 |
| Historia de la Medicina y la Pediatría |
V.M. García Nieto*, M. Zafra Anta**
*Coordinador del Grupo de Historia de la Pediatría de la AEP. Director de Canarias Pediátrica. **Servicio de Pediatría del Hospital Universitario de Fuenlabrada. Miembro del Grupo de Historia de la Pediatría de la AEP
Pediatr Integral 2022; XXVI (3): 189.e1 – 189.e4
Enfermedades pediátricas que han pasado a la historia (9). Hipercalciuria idiopática con nanismo y alteración renal en el niño
Prólogo
En un artículo reciente de esta serie, indicábamos que, al menos, los dos primeros casos iniciales que dieron nombre al término “Síndrome de De Toni-Debré-Fanconi” eran tubulopatías complejas distintas de la cistinosis que es el paradigma de enfermedad habitual con el que se equipara dicho síndrome en la actualidad(1). Esos dos casos iniciales, tanto el descrito por De Toni como el publicado por Debré, correspondían a enfermedades no descritas aún en los años treinta del pasado siglo. Algo similar ocurrió con otro trastorno tubular, la “Hipercalciuria idiopática con nanismo y alteración renal en el niño” descrito por Royer(2) (Fig. 1), Gentil(3) y sus respectivos colaboradores en 1962.

Figura 1. Pierryere Ro (1917-1995). Disponible en: https://histoire.inserm.fr/les-femmes-et-les-hommes/pierre-royer.
La asociación entre una excesiva excreción urinaria de calcio y la formación de cálculos renales fue descrita en población adulta por Flocks en 1940(4). En 1953, Albright et al. utilizaron por primera vez el término “Hipercalciuria idiopática”(5). Aunque inicialmente se creyó que esta entidad no existía en los niños, a finales de la década de los 50, Zetterström(6) y Rosenkranz(7) publicaron sendos casos pediátricos en los que coincidían la asociación de litiasis e hipercalciuria. Precisamente, en 1962, Valverde, urólogo que trabajaba en Córdoba, publicó los primeros casos pediátricos españoles de hipercalciuria idiopática, algunos de ellos con infección de vías urinarias asociada(8).
El objetivo de este artículo es repasar esa variante “grave” de hipercalciuria que, en realidad, se trataba de determinadas tubulopatías que eran desconocidas en los años sesenta del pasado siglo. Esta es la historia.
Hipercalciuria idiopática con nanismo
En 1962, dos grupos pediátricos radicados en París comunicaron seis casos de niños con nanismo, osteopenia o raquitismo y alteración renal (proteinuria, poliuria, hipercalciuria)(2,3). Los casos descritos eran clínica y bioquímicamente distintos de los calificados como hipercalciuria idiopática stricto sensu.
Pierre Royer indicó que la edad en la que se establecía el diagnóstico oscilaba entre los tres meses y los dieciocho años. El enanismo, armónico en todas las observaciones, era importante con una variación de 4 a 5 desviaciones estándar por debajo de la correspondiente a la edad. La maduración ósea, igualmente, estaba siempre retrasada en relación con la edad cronológica. La maduración dentaria era adecuada a la edad cronológica. Además, el examen radiológico del esqueleto puso de manifiesto, a menudo, una osteoporosis intensa con un descenso de la relación corticodiafisaria en los huesos largos y una reducción de la densidad del esqueleto. En otros casos, se apreciaron signos de raquitismo. Se observó que podía instaurarse una nefrocalcinosis en el curso evolutivo, aunque en uno de los casos se comprobó en el primer año de la vida. Podía acompañarse de litiasis renal.
La calciuria oscilaba entre 5,8 y 20 mg/kg/24 h. La hipercalciuria era estable o variable según un ritmo estacional: elevada en primavera y verano y más moderada en invierno. “La calciuria no descendía por efecto de la administración de bicarbonato sódico, contrariamente a lo que ocurre en la acidosis tubular crónica idiopática”(9,10) (el autor se refería a la acidosis tubular renal distal tipo 1). La inyección de calcitonina tampoco modificaba la calciuria. La prueba del cloruro amónico fue normal en todos los casos. La calcemia, las fosfatasas alcalinas y la citraturia eran normales. La fostatemia era normal o reducida, aunque el coeficiente de reabsorción tubular de fosfatos estaba descendido. En ocasiones, la calcemia se reducía y se acompañaba de tetania.
En todos los casos, existía poliuria, entre 600 y 2.500 ml por 24 h, con una “concentración osmolar máxima situada entre 170 y 778 mOsm/kg de agua”(9,10). Este defecto de concentración era resistente a la pitresina. La proteinuria no constante (dos casos sobre cinco) se situaba entre 0,4 y 3 gramos por día. Se trataba, en general, de una proteinuria de tipo tubular con globulinuria predominante(9,10). El aclaramiento de creatinina endógena era normal. No existía glucosuria ni hiperaminociduria, ni pérdida salina.
Dos de las biopsias renales realizadas por el grupo de Royer fueron normales. En tres casos existía una nefritis intersticial focal (Fig. 2).
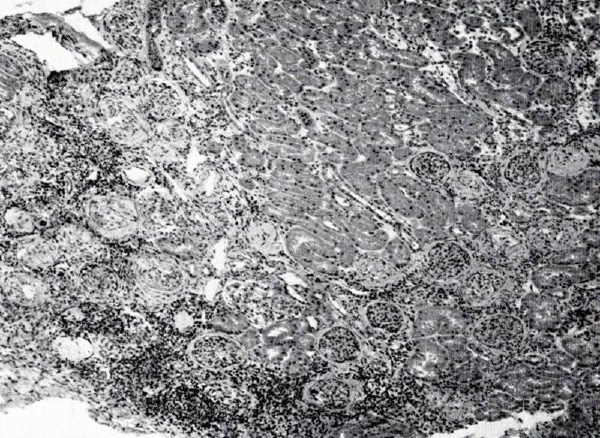
Figura 2. “Hipercalciuria idiopática. Zona segmentaria de nefritis intersticial con infiltración importante de células redondas; atrofia de los túbulos y lesiones más o menos importantes de esclerosis glomerular. Aparte de esta zona segmentaria, existen pequeños focos de nefritis intersticial esparcidos por el parénquima renal (Aumento x 100). Este niño de seis años presentaba un importante retraso estaturo-ponderal y proteinuria de 1 a 2 g/24 h. Su calciuria era elevada, alrededor de 7,5 mg/kg/24 h”(12).
En cuanto al tratamiento, la calciuria disminuía tras un régimen pobre en calcio o en cloruro sódico(11). Asimismo, podía reducirse tras el uso de fitato sódico o de hidroclorotiacida. Inicialmente se ensayó, incluso, el tratamiento con vitamina D y con corticoides(12).
Los casos publicados por Royer et al. en 1962. Intentar conocer qué enfermedad renal padecían los pacientes estudiados por Royer y sus colaboradores hace 60 años(2), es difícil entreverlo en la actualidad, puesto que muchas pruebas morfológicas y determinaciones bioquímicas actuales fundamentales no existían en ese momento. Un somero examen de los casos resalta las notables diferencias que existían en las características clínicas y bioquímicas entre los pacientes analizados.
El paciente que iniciaba la casuística tenía retraso póndero-estatural, una importante poliuria resistente a la pitresina (2-2,5 l en 24 h) e hipercalciuria. Los padres eran consanguíneos, descendientes de primos hermanos. Al ser un varón podría tratarse de una diabetes insípida nefrogénica; otra opción sería un síndrome de Bartter de la forma clásica (no tenía nefrocalcinosis), aunque la biopsia renal fue normal y en las revisiones hechas por los autores posteriormente, se indicaba que los niveles plasmáticos de potasio de los pacientes eran normales(9,10). Precisamente, los dos primeros casos de síndrome de Bartter se publicaron ese mismo año de 1962(13).
El niño incluido como Observación II tenía: hipofosfatemia (2 mg/dl), hipercalciuria (7,5 mg/kg/día) y proteinuria glomerular (albúmina; 1-2 g/día). El raquitismo hipofosfatémico con hipercalciuria, causado por mutaciones en el gen SLC34A3 que codifica el cotransportador sodio-fosfato(14), no cursa con proteinuria. El caso III había sido diagnosticado de ataxia-telangiectasia y, también, tenía proteinuria glomerular (0,4-3 g/día) e hipercalciuria (6 mg/kg/día). La ataxia-telangiectasia descrita por Denise Louis-Bar en 1941(15) tampoco cursa con proteinuria. Podría suponerse que ambos pacientes tuvieran una pérdida de parénquima renal asociada a hiperfiltración glomerular y albuminuria, pero la urografía endovenosa fue normal.
El caso IV era una niña con retraso estaturo-ponderal que había sido tratada en un centro “helio-marino” por aunar mal estado general y signos de raquitismo. Tenía una dismorfia facial con orejas displásicas, Pterygium colli y cubitus valgus que evocaba un síndrome de Turner. El cariotipo fue normal. Tenía hipercalciuria, pero la prueba de concentración fue normal (912-1.080 mOsm/kg) y no tenía proteinuria ni nefrocalcinosis. Podría, pues, tratarse de un síndrome de Noonan, aunque no había datos de cardiopatía ni conocemos que se haya descrito su asociación con hipercalciuria. El síndrome de Noonan fue publicado en 1963(16), es decir, un año después de la asociación que nos ocupa.
Artículos posteriores sobre “Hipercalciuria idiopática con nanismo y alteración renal”
Especialmente en las décadas de los años 60 y 70 del pasado siglo, se publicaron diversos casos de esa entidad(17-26). Los niños incluidos en los artículos escritos por Fovet-Poingt y Toursel(23) y por Tieder y Stark(26), con cierta probabilidad estaban afectos de “Raquitismo hipofosfatémico con hipercalciuria”.
El caso publicado por Jeune et al., junto a la asociación de retraso estáturo-ponderal, poliuria, hipercalciuria y nefrocalcinosis, contaba con la particularidad de tener anomalías oculares(21). Además, el estudio del fondo de ojo reveló “una despigmentación difusa de la retina junto a una lesión pigmentaria en forma de estrella en la macula” (Fig. 3).
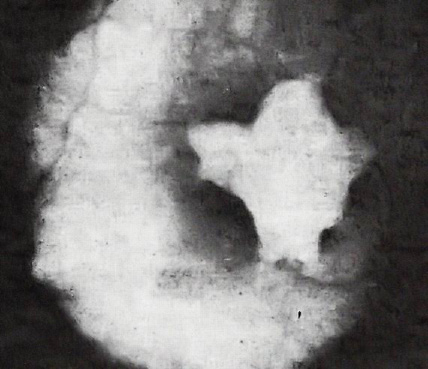
Figura 3. “Aspecto del fondo de ojo: despigmentación difusa de la retina, opacidad en estrella de la macula”(21). Esta imagen es muy similar a la descrita en otros artículos sobre el tema: Meier W et al. Helv Paediatr Acta 1979; 34: 257-9; Gil-Gibernau J et al. J Pediatr Ophthalmol Strabismus. 1982; 19: 7-11.
Más tarde, a los 15 meses de edad, se observaron cataratas de aspecto no congénito que estaban ausentes en los primeros exámenes. El paciente tenía un defecto de acidificación incompleto en la prueba realizada con cloruro amónico (pH mínimo 5,4). La “hipomagnesemia familiar con hipercalciuria y nefrocalcinosis” es una tubulopatía que se manifiesta en forma de infecciones recurrentes del tracto urinario, nefrolitiasis, poliuria, polidipsia, amelogénesis imperfecta o retraso en el crecimiento(27). El diagnóstico clínico se basa especialmente en la presencia de hipomagnesemia, hipercalciuria, nefrocalcinosis bilateral y defectos oculares severos; estos últimos aparecen en el tipo 2. Las anomalías oculares son, principalmente: miopía severa, coloboma macular y nistagmo y, en menor medida, estrabismo, coriorretinitis, despigmentación peripapilar, calcificación corneal e hipoplasia papilar. Algunos casos presentarán insuficiencia renal crónica en el momento del diagnóstico o a lo largo de la evolución(27). El diagnóstico se confirma con la detección de mutaciones en ambos alelos de los genes CLDN16 o CLDN19 que codifican las proteínas intercelulares claudina 16 y 19, respectivamente. Se ha descrito un defecto de acidificación en esta entidad(28) y, en alguna familia, se ha advertido la presencia de cataratas(29). A la vista de lo expuesto, es probable que el paciente descrito por Jeune et al. sea el primer caso conocido de esa tubulopatía.
El caso descrito por Sorel et al. fue estudiado a los cinco años de edad por retraso estatural (- 3DS), poliuria, polidipsia y proteinuria(22). La calciuria era elevada (12,5-18,5 mg/kg/24 h). La fosfatemia estaba reducida en dos de tres determinaciones (4,2; 2,8 y 2,6 mg/dl). En la electroforesis de la proteinuria, se observó la presencia de una proteína alfa-2 indicativa de una proteinuria tubular. En los exámenes radiológicos, existía una desmineralización intensa y generalizada. En el estudio histológico, se apreciaron “algunos depósitos cálcicos medulares” (nefrocalcinosis). La enfermedad de Dent fue descrita por Charles Dent y M. Friedman en 1964(30). Se trata de una tubulopatía proximal renal incompleta caracterizada por: proteinuria de bajo peso molecular, hipercalciuria, nefrocalcinosis y/o nefrolitiasis, raquitismo u osteomalacia e insuficiencia renal crónica. En algunos pacientes, también, se aprecian otros defectos tubulares como: hiperaminoaciduria, hiperfosfaturia, glucosuria, hiperuricosuria o incremento en la eliminación urinaria de potasio. Aunque la proteinuria de bajo peso molecular se observa de manera no específica en muchos defectos tubulares, no es común que coincida con nefrolitiasis, en ausencia de infección u obstrucción por cálculos recurrentes. La enfermedad es de herencia recesiva ligada al cromosoma X; de tal modo que, es más severa en los varones que en las mujeres. El diagnóstico definitivo se basa en la presencia de mutaciones en los genes CLCN5 (Dent-1)(31)
y OCRL (Dent-2)(32), que codifican el intercambiador 2Cl–/H+ ClC5 y la fosfatasa lipídica OCRL1, respectivamente. Con alta probabilidad, el paciente estudiado por Sorel et al. tenía una enfermedad de Dent(22), al igual que uno de los pacientes descritos por Gentil et al. (Observación 2)(3). El médico inglés Charles Dent nació en España, en concreto, en la ciudad de Burgos(33).
Epílogo
Pierre Royer (1917-1995) se especializó en Pediatría bajo la dirección de Robert Debré, con quien trabajó directamente durante veinte años. Fue un maestro de la Pediatría francesa e internacional en el sentido más amplio del término y, durante varias generaciones, un médico e investigador modelo, cualidades que supo combinar con perseverancia y lucidez. Su labor científica se centró en el metabolismo de los minerales y su regulación a nivel del esqueleto, las glándulas endocrinas y, sobre todo, el riñón. Contó con la colaboración de eminentes médicos como: Henri Lestradet, Renée Habib, Henri Mathieu y Michel Broyer(34). Pierre Royer creó una de las primeras escuelas de Nefrología pediátrica en la que se formaron pediatras franceses y europeos, que luego desarrollaron sus propios grupos en sus países de origen. Muchos de los nefrólogos pediátricos españoles actuales son “descendientes científicos” de la escuela parisina, puesto que los dos primeros nefrólogos pediátricos españoles, Luis Callís y Juan Rodríguez Soriano se formaron en el Hôpital des Enfants-Malades Necker de París a principios de los años 60 del pasado siglo(35,36).
Los autores que estudiaron a los pacientes pediátricos con hipercalciuria en la década de los 60, los subdividieron en dos subtipos, a saber, aquellos sin manifestaciones clínicas sustanciosas (hipercalciuria idiopática) y aquellos con manifestaciones clínicas evidentes (hipercalciuria idiopática con nanismo y alteración renal en el niño). De este segundo subtipo, como ya se ha indicado, se publicaron bastantes casos por parte de pediatras y de los primeros nefrólogos pediátricos europeos en los años 60 y 70 del pasado siglo, aunque no conocemos ninguna notificación de casos similares por parte de pediatras americanos. ¿Era un trastorno exclusivamente europeo? Los autores que publicaron los últimos pacientes con esa asociación eran alemanes (1987)(37) y españoles (1990)(38). Después, la enfermedad desapareció. La razón de ello es, seguramente, que los siguientes casos similares a los de los primeros pacientes descritos con ese cuadro fueron diagnosticados más tarde de unas tubulopatías bien definidas que han sido nombradas más arriba.
Uno de los autores de este articulo (VMGN) presentó en la IX Reunión Anual de Nefrología Pediátrica celebrada en Sevilla en 1982, un caso de “Hipercalciuria idiopática tipo Royer”(39) (Fig. 4). Una década después, mediante estudio genético se confirmó que, en realidad, la paciente estaba afecta de un síndrome de Bartter neonatal(40).

Figura 4. Frontal de una comunicación presentada por uno de los autores en un congreso celebrado a principios de los años 80 del pasado siglo(39).
Bibliografía
1. García Nieto VM, Zafra Anta M. Enfermedades pediátricas que han pasado a la historia (7). El síndrome de De Toni-Debré-Fanconi revisado. Pediatr Integral. 2022; 26: 59e1-59.e5.
2. Royer P, Mathieu H, Gerbeaux S, Fréderich A, Rodríguez-Soriano J, Dartois AM, et al. L’hypercalciurie idiopathique avec nanisme et atteinte rénale chez l’enfant. Ann Pediatr (París). 1962; 38: 767-83.
3. Gentil C, Habib R, Le Tan Vinh, Colin J, Gabilan JC, Courtecuisse V, et al. Nanisme avec rachitisme, hypercalciurie et protéinurie. (deux observations). Ann Pediatr (París). 1962; 38: 164-72.
4. Flocks RH. Calcium and phosphorus excretion in the urine of patients with renal or ureteral calculi. J Urol. 1940; 44: 183.
5. Albright F, Henneman P, Benedict PH, Forbes AP. Idiopathic hypercalciuria (A preliminary report). Proc R Soc Med. 1953; 46: 1077-81.
6. Zetterström R. Idiopathic hypercalcemia and hypercalciuria. Bibl Paediatr. 1958; 66: 478-87.
7. Rosenkranz A. Ein eigenartiges syndrom tubulärer nierenstörungen mit urolithiasis beim säugling; über die biologische vielfalt der tubulären nierenaffektionen im kindesalter. Helv Paediatr Acta. 1958; 13: 455-70.
8. Valverde A. A propos de lithiase urinaire infantile. Acta Urol Belg. 1962; 30: 568-72.
9. Royer P. Tubulopatías hereditarias. Hipercalciuria idiopática. En: Nefrología pediátrica (ed. esp.). Royer P, Habib R, Mathieu H, Broyer M, eds. Barcelona: Ediciones Toray; 1973. p. 46-8.
10. Royer P. L’hypercalciurie idiopathique avec nanisme et atteinte rénale chez l’enfant. Acta Paediatr Scand. 1967: 172: 186-91.
11. Royer P, Balsan S. Effet d’un régime pauvre en chlorure de sodium dans le “syndrome d’hypercalciurie idiopathique avec nanisme et troubles rénaux” de l’enfant. Schweiz Med Wschr. 1966; 96: 412-5.
12. Royer P, Mathieu H, Habib R. Hipercalciuria idiopática con nanismo y alteración renal. En: Problemas actuales de Nefrología Infantil, ed. esp. Barcelona: Ediciones Toray; 1965. p. 156-76.
13. Bartter FC, Pronove P, Gill JR Jr, MacCardle RC, Diller E. Hyperplasia of the yuxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. Am J Med. 1962; 33: 811-28.
14. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006; 78: 193-201.
15. Louis-Bar D. Sur un syndrome progressif comprenant des télangiectasies capillaires cutanées et conjonctivales symétriques, à disposition naevoïde et des troubles cérébelleux. Confinia Neurologica. 1941; 4: 32-42.
16. Noonan JA, Ehmke DA, Associated noncardiac malformations in children with congenital heart disease, J Pediatr 1963; 63: 468-70.
17. Fanconi A. Idiopathische hypercalciurie im kindesalter. Helv Paediatr Acta. 1963; 18: 306-22.
18. Beilin LJ, Clayton BE. Idiopathic hypercalcuria in a child. Arch Dis Child. 1964; 39: 409-14.
19. De Luca R, Guzzetta F. L’ipercalciuria idiopatica infantile. Observazione in quattro fratelli. Pediatria (Napoli). 1965; 73: 613-39.
20. Nordio S, Bertolotti E, Gatti R. Due casi con alterazioni non comuni del ricambio calcio fosforico. “Disparatiroidismo” (Ipo-iperparatiroidismo?). Difetto di concentrazione delle urine secondario ad “ipercalciuria idiopatica”. Minerva Pediatr. 1965; 17: 79-86.
21. Jeune M, Gilly R, Hermier M, Frederich A, Collombel C, Raveau J. L’hypercalciurie idiopathique de l’enfant. A propos d’une observation. Pediatrie. 1967; 22: 17-49.
22. Sorel R, Dalous A, Fabre J, Rochiccioli P, Ghisolfi J, Fabre MT. Hypercalciurie idiopathique avec nanisme. A propos d’une observation avec lésions rénales glomérulaires particulières en microscopie électronique. Arch Fr Pediatr. 1970; 27: 161-76
23. Fovet-Poingt O, Toursel F. Une observation d’hypercalciurie idiopathique. Pediatrie. 1972; 27: 295-302.
24. Verger P, Guillard JM, Grenier A, Fontan D, Cixous P, Laigle JL. Hypercalciurie idiopathique chez un nourrison. Arch Fr Pediatr. 1973; 30: 441-2.
25. Lama G, Perrone L. Considerazioni sull’ipercalciuria idiopatica. Descrizione di un caso. Pediatria (Napoli). 1974; 82: 583-96.
26. Tieder M, Stark H. Forme familiale d’hypercalciurie idiopathique avec nanisme, atteinte osseuse et rénale chez l’enfant. Helv Paediatr Acta. 1979; 34: 359-67.
27. Perdomo-Ramírez A, García Nieto VM, Claverie-Martín F. Hipomagnesemia familiar con hipercalciuria y nefrocalcinosis. Otras causas de hipomagnesemia. En: Nefrología Pediátrica. Exeni R, García-Nieto V, Medeiros M, Santos F, eds. Oviedo: Ediciones de la Universidad de Oviedo; 2021. p. 241-50.
28. Rodríguez-Soriano J, Vallo A. Pathophysiology of the renal acidification defect present in the syndrome of familial hypomagnesaemia-hypercalciuria. Pediatr Nephrol. 1994; 8: 431-5.
29. Al-Haggar M, Bakr A, Tajima T, Fujieda K, Hammad A, Soliman O, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: unusual clinical associations and novel claudin16 mutation in an Egyptian family. Clin Exp Nephrol. 2009; 13: 288-94.
30. Dent CE, Friedman M. Hypercalcuric rickets associated with renal tubular damage. Arch Dis Child. 1964; 39: 240-9.
31. Fisher SE, van Bakel I, Lloyd SE, Pearce SH, Thakker RV, Craig IW. Cloning and characterization of CLCN5, the human kidney chloride channel gene implicated in Dent disease (an X-linked hereditary nephrolithiasis). Genomics. 1995; 29: 598-606.
32. Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, et al. Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005; 76: 260-7.
33. De la Mata Franco G, García Nieto V. Charles Enrique Dent y Burgos. En: Cuadernos de Historia de la Pediatría Española nº 17. Madrid: Asociación Española de Pediatría. 2019; p. 22-33.
34. Chesney RW. The development of pediatric nephrology. Pediatr Res. 2002; 52: 770-8.
35. Rodríguez Soriano J. Nacimiento y desarrollo de la nefrología pediátrica. Una historia vivida. Bol Pediatr. 2002; 42: 313-6.
36. García Nieto VM, Grünberg J, Luis Yanes MI. Discípulos y maestros. Lo que aprendimos de Juan Rodríguez Soriano. Rev Esp Pediatr. 2011; 67: 324-31.
37. Kohout I, Bachmann H. Klinische varianten der “idiopathischen hypercalciurie” bie kindren. Monatsschr Kinderheilkd. 1987; 135: 847-50.
38. Baena Sáez J, Cañuelo Ortiz O, Cardenas Talaverón C, Garrido Palomo R, Paz Azcárate JL, Fernández Gutiérrez F, et al. Hipercalciuria renal idiopática asociada a nefrocalcinosis e hipocrecimiento. Rev Esp Pediatr. 1990; 46: 175-7.
39. García Nieto V, Melchor Pérez E, Souto Martínez I, Oliveros Pérez R. Manejo renal del sodio en la hipercalciuria idiopática tipo Royer. An Esp Pediatr. 1983; 18: 330.
40. García Nieto V, Müller D, van del Bliet W, Claverie-Martín F. Enfermedad de Bartter neonatal diagnosticada mediante la detección de una mutación en el gen NCNJ1 que codifica la síntesis del canal renal de potasio ROMK1. Nefrología. 2001; 21: 448-55.

 Juvenile Idiopathic Arthritis
Juvenile Idiopathic Arthritis