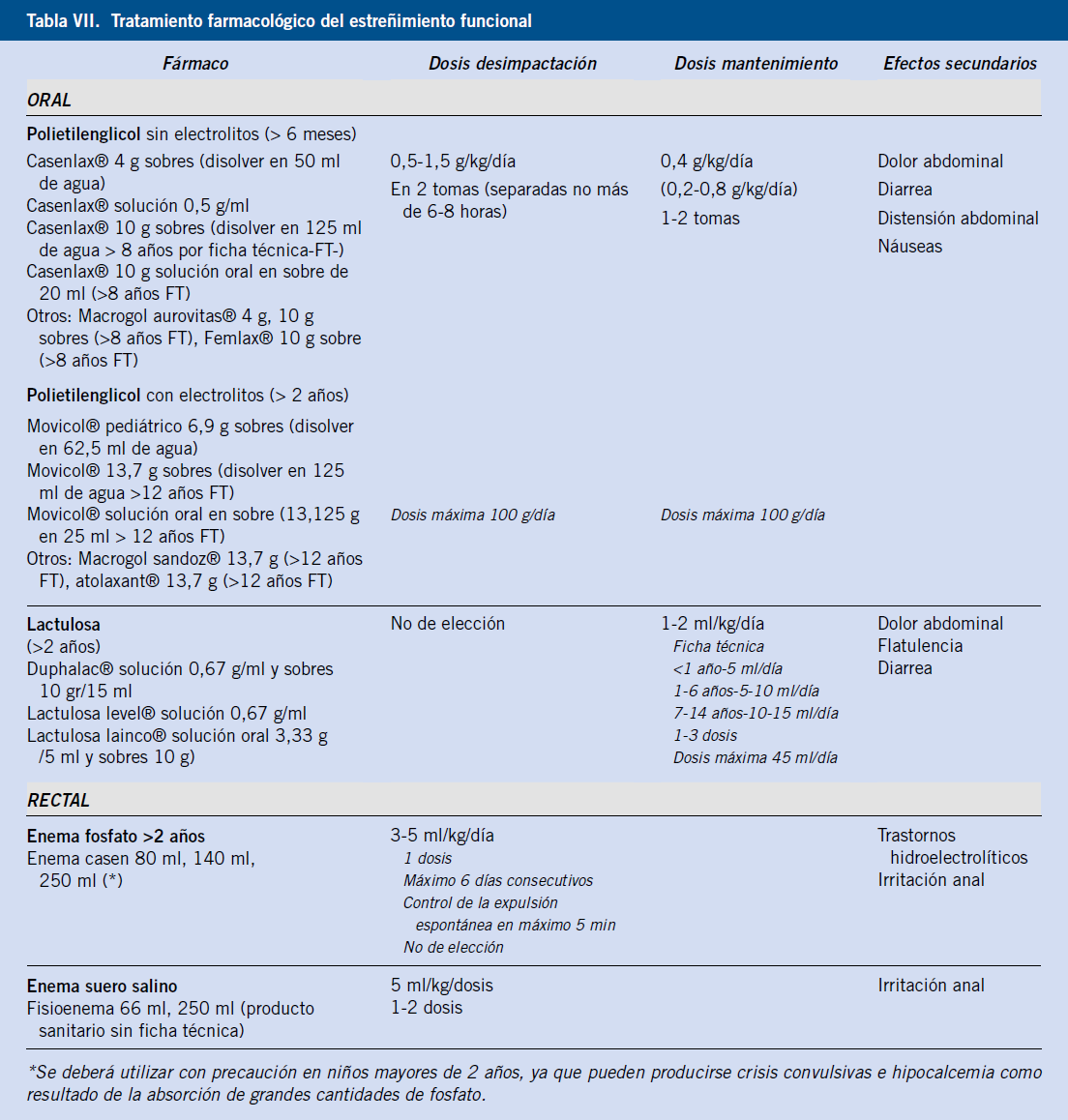
• Tratamiento farmacológico: los tratamientos específicos dependerán de la etiología de la diarrea. Los antibióticos son empleados fundamentalmente en la diarrea de causa infecciosa, aunque también pueden indicarse en el SIBO (rifaximina o metronidazol)(18). Los corticoides y los fármacos inmunomoduladores son ampliamente utilizados en la EII y en patologías menos frecuentes, como la colitis microscópica(15), los trastornos eosinofílicos gastrointestinales(14), la enteropatía autoinmune y la linfangiectasia intestinal(8). Otros tratamientos específicos incluyen: el tratamiento enzimático de sustitución en la insuficiencia pancreática exocrina; las resinas de intercambio iónico en la diarrea por ácidos biliares; y el octreótido en la linfangiectasia intestinal(8).
Función del pediatra de Atención Primaria
• Realizar la historia clínica y exploración física iniciales, detallando las características de las heces y la situación nutricional del paciente.
• Pedir las pruebas de primer nivel, con las que se pueden diagnosticar las causas más frecuentes de diarrea crónica.
• Derivación a la consulta de gastroenterología pediátrica, si no se ha llegado al diagnóstico y el paciente mantiene los síntomas.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. World Health Organization. Diarrhoeal Disease Fact Sheet N 330. 2017. Disponible en: http://www.who.int/mediacentre/factsheets/fs330/en/.
2.*** Tripathi PR, Srivastava A. Approach to a Child with Chronic Diarrhea. Indian J Pediatr. 2024; 91: 472-80.
3.** Thiagarajah JR, Kamin DS, Acra S, Goldsmith JD, Roland JT, Lencer WI, et al. Advances in Evaluation of Chronic Diarrhea in Infants. Gastroenterology. 2018; 154: 2045-59.
4. Benninga MA, Nurko S, Faure C, Hyman PR, Roberts IJ, Schechter NL. Childhood Functional Gastrointestinal Disorders: Neonate/Toddler. Gastroenterology. 2016; 150: 1443-55.e2.
5. Rajindrajith S, Hathagoda W, Devanarayana NM. Functional Diarrhea in Children. Indian J Pediatr. 2024; 9: 584-9.
6. Hyams JS, Di Lorenzo C, Saps M, Shulman RJ, Staiano A, van Tilburg M, et al. Childhood Functional Gastrointestinal Disorders: Child/Adolescent. Gastroenterology. 2016; 150: 1456-68.
7. Naim HY, Heine M, Zimmer KP. Congenital Sucrase-Isomaltase Deficiency: Heterogeneity of Inheritance, Trafficking, and Function of an Intestinal Enzyme Complex. J Pediatr Gastroenterol Nutr. 2012; 55: S13-20.
8.*** Shankar S, Rosenbaum J. Chronic diarrhoea in children: A practical algorithm-based approach. J Ped Child Health. 2020; 56: 1029-38.
9. Gu L, He XH, Zhu P. Analysis of similarities and differences between transient symptomatic zinc deficiency an acrodermatitis enterophatica in children: a case report of a Chinese Yi-ethnic infant. BMC Pediatr. 2024; 24: 338. Disponible en: https://doi.org/10.1186/s12887-024-04830-y.
10. Sissaoui S, Cochet M, Poinsot P, Bordat C, Collardeatu-Frachon S, Lachaux A, et al. Lipids reponsible for intestinal or hepatic disorder. When to suspect a familiar intestinal hypocholesterolemia? J Ped Gastroenterol Nutr. 2021; 73: 4-8.
11. Niu Y, Wu Q, Wang Y, Lu L. Feng Y, Cai W, et al. Primary intestinal lymphangiectasia in children: Twelve years of experience in the diagnosis and management. Asia Pac J Clin Nutr. 2021; 30: 358-64.
12. Jin M, Gong Y, Liu W, Zhong X. Clinical characteristics and management of autoimmune enteropathy in children: case reports and literature review. BMC Pediatr. 2023; 23: 601. Disponible en: https://doi.org/10.1186/s12887-023-04435-x.
13. Vandenplas Y, Broekaert I, Domellöf M, Indrio F, Lapillonne A, Pienar C, et al. An ESPGHAN Position Paper on the Diagnosis, Management, and Prevention of Cow’s Milk Allergy. J Ped Gastroenterol Nutr. 2024; 78: 386-413.
14. Papadopoulou A, Amil-Dias J, Auth MKH, Chehade M, Collins MH, Gupta SK, et al. Joint ESPGHAN/NASPGHAN Guidelines on Childhood Eosinophilic Gastrointestinal Disorders Beyond Eosinophilic Esophagitis. J Pediatr Gastroenterol Nutr. 2024; 78: 122-52. Disponible en: https://doi.org/10.1097/mpg.0000000000003877.
15. Khushal S, Hemker MO. Diagnosis and Management of Microscopic Colitis in Pediatric Patients. Pediatric Drugs. 2022; 24: 217-33.
16. Sankararaman S, Schindler T, Sferra TJ. Management of Exocrine Pancreatic Insufficiency in Children. Nutrition in Clinical Practice. 2019; 34: S27-42.
17.** Sathiyasekaran M, Ganesh R, Natarajan S. Other Causes of Chronic Diarrhea in Children. Indian J Pediatr. 2024; 91: 606-13.
18. Cho YK, Chang J, Paik CN. Prevalence, risks factors, and treatment of small intestinal overgrowth in children. Clin Exp Pediatr. 2023; 66: 377-83.
19. Lężyk-Ciemniaka E, Tworkiewicza M, Wilczyńskaa D, Szaflarska-Popławskab A, Krogulskaa A. Usefulness of Testing for Fecal Calprotectin in Pediatric Gastroenterology Clinical Practice. Med Princ Pract. 2021; 30: 311-9.
20.*** Shankar DS, Durairaj E. Diet and Management of Diarrhea. Indian Journal of Pediatrics. 2024; 91: 590-7.
21. Ramos Boluda E, González Sacristán R. Diarrea crónica. Pediatr Integral. 2019; XXIII: 386-91. Disponible en: https://www.pediatriaintegral.es/publicacion-2019-12/diarrea-cronica-2/.
Bibliografía recomendada
– Tripathi PR, Srivastava A. Approach to a Child with Chronic Diarrhea. Indian J Pediatr. 2024; 91: 472-80.
Artículo de revisión en el que se explican los distintos tipos clínicos de diarrea crónica. Asimismo, se actualizan los procedimientos diagnósticos de los que se dispone y se muestran algoritmos diagnósticos exhaustivos para efectuar el diagnóstico diferencial.
– Shankar S, Rosenbaum J. Chronic diarrhoea in children: A practical algorithm-based approach. J Ped Child Health. 2020; 56: 1029-38.
Artículo de revisión en el que, además de mostrar algoritmos diagnósticos, se valoran los tratamientos farmacológicos disponibles para algunos tipos de diarrea crónica.
– Shankar DS, Durairaj E. Diet and Management of Diarrhea. Indian Journal of Pediatrics. 2024; 91: 590-7.
Excelente artículo de revisión en el que se valora de forma detallada el tratamiento de las diferentes causas de diarrea crónica, cuya terapia es exclusivamente dietética.
| Caso clínico |
|
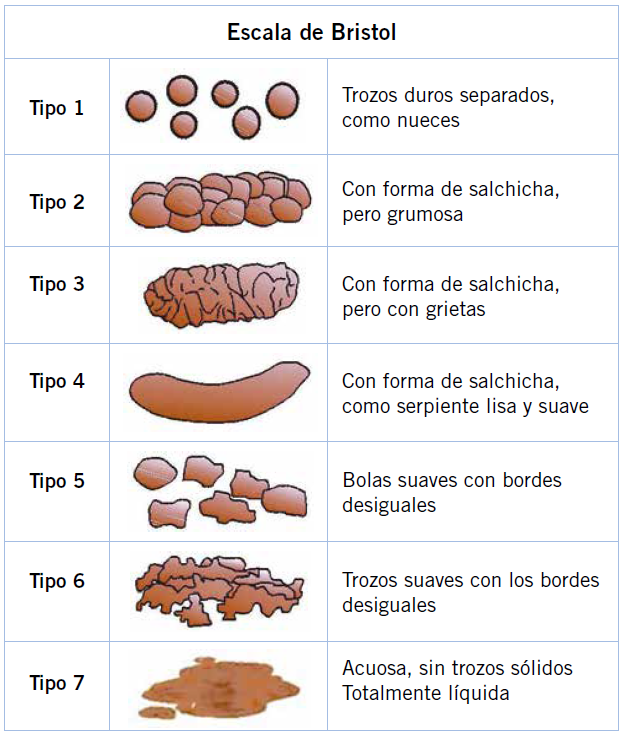
Niño de 3 años con antecedente de dermatitis atópica y alergia a huevo IgE mediada que acude a consulta por diarrea de 6 meses de evolución. Los padres refieren que previamente realizaba una o dos deposiciones diarias tipo 4 Bristol. Sin embargo, tras el inicio de guardería presentó múltiples procesos infecciosos, entre ellos una gastroenteritis aguda de 5 días de evolución, con diarrea, fiebre y vómitos. Desde entonces, el paciente realiza 2-3 deposiciones al día tipo 6 Bristol con abundante meteorismo. Asocia distensión abdominal tras las comidas y molestias periumbilicales. No ha presentado pérdida de apetito y tiene una curva ponderoestatural ascendente. No relacionan la diarrea y los síntomas acompañantes a la ingesta de ningún alimento específico. Los padres retiraron la lactosa de la dieta con mejoría parcial durante una semana. Sin embargo, posteriormente recayeron de nuevo los síntomas antes descritos. Presenta buen estado de nutrición y de hidratación, con exploración por aparatos normal, en la que solo destaca un abdomen globuloso, aunque blando y depresible, sin masas ni megalias, que no impresiona de doloroso a la palpación. Ante diarrea crónica, se realiza coprocultivo y parásitos en heces, que son negativos, y analítica de sangre con hemograma y bioquímica (con inmunoglobulinas) normales, salvo anti-péptido deaminado de gliadina IgG positivo (40 U/ml), con anti-transglutaminasa IgA y antiendomisio negativos.
|