Enfermedades pediátricas que han pasado a la historia (4): sífilis congénita
“El que conoce la sífilis, conoce la medicina”
William Osler (1894-1919)
“La santa ignorancia no es la Santa Inocencia”
Adolphe Pinard (1844-1934)
Generalidades. Apunte histórico. Definiciones
La sífilis es una enfermedad infecciosa transmitida principalmente por contacto sexual, producida por una bacteria espiroqueta denominada Treponema pallidum subespecie pallidum(1-3). Puede ir asociada a otras enfermedades de transmisión sexual y VIH, en heterosexuales y en hombres que tienen sexo con hombres. También se transmite por vía transplacentaria y contacto estrecho a través de mucosas. Está extendida por todo el mundo. La evolución natural es hacia un curso crónico, que puede producir discapacidad progresiva y ser mortal. Sus manifestaciones son fluctuantes, versátiles, en características clínicas e intensidad, apareciendo y desapareciendo en las distintas etapas de la enfermedad, y puede simular signo-sintomatología de diversas enfermedades.
Se conocen(1-3) tres subespecies de T. pallidum:
• Treponema pallidum pallidum. Causa la sífilis o sífilis venérea.
• T. p. endemicum. Causa bejel o sífilis endémica.
• T. p. pertenue. Causa frambesia, buba o pian (en inglés yaws).
A veces se incluye como subespecie al Treponema herrejoni (carateum), que causa el Pinta o Mal de Pinto.
Las subespecies de T. pallidum son morfológica y serológicamente indistinguibles.
Desde mediados de los años 40 del siglo XX hasta finales de siglo, la sífilis había experimentado un notable descenso en las tasas de incidencia debido al tratamiento con la penicilina, que es muy eficaz y de primera línea, así como con el uso de preservativo y otras medidas de prevención de enfermedades venéreas. El acceso a los sistemas sanitarios públicos y la generalización del cribado diagnóstico en las embarazadas, abrió una ventana para que la sífilis congénita (SC) pudiera desaparecer. La OMS, desde el año 2006, organiza planes para erradicar la SC(4,5). Los casos de enfermedad crónica de larga evolución podrían parecer reliquias de la era de una medicina pasada. O quizás no lo sea. Se afirma que está teniendo un resurgimiento en estos principios del siglo XXI; todavía afecta a un millón de mujeres embarazadas en todo el mundo(2,4,5).
La sífilis es conocida desde hace varios siglos y está presente, en numerosas publicaciones impresas desde finales del siglo XV. Su estudio histórico nos ofrece muchas enseñanzas sobre todas las áreas de la medicina: infectología, epidemiología, higiene, inmunología, clínica, diagnósticos de laboratorio, anatomía patológica, tratamiento farmacológico, salud pública e historiografía. Ha contribuido decisivamente en el aprendizaje en áreas de ética en la investigación. La enfermedad ha tenido un impacto en todas las actividades humanas: social, económico, en los estilos de vida, político, ideológico, periodístico, etc.
El manejo de la SC en la historia nos ha aportado direcciones decisivas en el valor social de la pediatría. La sífilis congénita entró en la historia y en la cultura; aunque aún no sea historia definitivamente.
Sífilis congénita. Clínica y datos diagnósticos
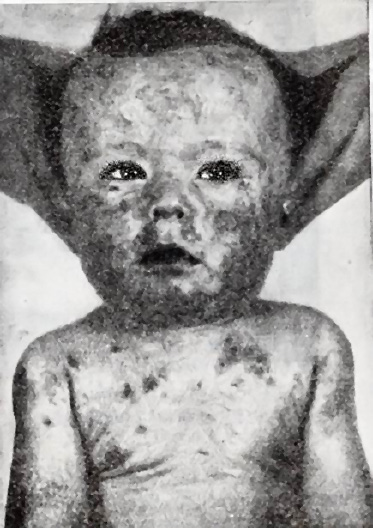
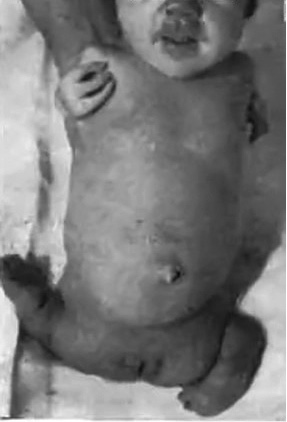
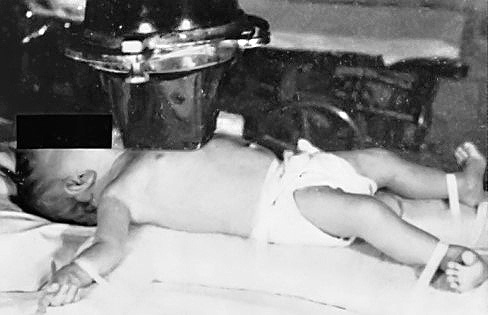
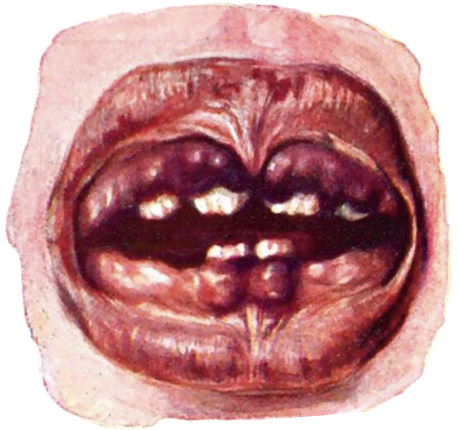
La sífilis congénita es una infección multisistémica por T. pallidum transmitida al feto a través de la placenta. Para una revisión actual, remitimos a otras referencias(1,5-7). El riesgo global de transmisión es hasta un 60-80% en la sífilis materna primaria o secundaria no tratada, disminuye hasta un 10-20% en la sífilis latente o terciaria. La amplia variedad de manifestaciones de sífilis congénita es determinada por el estadio de la sífilis materna en el embarazo al momento de la infección, el diagnóstico materno temprano, las reacciones inmunológicas y el tratamiento de la mujer y del feto. La sífilis no tratada puede producir muerte fetal (hasta un 40% de los afectados) y neonatal o manifestaciones congénitas precoces o tardías. La sífilis congénita precoz, definida por convenio, como la que ocurre desde el nacimiento hasta los 2 años de edad, se manifiesta con lesiones cutáneas vesiculoampollosas típicas o exantema maculoso en palmas y plantas, lesiones papulares alrededor de boca-nariz y zona de pañal (Fig. 1).

Figura 1. Sífilis papulo-pustulosa, descrita por Friedinger and Mayr in 1859. Tomado de Obladen(8).
También, pueden aparecer petequias en piel, rinitis mucopurulenta característica, linfadenopatías y hepatoesplenomegalia; en algunos casos, meningitis, hidrocefalia o convulsiones, discapacidad intelectual, retraso de crecimiento y osteocondritis, con alteraciones radiológicas distintivas. Inmediatamente al nacer y durante días o semanas, el recién nacido puede estar aparentemente asintomático. En la sífilis congénita, el compromiso pulmonar es poco frecuente, aunque la neumonía alba es un hallazgo necrópsico habitual en pacientes fallecidos de esta enfermedad.
La sífilis congénita tardía es la que se presenta después de los 2 años de edad, con úlceras gomosas en nariz, en el tabique nasal o en el paladar duro; también, con lesiones óseas periósticas que dan lugar a alteración morfológica, tibias “en sable” y abombamiento del cráneo a nivel frontal y parietal. Puede haber alteraciones faciales, como queratitis intersticial, sordera neurosensorial, incisivos de Hutchinson (la llamada tríada de Hutchinson), molares en mora o fisuras peribucales. Además, puede producirse neurosífilis, asintomática o bien con paresias y tabes, o atrofia óptica.
En este artículo histórico, no entraremos en profundidad en la actualización diagnóstica y en el tratamiento. Véase las referencias siguientes(5-7,9). El diagnóstico de SC se sospechará por la serología materna realizada en el primer trimestre del embarazo (o si se repiten, en el tercer trimestre y en el parto), por la clínica y por pruebas serológicas no treponémicas, como: RPR (reagina plasmática rápida) o VDRL (Venereal Disease Research Laboratory). Las pruebas no treponémicas positivas deben confirmarse con pruebas treponémicas confirmatorias, pero sin demorar por ello el tratamiento antibiótico y el dirigido asimismo a las complicaciones. Los test no treponémicos negativizan hacia los 6 meses de tratamiento. Resulta decisivo que el pediatra conozca los posibles escenarios de presentación neonatal-lactante para su adecuado abordaje. Según la clínica, en recién nacidos y lactantes, se realizarán: pruebas serológicas, punción lumbar para estudio, analítica, radiología y pruebas oftalmológicas, de neuroimagen y potenciales evocados auditivos.
En la edad pediátrica, también puede producirse infección adquirida por sífilis por contacto sexual y, en raras ocasiones, por contacto con tejidos infectados o hemoderivados contaminados.
Denominaciones de la enfermedad
El nombre más extendido es sífilis. El nombre de sífilis proviene de un poema de Fracastorius –Syphilis sive morbus Gallicus–, escrito al modo de Ovidio y publicado en 1530. Girolamo Fracastorius (1478-1553) fue un médico, poeta, astrónomo, geólogo y erudito italiano, de bastante fama en su época. El protagonista del poema tiene por nombre Syphilis, un pastor que recibe la enfermedad como un castigo del dios Apolo por desobedecer a los dioses y seguir a un rey mundano. Este concepto de “castigo divino a un pecado” era propio de la época, pero se mantuvo durante siglos. De Fracastoro fue significativo, históricamente hablando, que en su trabajo De contagione et contagiosis morbis (1546), por primera vez, se habla de la transmisión de enfermedades por “semillas”, seminaria. Aunque esto no recibió interés hasta cientos de años después(8,10,11).
La sífilis también es denominada como lúes, dentro de las enfermedades venéreas o de transmisión sexual. Se la conoció(11) por: búa, buba (bubas), gálico o gallico, lúes venérea y avariosis.
Lúes venérea procede del latín: epidemia-peste más Venus (diosa del amor-sexo). Hubo cultismos “innecesarios”, para evitar citarla u ocultar en el lenguaje médico el nombre común, como fue “avariosis”, cuyo origen fue a partir de Les Avariés (1901), una obra teatral francesa, de Eugene Brieux. Se utilizó a principios de XX, en Francia, también en nuestro país(12); en 1906, se cita así en la Revista Íbero-Americana de Ciencias Médicas (Madrid). La SC se denominó igualmente heredosífilis, antes de principios del XX; nombre condicionado inicialmente por la hipótesis de transmisión “hereditaria” a través de espermatozoides afectados.
Históricamente, las enfermedades de transmisión sexual y, sobre todo, la sífilis, estigmatizaban a las personas que las sufrían. En el nombre evidenciaban cierta xenofobia, pues su origen o extensión se atribuyó a extranjeros: gálico o mal francés, mal napolitano o “cristiana” por los turcos.
La bacteria de la sífilis fue descubierta en 1905 por Fritz Schaudinn (1871-1906) y Erich Hoffman (1868-1959). El término Treponema es un neologismo latinizado, que adoptó su descubridor Schaudinn, fusionando los léxicos griegos trépein (girar) y nēma (filamento). Inicialmente, lo denominaron Spìrochaeta pallida, y en carta a Hoffman lo cambió por Treponema pallidum. La historia del descubrimiento del Treponema es fascinante(13).
Origen de la enfermedad. Primeras publicaciones
A finales del siglo XV, se registró por escrito en Europa el inicio de una enfermedad que actuaba como nunca se había experimentado. El origen y antigüedad de la sífilis representan una de las controversias no resueltas más importantes en la Historia de la Medicina(10,11,14). Se resume en: a) hipótesis colombina, ¿llegó la sífilis al Viejo Mundo desde el Nuevo Mundo a través de Colón y su tripulación? Cristóbal Colón (hacia 1451-1506), descubridor de América, hizo sus dos primeros viajes entre 1492 y 1496; el primero en 1492, volvió en 1943, atracando en Lisboa, Palos y fue hasta Barcelona, abril 1493; y el segundo viaje (1493, volvió en 1496); b) hipótesis pre-colombina e hipótesis unitaria, ¿se originó la sífilis en el Viejo Mundo y permaneció como una enfermedad no identificada hasta que a finales del siglo XV se hizo notoria?
La sífilis es la primera pandemia de la Historia con publicaciones escritas y difundidas desde la “primera ola” aparentemente, desde la aparición como erupción brusca y grave en Nápoles, Italia, tras la toma de la ciudad entre febrero y mayo de 1495, por las tropas mercenarias del rey Carlos VIII de Francia(3,10,11,14). Los tres primeros autores fueron:
• Joseph Grunpeck (1496), publicó en alemán y en latín: Tractatus de Origene pestilentia Scorra sive Mala de Franzos (Tratado de los orígenes de la plaga conocida como enfermedad francesa). Era un clérigo que estudió Medicina. Él mismo cayó enfermo. Creía que la causa era una conjunción de planetas.
• Nicolo Leoniceno (1497), italiano. Asegura que la enfermedad ya era conocida desde Hipócrates y Galeno.
• Juan Gaspar Torella (1497), español, valenciano. Médico requerido por el Papa Alejandro VI ante la alarmante diseminación de la “enfermedad francesa” en Roma.
• Por descontado que la imprenta contribuyó a difundir mucho más la información sobre la enfermedad por toda Europa.
Otros autores posteriores significativos fueron:
• Francisco López de Villalobos (1498), médico y humanista español, publicó en Salamanca el “Sumario de la medicina con tratado sobre las pestilíferas buvas”. Describió la clínica cutánea, incluso neurológica en los estados tardíos. Afirmaba que era consecuencia de conductas licenciosas.
• Nicolás Poll, en 1517, profesor de universidad y médico de Carlos V, publicó sobre el tratamiento de la “enfermedad francesa” con la madera de Guayacán o Guayaco. Se usó el guayaco o palo santo o “lignum vitae” en la primera mitad del XVI, luego se desechó su uso. En 1526, González de Oviedo aseguró que el origen de la enfermedad estuvo en América, las Indias Occidentales. Insistió en el tratamiento con guayaco.
En seguida, desde principios del XVI, se difundieron ampliamente las curas mercuriales, tópicas y en “estufas” o vapores, pero se vio que tenían un alto riesgo de toxicidad y accidentes en el tratamiento.
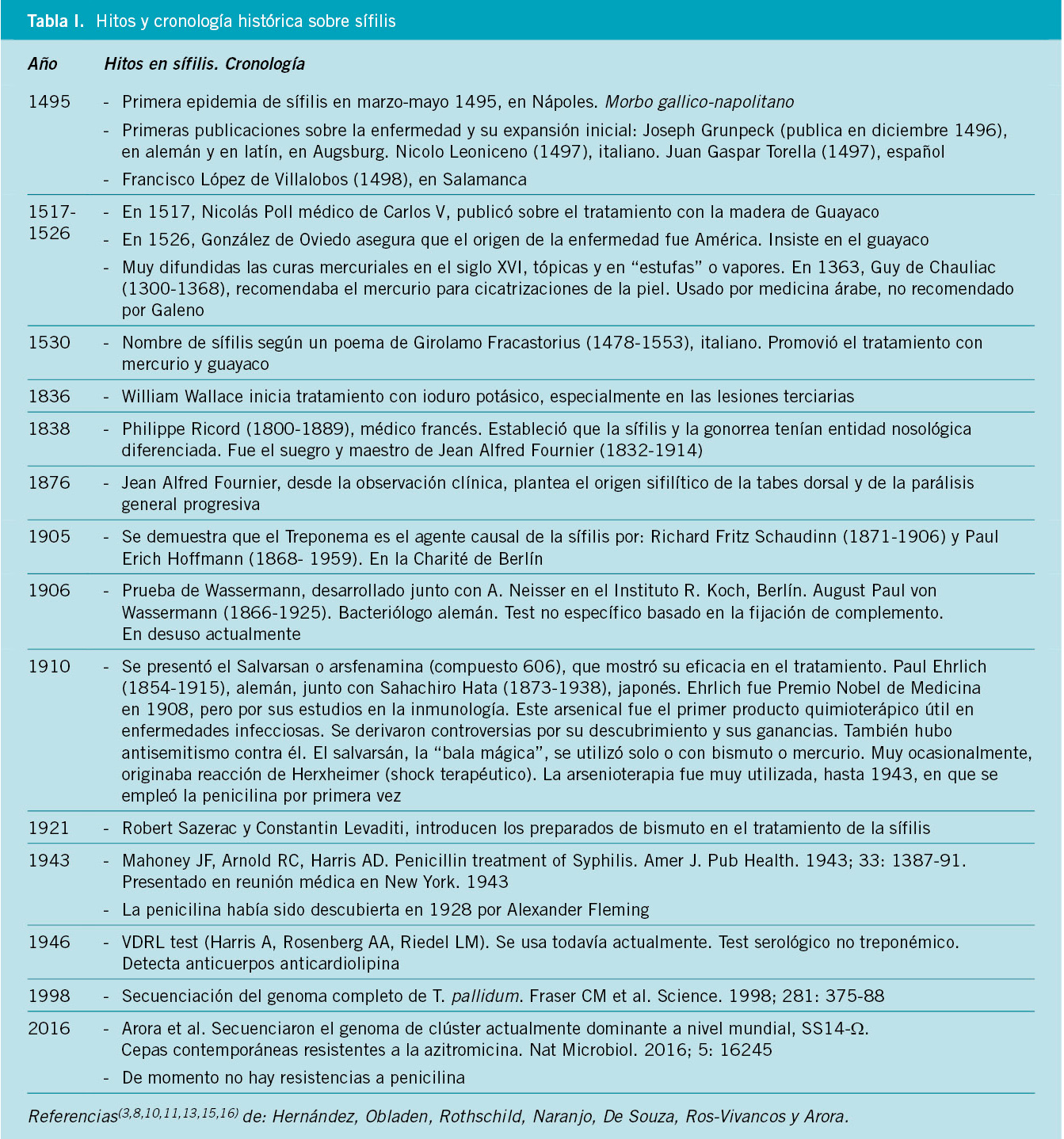
Hay publicaciones que revisan la historia de la sífilis(3,8,10,11,15,16). Se ha elaborado un resumen en la tabla de hitos en sífilis (Tabla I), para posteriormente centrarnos en la SC.
Historia de la sífilis congénita
La primera publicación sobre recién nacidos-lactantes con el mal venéreo fue realizada en 1497, por Gaspar Torella (1452-1520). Médico español, nacido en Valencia, doctorado en Siena (Italia), ordenado sacerdote en 1487. Estuvo como médico al servicio de los Papas Alejandro VI y Julio II. Adscrito al galenismo avicenista predominante entonces. Fue de los tres primeros médicos en describir la sífilis por escrito, junto con Grünpeck, alemán, y Leoniceno, italiano. Tiene una primera publicación donde cita casos de SC como alteraciones en la boca transmitidas “desde el pecho materno o la nodriza”: Tractatus cum consiliis contra pudendagram seu morbum gallicum. Romae, Petrus de la Turre, 1497(8,11,14). Muchos médicos hablaban de la noción transmitida, aunque no se supiera el mecanismo. En esa época y hasta siglos después, no se pensó en la transmisión materno-fetal, sobre todo, al estar la madre sin diagnóstico o enfermedad aparente. Se consideraba especialmente decisiva la “semilla del padre”, y quizá a través de la leche materna o los pechos “infectados” o alterados.
Paracelso (1493, 1541), escribió sobre los tres 3 modos de transmisión de la “materia francesa” al neonato: a) durante la concepción; b) después de la concepción en un segundo coito infeccioso; c) fuera del vientre de la madre por la “leche envenenada”. Fue gran defensor del mercurio para el tratamiento(8).
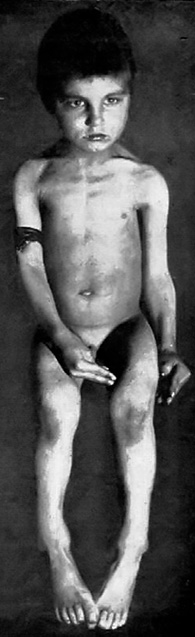
En el siglo XIX, John Burns (1774-1850), obstetra inglés, describe detalladamente, en 1811, las alteraciones cutáneas diseminadas del neonato (manchas color cobre, úlceras, pápulas); también en los que los desarrollan tras nacer sin enfermedad aparente. Resalta el aspecto del lactante, en ocasiones, como un “pequeño anciano”.
El diagnóstico clínico precoz y “con seguridad” de SC se consideró muy decisivo en los siglos XVIII y XIX, puesto que ello llevaba a tomar decisiones sociales y de crianza. Hay algunas descripciones, como la de Underwood en Londres o la de Maunsel en Dublín, que subrayan la “rinitis” o “snuffles”(8); pero históricamente, fueron determinantes las descripciones clínicas de 3 médicos(8,17,18): Hutchinson, Parrot y Fournier.
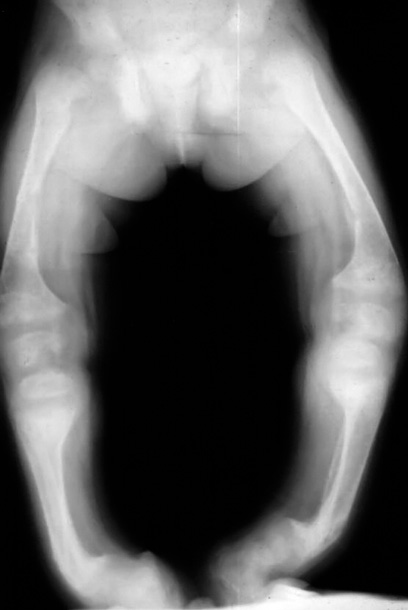
• Jonathan Hutchinson (1823-1913), médico y patólogo inglés, realizó diversos estudios y publicaciones, destacando, en 1856, On the influence of hereditary syphilis on the teeth (Lancet). En 1863, Hutchinson describe la tríada de la SC, alteraciones oculares, auditivas y dentales, en Syphilitic Diseases of Ears and Eye (Fig. 2). Es el “médico de los epónimos”, pues tiene al menos 17. En 1871, operó con éxito por primera vez una invaginación intestinal. Describió la progeria.
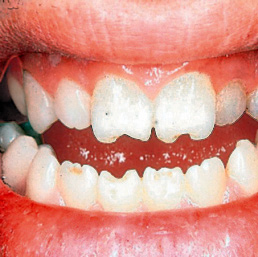
Figura 2. Dientes de Huchinson. Tomado de(17).
• Jules Parrot (1829-1883), médico francés. Fue médico del Hospicio de niños asistidos de París, profesor de la Facultad de Medicina y de Historia de la Medicina y académico. Describió en 1871, la pseudoparálisis, por falta de movilidad de los lactantes debido al dolor óseo, no a incapacidad neurológica. La alteración ósea en necropsias fue descrita en 1870 por Georg Wegner; este entonces era asistente de Virchow en Berlín. Acuñó además los términos de acondroplasia y el de atrepsia (desnutrición, atrofia general), en desuso ahora.
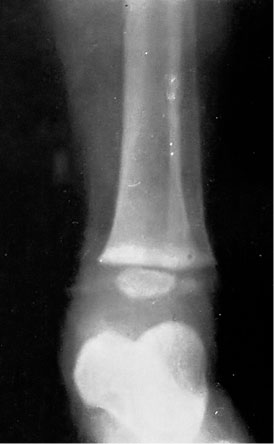
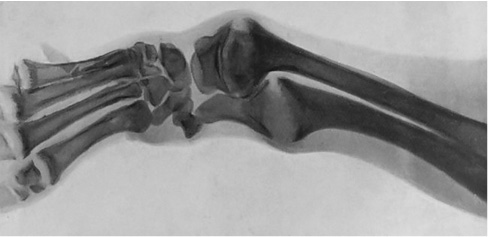
• Jean Alfred Fournier (1832-1914) fue un médico francés, dermatólogo y una de las figuras más destacada de la sifiliografía de la época. Describió en 1883, la “tibia en sable”, con esa deformidad por la alteración ósea (Fig. 3).

Figura 3. Tibia en sable. Tomado de Dobson SR, Kaplan S, Weisman LE, Armsby C. Congenital syphilis: Clinical features and diagnosis(1).
También describió el signo del ómnibus de Fournier: alopecia o pérdida de cabello en la porción lateral de la ceja (cola de la ceja) que impresiona truncada. Está presente en la sífilis secundaria, lepra, hipotiroidismo o como consecuencia del rascado en la dermatitis atópica. Otro signo característico son las cicatrices en la boca que son secuelas de lesiones sifilíticas en la enfermedad adquirida congénitamente. Dan aspecto de “pequeño anciano”.
Durante muchos años investigó sobre sífilis y SC. Realizó publicaciones precoces: “De la contagion syphilitique” (1860). Fournier acuñó el epónimo “tríada de Hutchinson” con ese nombre, para rendirle homenaje. Tiene un texto muy completo: “Textbook of syphilis” (1899). Fue decidido defensor del tratamiento hidrargírico, mercurial(15).
En 1875, Fournier postuló que la sífilis podía ser la causa de la tabes dorsal. Esto no se aceptó por los neurólogos en esas fechas. Le acusaban de “ver sífilis por todas partes”. Esto ha sido y es una de las complejidades de esta enfermedad infecciosa: por un lado es una enfermedad sistémica y frecuente, con la que se tardó siglos en reconocerse sus afectaciones viscerales. Es la “gran simuladora”, con difícil diagnóstico diferencial en sus muy poliédricas presentaciones. Por otro lado, lo de ver sífilis por todas partes, pensamos que quizá pueda tener otro significado socio-cultural-periodístico, bien distinto. Actualmente, hay muchos personajes de la historia que pudieron tener sífilis (o no) a los que ahora se atribuyen sus acciones geniales (o demenciales) a una posible neurosífilis. Las enfermedades en la historia no tienen tan fácil diagnóstico (basándose solo en imágenes o descripciones de época). Incluso si padecieron sífilis, no necesariamente todos evolucionarían a una neurosífilis, podrían tener además otras enfermedades y sufrir los efectos adversos del tratamiento médico de la época. Un ejemplo, Goya (1746-1828), genial pintor español, que tuvo una enfermedad grave a los 47 años, que para muchos autores, señalan como un antes y un después en su trayectoria vital y artística(19); es difícil llegar a conclusiones definitivas: sífilis, malaria, intoxicación por mercurio, por plomo, por quinina (cinconismo), depresión, psicosis, otras…
La transmisión de la SC tuvo grandes controversias en el siglo XIX. Los contagiadores postulaban un origen exclusivamente materno de la infección (a través del canal del parto, sobre todo); mientras que los hereditaristas creían en la transmisión germinal a través del esperma paterno, obviando a la madre(8). Hay que tener en cuenta que, en ese siglo, había todavía planteamientos y tratamientos desde tiempos hipocráticos o de Galeno. Lamarck reactivaría las ideas sobre la herencia de los caracteres adquiridos (epigénesis). Se hablaba de la “degeneración de la raza”, por ejemplo.
El diagnóstico de la SC durante el siglo XIX fue clínico. Solo la radiología pudo aportar alteraciones epifisarias. Las primeras radiografías en SC(8) fueron de OV. Schjerning y F. Kranzfelder en 1896, en Alemania (Dtsch Med Wschr), tres meses después de que Roentgen comunicara su descubrimiento. Los experimentos se realizaron en el Departamento de Medicina del Ministerio de Guerra.
Las pruebas serológicas, desde la detección de la reacción de Wasserman en 1906, fueron útiles, especialmente para detectar a la madre con sífilis.
El Hospital Infantil de Vaugirard 1780-1793, fue una institución pionera en el cuidado de los niños con SC. Fue un hospicio, a expensas del gobierno municipal, dedicado a los niños pobres que habían nacido afectados por enfermedades venéreas, junto con sus madres o nodrizas. Lo inauguró en julio de 1780, Jean Lenoir (1732-1807), abogado, que fue “teniente general de policía” de la prefectura de París, en el periodo inmediatamente anterior a la Revolución Francesa. Lenoir se preocupó por muchos temas de orden público, reducción de enfermedades y de la suciedad. Abrió el hospicio de Vaugirard, París(8); allí las mujeres con sífilis recibieron tratamiento con mercurio oral y por fricción, se creía entonces que los bebés se curaban con la leche de la mujer tratada. Se cita un cierto porcentaje de curación, hasta de un 21% según datos de la época (o quizás también mejoraba la supervivencia al recibir cuidados y leche de nodriza). El hospital se cerró tras la Revolución, y los niños y mujeres con sífilis se trasladarían al nuevo Hôpital des Vénériens en el Couvent des Capucins.
Durante el siglo XIX y principios del XX, se organizó el tratamiento de la sífilis con centros específicos para las enfermedades venéreas: los dispensarios. Tenían función de educación para la salud.
Para el tratamiento de la sífilis congénita, se utilizó el mercurio durante el siglo XVIII y el XIX, pero siguiendo la máxima hipocrática de curar a los niños de pecho tratando a la nodriza o a la madre lactante. No resultaba curativo salvo en circunstancias excepcionales. Debemos tener en cuenta que se decía que el bebé podía transmitir la sífilis a la nodriza; con lo cual retirar la lactancia humana significaba una sentencia para ese bebé(8,15). En 1837, Colles postuló que cuando nace un niño sin síntomas y presenta sífilis, tras unas semanas puede infectar a la nodriza, pero no a su madre. Colles ignoraba en este tiempo, que la madre ya tenía sífilis (asintomática o latente, no se conocía la inmunidad entonces).
Desde 1910, se empezó a utilizar la arsfenamina, el salvarsán (compuesto 606) o neosalvarsán, posteriormente, en recién nacidos afectados. Carl T. Noeggerath (1876-1952) aportó la primera investigación, en Berlín, publicado en 1912. También E. Welde (1911) y Ernst Dünzelmann en Lepizig (1912); pero tenía efectos locales importantes por la inyección. Se preconizó al principio usar mercurio y después salvarsán. La “bala de Erlich” no fue lo eficaz que se pretendió, lo fue relativamente con el tratamiento a las madres durante el embarazo(8,15). Al neosalvarsán, a veces, se asociaban otros tratamientos, como el bismuto.
El primer uso de penicilina en prevención y tratamiento de la SC fue realizado en 1944 por Lentz JW et al, publicado en JAMA(20). Este estudio fue una serie en 4 meses, no se hizo con controles (p. ej., frente a arsenioterapia). Se detectó una reacción de Herxheimer. La penicilina había sido descubierta en 1928 por Alexander Fleming, pero tuvo que ser “redescubierta” durante los años 40. La reacción de Jarisch-Herxheimer se observó hasta en el 40% de las mujeres que reciben tratamiento para sífilis durante el embarazo, pero no es letal actualmente. Véase un resumen de historia de la SC en las tablas II y III.


Historia reciente de la sífilis congénita en España
Para reflejar la importancia historiográfica del tema en la Pediatría española en el siglo XX, citaremos la presencia de la SC en revistas pediátricas españolas, el trabajo en Sanidad en el Protectorado español de Marruecos, iconografía de la lucha antivenérea en la Segunda República y las últimas publicaciones en la revista de la Asociación Española de Pediatría, Anales de Pediatría. Por último, una investigación de Paleopatología en España, con casos de SC en el siglo XVI.
Durante la primera mitad del siglo XX, publicaron trabajos sobre SC numerosos autores. En el magno texto de Juan Luis Morales(21), hasta 1960 hay más de 100 autores, con 268 referencias. Escriben catedráticos, figuras de la Pediatría desde el primer catedrático de Pediatría, Criado Aguilar F, Martínez Vargas A, Suárez Pediguero M, Suñer Ordónez E, Romeo Lozano A, Duarte Salcedo R. Destacan, con 8 artículos, Velasco Pajares J (1878-1954), pediatra dermatólogo del Hospital del Niño Jesús de Madrid, que organizó un Cursillo de sífilis congénita (1923) en dicho hospital; Álvarez Sainz de Aja E (8 artículos) y González Álvarez M (1884-1956) con 6 artículos.
Otras referencias de gran valor(21) en esa época: Muñiz C. Tratamiento con yodobismutato de quinina y lecitina; Muñoz Arbat J. Terapéutica específica por transfusión; Noguer More S. Reivindicación de las fricciones mercuriales en el tratamiento de la heredosífilis infantil; Bravo Frías J. Tratamiento con salvarsán; Blázquez Montoro E. Tratamiento de la sífilis en el Dispensario de lucha contra la sífilis congénita; Conde Muñoz JM. Accidentes salvarsánicos en los dispensarios de lucha contra la sífilis congénita. Incluso propuestas de epónimos, como Cavengt Gutiérrez S. con el síntoma de Genaro Sixto en la heredosífilis. Genaro Sixto fue un reconocido pediatra e higienista argentino, director del Cuerpo Médico Escolar en Buenos Aires, Argentina.
Citaremos los artículos en las siguientes revistas:
• La Medicina de los Niños. Revista pediátrica de la cátedra de Barcelona, dirigida por Andrés Martínez Vargas, desde 1900 a 1936. En ella, hay muchas publicaciones y referencias sobre la sífilis, la etiología, el manejo terapéutico y la importancia social. Hay hasta 37 citas sobre el tema(12). Concretamente, un primer artículo: “Distrofia ósea sifilítica congénita”. 1906; 1: 30; y otro ya en 1926 “Inoclusión del agujero de Botal de origen sifilítico. 1926; 7:,195. Otros temas en esta revista:
- En 1928 se cita sobre la construcción del destacado Jardín de Infancia en Santander, adosada a una Maternidad y Escuela de Puericultura y un Departamento de Servicio Social. Se refiere la asistencia de Pabellón de rayos X, sol artificial, sala para niños sifilíticos. Se organizó también una Gota de Leche.
- Llanto en la SC, Antonio Palacios. “Grito en los heredosifilíticos”. 1930; 7: 205-8.
- Martínez Vargas desarrolló el tema de las alteraciones dentales en las enfermedades sistémicas. Publicó una conferencia dada en la Cruz Roja de Barcelona: “Síndrome dentario luético”. 1932; 6: 153-61.
• El tratamiento lo revisa Martín González Álvarez en la Ponencia Oficial en el Congreso Nacional de Pediatría, de Granada, en abril de 1933. “Diagnóstico y tratamiento de la sífilis congénita” (Disponible el programa en: https://dialnet.unirioja.es/servlet/articulo?codigo=6249970&orden=0&info=link). 1933; 9: 273-80.
• Martínez Vargas defendió, en el primer tercio del siglo XX, y no fue el único en España, la idea de matrimonio eugénico, al modo de EE.UU. con un examen previo de los cónyuges y denegando el permiso a las parejas que no reunieran las condiciones adecuadas de salud, neurológicas o hasta sanar de enfermedades infecciosas.
• Revista Española de Pediatría. Se han localizado 4 artículos entre 1945 y 1954(22). Destacan Aldecoa (1946; 4: 1541-52) y Gómez Pedreira (1948; 6: 351-62), en el tratamiento con penicilina, Schachter M en 1949 (5: 246-52).
• Acta Pediátrica Española. Cuenta con 10 artículos entre 1943 y 1952(22). Escriben Aldecoa y Gómez Pedreira sobre penicilina.
Sobre el trabajo de Sanidad en el Protectorado Español de Marruecos (1913-1956), Miret Cuadras(23) expone sus memorias con abundante iconografía y citas. En Tetuán, además, trabajaron en dispensarios las médicos españolas Nieves González Barrio y María del Monte López Linares. Cita que la sífilis era muy frecuente en esta zona y describe la afectación que se veía entre los niños.
En el Protectorado se organizó una Lucha Antivenérea y se establecieron Clínicas Especiales o Sifilicomios en las cinco ciudades cabeza de provincia. El Neosalvarsán, que mejoraba, “contribuyó a prestigiar al médico y a la medicina española, y facilitó que acudieran los afectados”. Por ejemplo, se comunicó en 1939, que en estadísticas de los cuatro años previos, en los consultorios médicos de Yebala se encontraron hasta un 54,42% de sifilíticos entre todos los enfermos que habían acudido. El Servicio de Profilaxis Social antivenéreo, controlaba, trataba y retiraba del servicio, teóricamente, a las prostitutas afectadas, hasta su curación. Véase la figura 4 sobre lucha antivenérea durante la Guerra Civil española.

Figura 4. Cartel anarquista de lucha contra enfermedades venéreas durante la Guerra Civil española. Utiliza una imagen de infancia afectada, sin vincular androcentrismo ni moralismo. Disponible en: https://www.pinterest.es/pin/230246599672148296/.
En la revista española Anales de Pediatría, encontramos publicaciones muy recientes de casos de SC, el último en julio 2021. Hay un caso del año 2009 por diagnóstico clínico a las 7 semanas de edad, con cribado negativo durante el primer trimestre de la gestación(24). También, se encuentran trabajos de Tesis en España, como la de Hernández en Canarias, disponibles en Internet(3).
• Paleopatología, un estudio en España. Citemos el trabajo de Montiel y cols. (2012)(25), que aportan un análisis exitoso del ADN antiguo (ADNa), concretamente en cuatro especímenes de recién nacidos recuperados de la cripta de ‘’La Ermita de la Soledad’’ (siglos XVI-XVII), situada en Huelva (España). Mientras que la recuperación de Mycobacterium spp. ha sido ampliamente exitosa, varios autores informan de resultados infructuosos en relación con el ADN treponémico antiguo, poniendo en duda la utilidad de esta técnica para el diagnóstico de la sífilis antigua. Pero los datos de Montiel apoyan la hipótesis de que, en los neonatos, el número de espiroquetas en el esqueleto es lo suficientemente alto como para garantizar la conservación de su ADN, y que cuanto más joven es el individuo afectado por la sífilis congénita, mayor es la probabilidad de conservación amplificable del ADN a efectos de estudios históricos. Por el contrario, en los adultos afectados por la sífilis venérea, el número de espiroquetas en los huesos se reduce a medida que la enfermedad es más avanzada.
Actualidad de la sífilis congénita
En países occidentales, se constata un cierto resurgir de la sífilis en este siglo XXI.
Los trabajos de Cooper et al. y otros(1,4-7), son excelentes para estudiar la situación actual de la SC.
La desaparición de la SC, como se la ha conocido durante siglos, no quiere decir erradicación. Si bien, la OMS, en su búsqueda de erradicación, declara la eliminación de SC cuando la tasa de transmisión madre a hijo de sífilis es menor a los 50 casos por 100.000 nacidos vivos (quizá sean muchos, parece). El término eliminación no lo considera aquí como para otras enfermedades infecciosas (v. noticia de 2015, disponible en: https://www.who.int/es/news/item/30-06-2015-who-validates-elimination-of-mother-to-child-transmission-of-hiv-and-syphilis-in-cuba).
La SC es uno de los problemas de salud pública en el mundo y su incidencia ha aumentado en las últimas décadas. Cada caso de sífilis congénita debería ser considerado como un fallo en el sistema público de salud en la atención óptima a los cuidados de las mujeres embarazadas, una alerta para detectar opciones de mejora. Debe insistirse en la prevención con cribado serológico a las madres, repetido a lo largo del embarazo en los casos de riesgo o que considere la autoridad sanitaria, en el tratamiento con penicilina a las mujeres con infección, a sus contactos sexuales y a sus hijos, garantizando un seguimiento protocolizado(1,7).
En el artículo de Arora (2016)(16), se investiga por qué ha reaparecido a nivel mundial la sífilis en las últimas décadas (“estimación de 10,6 millones de casos en 2008 en todo el mundo”). Aunque todavía no se ha identificado la resistencia a la penicilina, su tratamiento de primera línea, un número creciente de cepas no responden a un antibiótico de segunda línea, concretamente la azitromicina. Las cepas contemporáneas son miembros de un clúster globalmente dominante, denominado SS14-Ώ. Dicho clúster se diversificó a partir de un ancestro común a mediados del siglo XX, tras el descubrimiento de los antibióticos. Su reciente divergencia filogenética y su presencia global apuntan a la aparición de un clúster de cepas pandémicas.
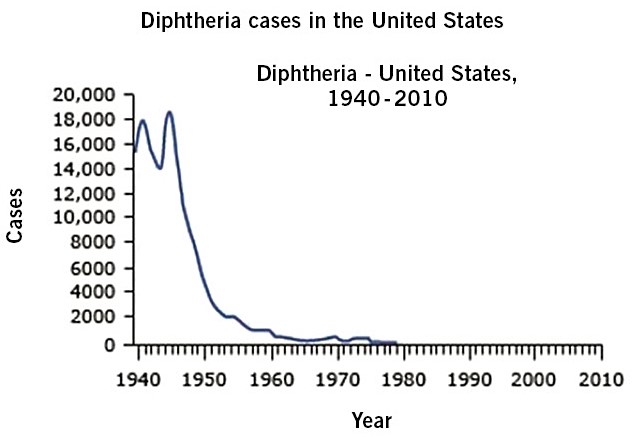
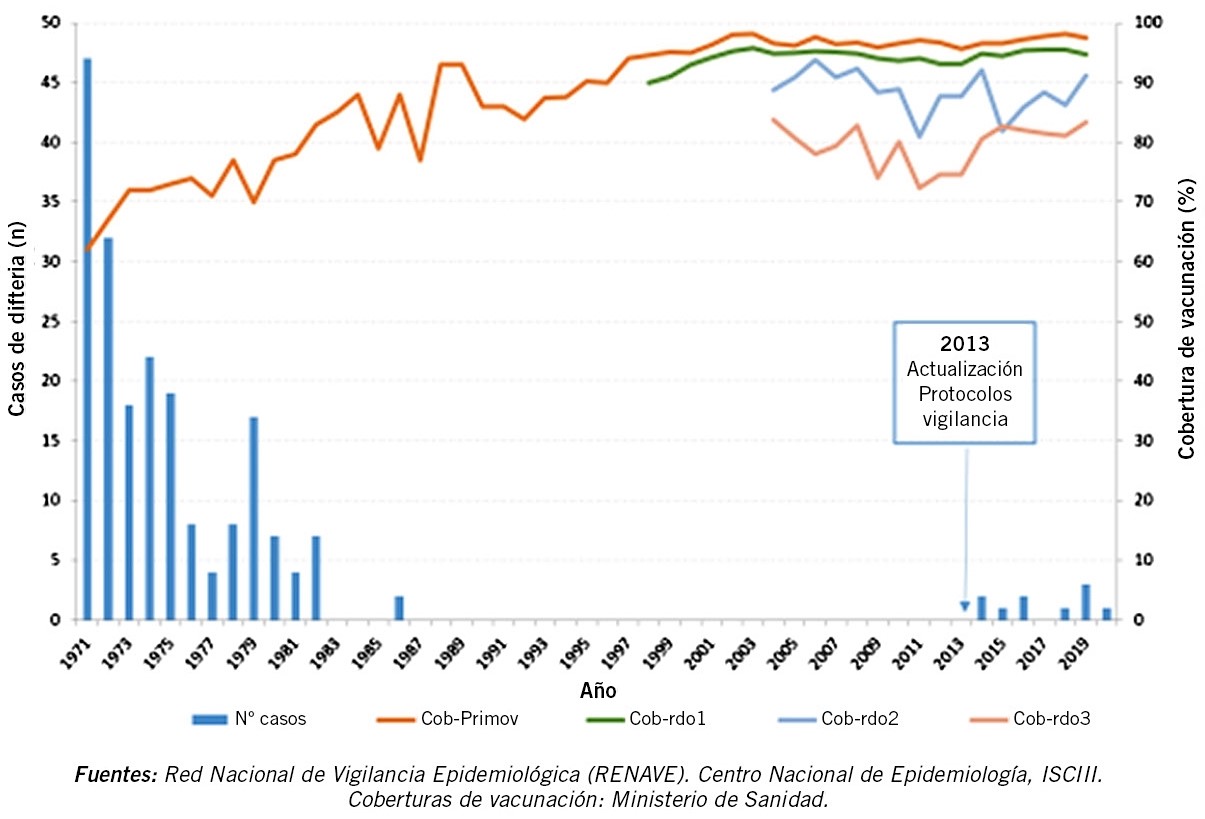
Han aumentado los casos de SC, de muertes atribuidas a ella, y se ha extendido por casi todos los EE.UU. En EE.UU., en 2020, ya se han notificado más de 2.020 bebés como casos de SC. Ver figura de la evolución de últimos 10 años. Los datos actualizados en Europa y España, se pueden ver en las figuras 5 y 6.

Figura 5. Casos confirmados de sífilis congénita en Europa(6) del ECDC.
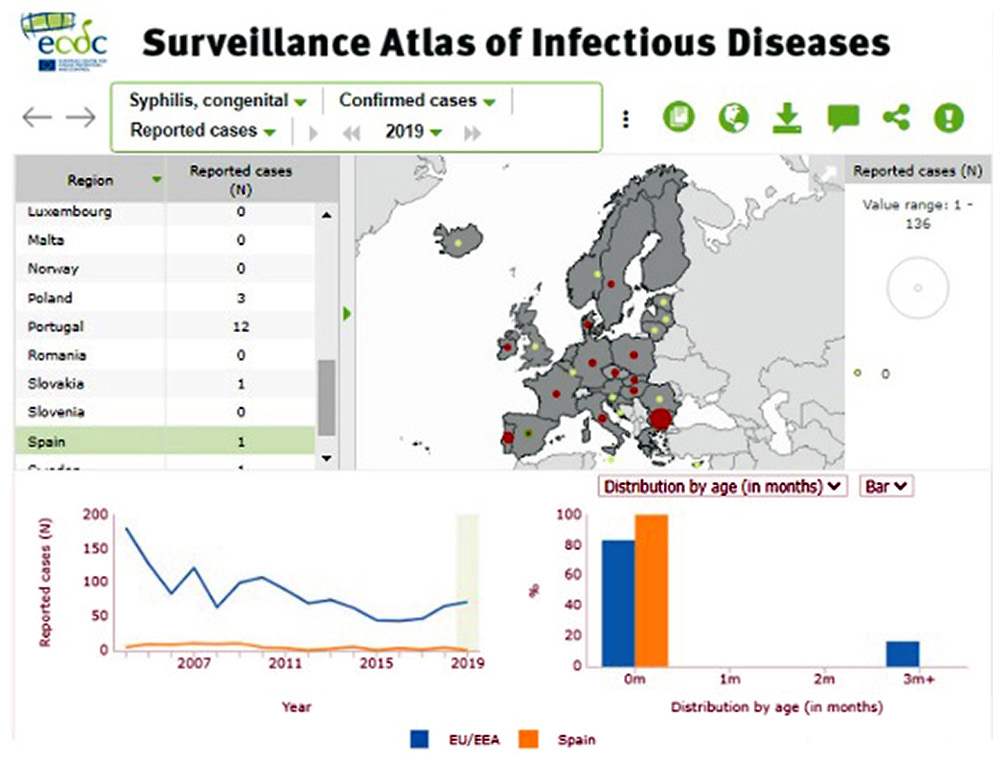
Figura 6. Casos notificados de sífilis congénita en España(6) del Centro Europeo para la Prevención y el Control de las Enfermedades (ECDC).
En España, según el European Centre of Disease Control (ECDC), se notificaron los siguientes casos de SC: 11 en 2007 (2,24:100.000 recién nacidos vivos); 6 en 2014; 5 en 2018 (1,35 por 100.000); y 1 caso (0,28 por 100.000) en 2019.
Se siguen publicando actualmente casos de SC en EE.UU., España(24,26,27).
Respecto a la investigación en vacunas para sífilis y para enfermedades de transmisión sexual, aquella ha venido condicionada por el impacto mundial de la enfermedad en la población, pero además por las posibilidades de profilaxis y tratamiento eficaz(28). La vacuna frente al Treponema tendría la ventaja de actuar sobre una enfermedad que, en ocasiones, evade el cribado estándar, que tiene un tratamiento muy eficaz; pero está desarrollando resistencias a fármacos de segunda línea(16). Los avances en bioinformática están permitiendo caracterizar las proteínas de membrana con el objetivo de diseñar vacunas(28). Queda un tiempo aún para disponer de ellas.
Algunas enseñanzas históricas de la sífilis
Comentaremos dos temas: sobre la polémica del origen de la epidemia, y sobre las investigaciones históricas científicas (o no tanto) sobre la enfermedad.
1. Sobre la polémica del origen de la epidemia: tras hacer búsquedas historiográficas, realmente sorprende la enorme investigación sobre el origen geográfico de los primeros brotes de sífilis, a finales del siglo XV. Ello debe obedecer a un interés científico y de estudio de diseminación de pandemias; aunque también puede ofrecernos enseñanzas sobre otro tipo de intereses o quizá sesgos. Incluso, nosotros mismos podemos tener un sesgo al escribir desde España, donde se respaldó e inició el viaje de Colón.
Citaremos dos estudios muy interesantes, historiográfico de Naranjo P(11), y otro, histórico filológico de Colón Domenech(11), que no es descendiente del almirante descubridor.
Naranjo, miembro de la Academia de Medicina y de Historia de Ecuador, tras realizar un detallado estudio de las primeras publicaciones sobre sífilis, (véase la tabla de Hitos y cronología histórica sobre sífilis), cita(11) que las publicaciones de finales del XV y principios del XVI no se plantean que el origen fuera en América. No fue hasta en 1526, con González Fernández de Oviedo (1478-1557), militar, colonizador y cronista, que publicó un sumario de plantas medicinales encontradas en el Nuevo Mundo (las Indias), que insiste que la virtud de la madera del guayaco era curar por los Indios las bubas que tenían, que “eran comunes entre los Indios, pero no tan peligroso como en nuestras tierras”. Apoya la antigua máxima de que donde Dios pone la enfermedad o el castigo, envía también la cura… por qué este personaje contradice a los médicos de la época. El comercio del guayaco fue uno de los primeros negocios sobre tratamiento “natural” de una enfermedad, que enriqueció enormemente a los banqueros alemanes Fugger, que financiaron a Carlos V. Las crónicas sobre las historias de América se publicaron rápidamente tras el primer viaje de Colón. Durante 2 décadas se cita que los españoles contrajeron unas bubas y una enfermedad, en la que todos los que escribieron estaban de acuerdo en su forma benigna, similar a Pian; pero no se cita que hubiera enfermedad de la virulencia de Nápoles entre los españoles procedentes de América, ni en Sevilla, ni en Barcelona, ni al volver a las Indias. En cambio, sí se hizo mención precisa de las enfermedades en el nuevo mundo, viruela, varicela, sarampión, gripe. En Europa, se comienza a hacer mención de casos aislados de sífilis hereditaria a principios del XV; pero no se describen en América en esos años(11,25).
Naranjo comenta finalmente(11) que: “la teoría americana del origen de la sífilis necesita referencias históricas (muchas eran tardías y se aceptaron sin discusión), pero, sobre todo, etnológicas, genéticas e inmunológicas. Es evidente que la teoría del origen americano de la sífilis podría estar basada en errores históricos que han sido repetidos por autores del pasado y del presente, como si estos errores fueran hechos probados”.
El estudio historiográfico y filológico de Colon Domenech, concluye(14) que sus datos filológicos reunidos “muestran que la enfermedad no la debieron de traer de América las huestes colombinas (¿ya en el primer viaje?), aserción propalada sin pruebas fehacientes, y que tiene la vida muy dura”. “Que hubiera sífilis en América no significa que dejara de haberla en Europa”. Es extremadamente difícil separar en los textos antiguos las enfermedades cutáneas que simulan sífilis de esta misma; pero ya antes del descubrimiento “tenemos documentos que avalan la existencia de esta última en el Viejo Mundo”.
Hay estudios histológicos óseos de Paleopatología y de ADN, como los de von Hunnius et al.(29), que evidencian la existencia de treponematosis prácticamente en todos los continentes en la época precolombina.
Hunnius y cols.(29), efectúan un estudio realizado con un enfoque multimetodológico histológico para realizar el diagnóstico paleopatológico. Se tomaron muestras óseas de cuatro esqueletos tomados de un cementerio medieval desenterrados en 1994, se dataron en 1300-1450 d.c., fechados a través de la dendrocronología de los ataúdes, la estratigrafía y el 14C. Las pruebas histológicas sugieren que la sífilis venérea, como enfermedad crónica ósea, estaba presente en Inglaterra antes de 1492.
Afirman que “la segura datación precolombina, junto con la identificación macroscópica y microscópica de la sífilis en algunas de las muestras, contribuyó a aportar más pruebas de que la hipótesis precolombina no puede seguir siendo ignorada. Estos resultados coinciden con los de otros investigadores que descubrieron restos óseos humanos con enfermedad treponémica en otras regiones del Viejo Mundo, fechados antes del viaje de Cristóbal Colón en 1492 (Escocia: Cardy, 1997; Italia: Henneberg y Henneberg, 1994; Ipswich y Rivenhall, Inglaterra: Mays et al., 2003; Francia: Pálfi et al., 1992; Gloucester, Inglaterra: Roberts, 1994; Kazajistán: Schultz et al., 2003; Norwich, Inglaterra: Stirland, 1991)”.
2. Las investigaciones históricas científicas (o no tanto) sobre sífilis se resumen en la tabla IV.

Epílogo
Actualmente, podemos compartir la frase de William Osler, padre de la medicina interna moderna: “el que conoce la sífilis, conoce la medicina”. Era una enfermedad tan frecuente en el siglo XIX y con tanta miríada de síntomas, que había que conocer toda la patología médica. Actualmente, es una enfermedad con tanta historia y tanto impacto cultural y social que puede ayudar a conocer la historia de la medicina. También puede colaborar en la historia del arte, de las colonizaciones, de la cultura, de la religión, de las ideologías, de la política, del periodismo y de las redes sociales.
Al examinarla, se puede aprender lo fácil que fue estigmatizar, no solo a los individuos afectados por la enfermedad, sino a poblaciones o naciones enteras. A lo largo de los siglos, la sífilis ha afectado o ha podido afectar a individuos de diversos orígenes, desde monarcas, políticos, músicos, pintores y filósofos, hasta personas de bajos recursos, principalmente, debido a la promiscuidad o a los estilos de vida. Las enfermedades generan pacientes y afectados, también víctimas, como el cuadro de “La herencia” de Munch (Fig. 7).

Palabras clave
Enfermedades de transmisión sexual; Enfermedades venéreas; Sífilis; Sífilis congénita; Paleopatología; Historia. Sexually transmitted diseases; Venereal diseases; Syphilis; Congenital syphilis; Paleopathology; History.
Bibliografía
1. Dobson SR, Kaplan S, Weisman LE, Armsby C. Congenital syphilis: Clinical features and diagnosis. UpToDate 2021. Disponible en: www.uptodate.com.
2. Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020; 382: 845-54.
3. Hernández Betancort MA. Sífilis en el Área Sur de Gran Canaria (análisis de 5 años). Epidemiología, diagnóstico, prevención, Tesis doctoral. Universidad Las Palmas de Gran Canaria. Facultad de Ciencias de la Salud. Departamento de Ciencias Clínicas. Disponible en: https://dialnet.unirioja.es/servlet/tesis?codigo=154571. Consultado el 28 de julio de 2021.
4. OMS-WHO. Detección y tratamiento de la sífilis en embarazadas. 2019. Disponible en: https://iris.paho.org/bitstream/handle/10665.2/51791/9789275321744_spa.pdf?ua=1.
5. Centers for Disease Control and Prevention (CDC). Congenital Syphilis Preliminary 2020 Data. Atlanta: U.S. Department of Health and Human Services; 2021. Disponible en: https://www.cdc.gov/std/syphilis/default.htm.
6. European Centre for Disease Prevention and Control (ECDC). Congenital syphilis. 2021. Datos de 2019. Disponible en: https://www.ecdc.europa.eu/en/congenital-syphilis.
7. Cooper JM, Sánchez PJ. Congenital syphilis. Semin Perinatol. 2018; 42. 176-84.
8. Obladen M. Curse on two generations: A history of congenital syphilis. Neonatology. 2013; 103: 274-80.
9. Dobson SR, Kaplan S, Weisman LE, Armsby C. Congenital syphilis: Evaluation, management, and prevention. UpToDate 2021. Disponible en: www.uptodate.com.
10. Rothschild. History of syphilis. Clin Infect D. 2005; 40: 1454-63.
11. Naranjo P. On the american indian origin of syphilis: fallacies and errors. Allergy Proc. 1994; 15: 89-99.
12. Reche Andrés J. La Pediatría española a través de la revista “La Medicina de los Niños”, 1900-1936. Tesis doctoral. Universidad Complutense de Madrid. Facultad de Medicina, Area de Historia. Disponible en: https://eprints.ucm.es/52396/1/5309854783.pdf. Consultado el 28 de julio de 2021.
13. De Souza EM. A hundred years ago, the discovery of Treponema pallidum. An Bras Dermatol. 2005; 80: 547-8.
14. Colón Domenech G. Filología y sífilis. Sobre el mal de simiente o mal de sement. Rev Filología Esp. 1998; LXXVIII: 275-308. Disponible en:
https://revistadefilologiaespañola.revistas.csic.es/index.php/rfe/article/download/309/497/0.
15. Ros-Vivancos C, González-Hernández M, Navarro-Gracia JF, Sánchez-Payá J, González-Torga A, Portilla-Sogorb J. Evolución del tratamiento de la sífilis a lo largo de la historia. Rev Esp Quimioter. 2018; 31: 485-92.
16. Arora N, Schuenemann VJ, Jäger G, Peltzer A, Seitz A, Herbig A, et al. Origin of modern syphilis and emergence of a pandemic Treponema pallidum cluster. Nat Microbiol. 2016; 5: 16245.
17. Van Ruth S, Toonstra J. Eponyms of Sir Jonathan Hutchinson. Int J Dermatol. 2008; 47: 754-8.
18. Vashisht D, Baveja S. Eponyms in syphilis. Indian J Sex Transm Dis AIDS. 2015; 36: 226-9.
19. Cipriani G, Ciprini L, Picchic L, Di Fiorino M. Art is long, life is short. Francisco José de Goya y Lucientes (1746-1828), the suffering artista. Medical Hypotheses. 2018; 117: 16-20.
20. Lentz JW, Ingraham NR, Beerman H, Stokes JH. Penicillin in the prevention and treatment of congenital syphilis. JAMA. 1944; 126: 408-13.
21. Morales JL. El Niño en la Cultura Española. III tomo. Alcalá de Henares, Imprenta de los Talleres Penitenciarios. 1960.
22. Zafra Anta, MA, García Nieto VM. Las nuevas publicaciones pediátricas de los años 40. En: García Nieto, VM, et al. La Pediatría española en la postguerra. Cuadernos de Historia de la Pediatría Española. Asociación Española de Pediatría, 18. 2019. p. 45-63. ISBN: 978-84-09-17421-8.
23. Miret Cuadras P. Memorias de un médico en el Protectorado Español de Marruecos a mediados del siglo XX. Rev Aldaba. 2014; 39: 207-70.
24. Madrigal Díez C, Lozano de la Torre MJ, Madrigal Díez V. Sífilis congénita en un niño de 7 semanas: ¿se hubiera evitado con otro protocolo? An Pediatr. 2009; 70: 190-1.
25. Montiel R, Solórzano E, Díaz N, Álvarez-Sandoval BA, González-Ruiz M, Cañadas MP, et al. Neonate human remains a window of opportunity to the molecular study of ancient syphilis. PLoS ONE. 2012; 7: e36371.
26. Arrieta AC. Congenital Syphilis. Images in clinical medicine. N Engl J Med. 2019; 38: 22.
27. Ordoño Saiz MV, Chacón Flocos S, Rodríguez-Ramos M, Gómez Zafra. Sífilis congénita precoz sintomática en el recién nacido. An Pediatr (Barc). 2021; 94: 341-2.
28. Singh T, Otero CE, Li K, Valencia SM, Nelson AN, Permar SR. Vaccines for Perinatal and Congenital Infections – How Close Are We? Frontiers Pediatr. 2020; 8: 569.
29. Von Hunnius TE, Roberts CA, Boylston A, Saunders SR. Histological Identification of Syphilis in Pre-Columbian England. Am J Phys Anthropol. 2006; 129: 559-66.
30. Beecher HK. Ethics and Clinical Research. New England Journal of Medicine. 1966; 274: 1354-60.
31. Reverby SM. The Dark History of Medical Experimentation from the Nazis to Tuskegee to Puerto Rico. 2010. Disponible en:
https://www.democracynow.org/2010/10/5/the_dark_history_of_medical_experimentation.