 |
| Temas de FC |
J. Antón López*, S. Carriquí Arenas**
*Jefe de sección de Reumatología pediátrica. Servicio de Pediatría. Hospital Sant Joan de Déu. Profesor asociado. Universitat de Barcelona. **Sección de Reumatología pediátrica. Servicio de Pediatría. Hospital Sant Joan de Déu. Barcelona
| Resumen
La vasculitis es una inflamación de la pared de los vasos sanguíneos. Las vasculitis pediátricas engloban un grupo heterogéneo de enfermedades. En Ankara 2008, con el consenso de EULAR/PRINTO/PRES, se validaron criterios clasificatorios pediátricos para intentar agruparlas. La incidencia en edad pediátrica es baja, a excepción de la vasculitis por IgA (VIgA) y la enfermedad de Kawasaki (EK). La prueba diagnóstica considerada gold estándar es la anatomía patológica, aunque en la VIgA y la EK, al diagnóstico se llega mediante criterios clínicos. Existen hallazgos clínicos y analíticos que deben hacernos sospechar vasculitis, como pueden ser: fiebre prolongada de origen desconocido, lesiones cutáneas sugestivas, elevación de parámetros inflamatorios, etc. La VIgA es la vasculitis pediátrica más común. Afecta principalmente a piel, intestino y riñón, siendo la afectación renal el principal factor de morbimortalidad a largo plazo. Su curso es autolimitado y el tratamiento es conservador. La EK es la segunda vasculitis más frecuente tras la VIgA. El curso es autolimitado, puede producir complicaciones graves, siendo la primera causa de enfermedad cardíaca adquirida en la infancia en países desarrollados. El tratamiento de elección son las inmunoglobulinas (IVIG), cuya administración dentro de los 10 primeros días disminuye de forma significativa el riesgo de complicaciones cardíacas. |
| Abstract
Vasculitis is an inflammation of the blood vessel wall. Pediatric vasculitis encompass a heterogeneous group of diseases. In Ankara 2008, with the consensus of EULAR/PRINTO/PRES, pediatric classification criteria were validated in order to group them. Its incidence is low in children with the exception of IgA vasculitis (VIgA) and Kawasaki disease (KD). Pathological anatomy is considered the gold standard diagnostic test, although in VIgA and KD the diagnosis is reached by means of clinical criteria. There are clinical and analytical findings that should raise suspicion of vasculitis, such as prolonged fever of unknown origin, suggestive skin lesions, elevated inflammatory parameters… VIgA is the most common pediatric vasculitis. It mainly affects the skin, bowels and kidneys. The renal involvement is the main factor in long-term morbidity and mortality. Its course is self-limited and treatment is conservative. KD is the second most common vasculitis after VIgA. Its course is self-limited and can generate serious complications. In developed countries it is the first cause of acquired cardiac disease in childhood. Immunoglobulin (IVIG) is the mainstay treatment, as its administration within the first 10 days significantly reduces the risk of cardiac complications. |
Palabras clave: Vasculitis pediátricas; Vasculitis IgA; Nefritis por IgA; Enfermedad de Kawasaki; Vasculitis monogénicas.
Key words: Pediatric vasculitis; IgA vasculitis; IgA nephritis; Kawasaki disease; Monogenic vasculitis.
Pediatr Integral 2022; XXVI (3): 151 – 162
OBJETIVOS
• Conocer la clasificación general de las vasculitis pediátricas y cuándo sospecharlas.
• Saber que la vasculitis por IgA y la enfermedad de Kawasaki son las 2 vasculitis más frecuentes en la infancia, cuyo curso es autolimitado.
• Aprender la presentación clínica de la vasculitis por IgA y de la enfermedad de Kawasaki, y cómo realizar el diagnóstico y tratamiento de las mismas.
• Comprender el tratamiento específico de la vasculitis por IgA y de la enfermedad de Kawasaki.
Púrpura de Schönlein-Henoch, enfermedad de Kawasaki y otras vasculitis
Introducción
Las vasculitis se definen por la inflamación de la pared de los vasos sanguíneos, cuyo origen puede ser primario o secundario a otras patologías. Se trata de un grupo heterogéneo de enfermedades cuyas manifestaciones clínicas dependen del tipo, localización y tamaño del vaso afecto(1).
La incidencia estimada de las vasculitis primarias pediátricas es aproximadamente de unos 50 casos/100.000 niños/año. Se trata de patologías raras en la infancia, a excepción de dos tipos de vasculitis agudas y autolimitadas, que son casi exclusivas de la edad pediátrica: vasculitis IgA/Púrpura de Schönlein-Henoch (PSH) y enfermedad de Kawasaki (EK)(2).
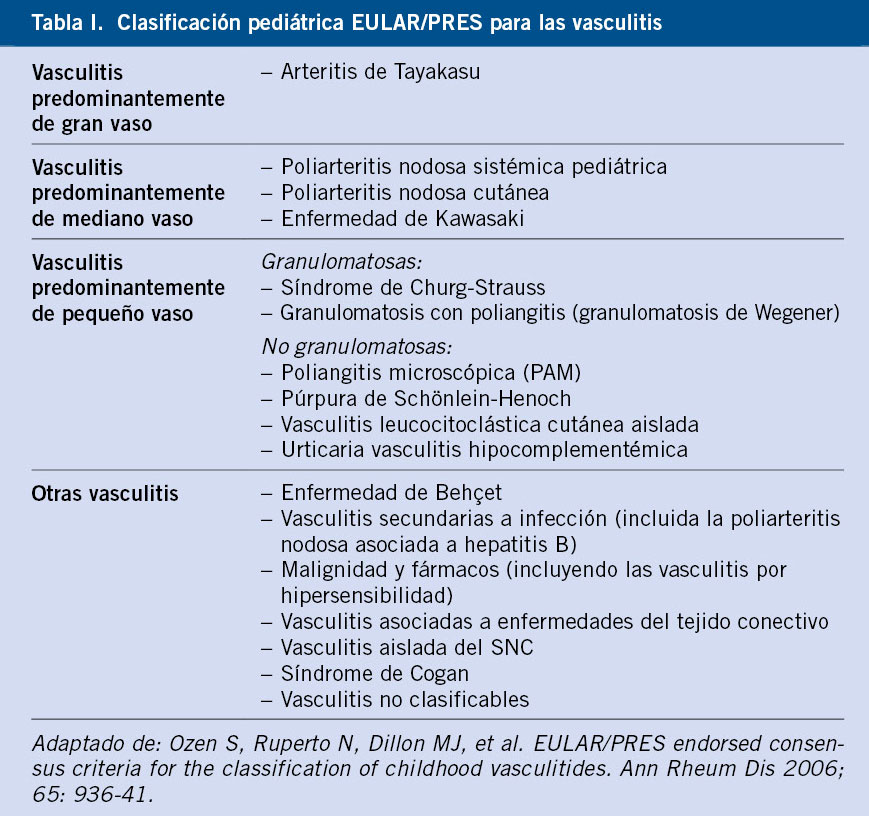
Criterios de clasificación pediátricos de las vasculitis sistémicas
Se prefiere utilizar los criterios clasificatorios del 2008 validados con el consenso de EULAR/PRINTO/PRES, ya que presentan una elevada sensibilidad y especificidad.
El término vasculitis agrupa a un grupo heterogéneo de enfermedades. Disponemos de criterios clasificatorios con el fin de poder agrupar grupos bien definidos, para poder realizar estudios y desarrollar protocolos. También para la enfermedad de Kawasaki disponemos de criterios diagnósticos(3).
Desde la década de 1990, los criterios de clasificación más aceptados para población pediátrica eran los propuestos por el ACR (American Collegue of Reumathology), basados en los criterios utilizados en población adulta.
En 2006, se propusieron los criterios clasificatorios de consenso EULAR/PRES. Utilizando la metodología Delphi, se elaboraron criterios para: PSH, arteritis de Takayasu, panarteritis nodosa (PAN) y granulomatosis con poliangeitis.
A pesar de que en el 2012, la conferencia internacional de Chapel Hill actualizó las categorías de vasculitis según el tamaño del vaso afecto, en Pediatría se prefiere utilizar los criterios clasificatorios del 2008, validados con el apoyo del EULAR/PRINTO/PRES, que presentan una elevada sensibilidad y especificidad(2-4) (Tabla I).
Diagnóstico general de vasculitis
El diagnóstico gold estándar de las vasculitis continúa siendo la anatomía patológica, a excepción del diagnóstico, mayormente clínico, de la púrpura de Schönlein-Henoch y de la enfermedad de Kawasaki.
Actualmente, la anatomía patológica sigue considerándose el gold estándar para el diagnóstico de la mayoría de las vasculitis, con la excepción de la púrpura de Schönlein-Henoch y la enfermedad de Kawasaki, cuyo diagnóstico en la edad pediátrica es mayormente clínico. Las pruebas de imagen son útiles, sobre todo en las vasculitis de mediano y gran vaso.
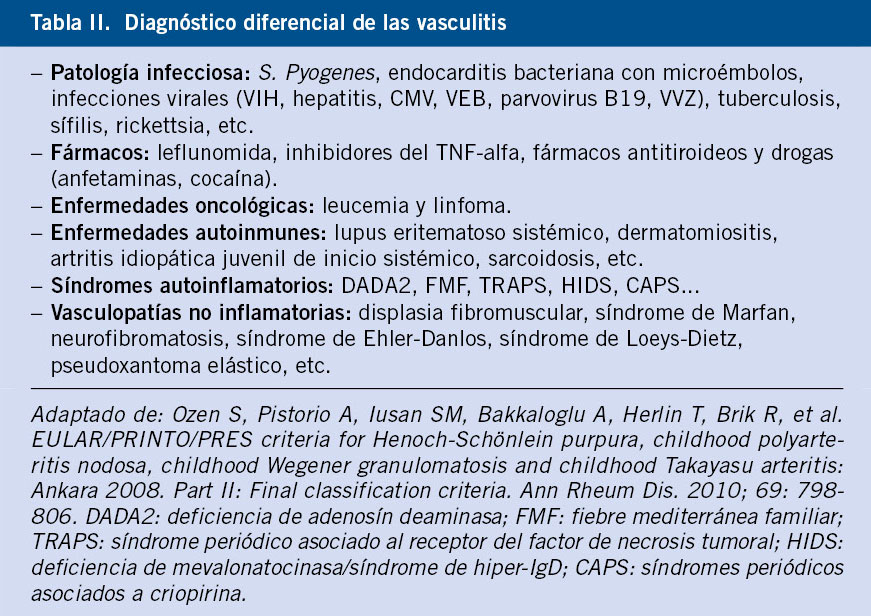
Existen una serie de hallazgos clínicos y de laboratorio que deben hacernos sospechar sobre la posibilidad de una vasculitis, son: fiebre prolongada de origen desconocido, lesiones cutáneas sugestivas y afectación renal, pulmonar o cardiovascular desconocida, junto con afectación sistémica, neuropatía periférica o afectación del SNC, serositis, artritis o elevación de parámetros inflamatorios (VSG, PCR, leucocitosis…)(3).
Ante la sospecha diagnóstica, lo primero es realizar una correcta anamnesis y exploración por órganos y sistemas, dirigiendo los estudios complementarios según la sospecha inicial y descartando patologías más frecuentes que cursen con clínica similar(3) (Tabla II).
Vasculitis IgA (púrpura de Schönlein-Henoch)
Introducción
La vasculitis por IgA es una vasculitis de pequeños vasos, anteriormente conocida como púrpura de Schönlein-Henoch.
La vasculitis por IgA (VIgA), anteriormente conocida como púrpura de Schönlein-Henoch (PSH), es una vasculitis de pequeños vasos (vénulas, capilares y arteriolas) en los que se depositan inmunocomplejos IgA. Los órganos afectados con más frecuencia son piel, intestino y riñón, principalmente a nivel glomerular. Su curso es agudo y autolimitado, llegándose al diagnóstico en la mayoría de los casos, por los criterios clínicos validados por EULAR/PRINTO/PRES en Ankara 2008(5).
Epidemiología
La vasculitis por IgA es la vasculitis pediátrica más frecuente. El 50% de los casos ocurren en niños de 5 años o menores.
La VIgA supone la vasculitis sistémica más frecuente en edad pediátrica (90%). La incidencia oscila entre 3-26,7 casos por 100.000 niños/año(5). La VIgA tiene un pico de presentación entre los 3 a 12 años, siendo raro que ocurra por debajo de los 2 años. El 50% de los casos ocurre en niños de 5 años o menores, y el 75-90% en menores de 10 años. Existe un discreto predominio en el sexo masculino (1,2:1 hasta 1,8:1 según series publicadas). El 30-50% de los casos ocurren durante los meses de otoño e invierno, normalmente precedido por infecciones respiratorias de vías altas(1-6).
Etiopatogenia
Los depósitos granulares patognomónicos de IgA y C3 en el mesangio en la VIgA/PSH son indistinguibles de los que se observan en la nefropatía por IgA.
La etiopatogenia de la enfermedad sigue siendo desconocida, se cree que la actuación de ciertos desencadenantes ambientales (infecciones como: estreptococo del grupo A betahemolítico, Mycoplasma pneumoniae, Bartonella Henselae, fármacos, etc.) sobre pacientes genéticamente predispuestos, daría lugar a la formación y depósito de inmunocomplejos en la pared vascular, generando una inflamación local que desencadenaría una vasculitis leucocitoclástica con necrosis en vasos de pequeño calibre(6).
La mayoría de los casos son esporádicos, pero se han descrito asociaciones intrafamiliares y la susceptibilidad de padecerla en presencia de determinados polimorfismos de genes del complejo mayor de histocompatibilidad (HLA), incluyendo: HLA-DRB1 y HLA-B*4102. El HLA-B35 y DQA1 se han relacionado con el riesgo de padecer nefritis en pacientes con PSH(2-3). Según estudios realizados en Turquía e Israel, existe un aumento significativo de la prevalencia de mutaciones en el gen MEFV (Mediterranean fever) en niños con vasculitis por IgA respecto a la población general, y hasta un 5-7% de pacientes con fiebre mediterránea familiar presentan una VIgA(6).
La glicosilación aberrante de la región bisagra de la IgA1 predispone a la formación de inmunocomplejos y, por tanto, se ha descrito como factor de riesgo para desarrollar vasculitis por IgA y nefritis por IgA (IgAN). La activación del complemento en la VIgA es un importante factor de daño tisular, ya que el aclaramiento defectuoso de inmunocomplejos de IgA tiene un papel importante en la patogenia de la nefritis por IgA. Los depósitos granulares patognomónicos de IgA y C3 en el mesangio en la VIgA/PSH son indistinguibles de los que se observan en la nefropatía por IgA(1).
La concentración y actividad del factor XIII podría constituirse como indicador pronóstico de la VIgA, ya que una disminución marcada de la actividad del factor XIII podría dar lugar a complicaciones severas, como hemorragia intracraneal, abdominal o pulmonar(1).
Clínica
La VIgA es una vasculitis sistémica. Las manifestaciones típicas son: púrpura, dolor articular y abdominal y afectación renal.
La VIgA es una vasculitis sistémica, por lo que puede afectar a cualquier órgano, siendo típica la afectación cutánea, articular, digestiva y renal. La sintomatología suele aparecer de forma progresiva en días o semanas, sin seguir un orden específico. La presentación inicial típica es la púrpura y el dolor articular.
Manifestaciones cutáneas
La afectación cutánea ocurre aproximadamente en el 75% de los niños con VIgA. La lesión más característica es la púrpura palpable en formas de petequias que pueden confluir, dando lugar a grandes equimosis. En ocasiones, puede presentarse como un exantema maculopapular eritematoso o urticarial. Las lesiones suelen aparecer en zonas declives (extremidades inferiores, glúteos), aunque también pueden afectarse las extremidades superiores, cara y tronco. El inicio del cuadro clínico, sobre todo en los más pequeños, puede acompañarse de edema del cuero cabelludo, cara, manos, pies y escroto. Las lesiones ampollosas, hemorrágicas o necróticas son raras en niños (2%), ocurriendo hasta en el 60% en adultos(1-6).
Manifestaciones articulares
Suelen aparecer en el 50-80% de los casos, principalmente en forma de artralgias de grandes articulaciones (tobillos y rodillas) y excepcionalmente como artritis. Suponen la primera manifestación de la enfermedad en el 15-25% de los pacientes. Clínicamente, se manifiesta como inflamación periarticular sin eritema ni aumento de temperatura local, con dolor que limita la articulación afecta. Son transitorias y no migratorias, resolviendo en pocos días sin secuelas(1-6).
Manifestaciones digestivas
Se describen en el 50-75% de los pacientes dentro de la primera semana de aparición de las lesiones cutáneas y dentro del primer mes de enfermedad, solo en el 11-20% suponen el primer síntoma. Los síntomas varían desde dolor abdominal con náuseas y vómitos, hasta complicaciones graves como: invaginación intestinal (2-3% de los casos), perforación, enteropatía pierde proteínas, pancreatitis, obstrucción u estenosis, hidrops vesicular, hemorragia gastrointestinal masiva, etc.(1-6).
La localización más frecuente es la zona proximal del intestino delgado, en forma de lesión isquémica, presentando sangre oculta en heces hasta en el 56% de los pacientes con afectación intestinal. En una publicación reciente se observó que el 71% de los pacientes presentaban manifestaciones digestivas; de ellos, el 7,6% sin afectación cutánea, llegándose al diagnóstico por biopsia vía endoscópica. La determinación de calprotectina fecal podría ser un marcador útil de afectación gastrointestinal en la VIgA/PSH(1).
Manifestaciones renales
Un tercio de los pacientes presentará afectación renal, que suele manifestarse entre la 4ª y 6ª semana tras la aparición de las lesiones cutáneas. Se puede manifestar de diversas formas como: hematuria microscópica/macroscópica con o sin proteinuria, nefritis o síndrome nefrótico, hipertensión, insuficiencia renal, etc. En la mayoría de las ocasiones, la afectación es leve y autolimitada, pudiendo ser grave en un 10%, aproximadamente; pudiendo cronificarse, dando lugar a un daño renal permanente. Por ello, la afectación renal es el principal factor pronóstico a largo plazo de la enfermedad(1-6).
En el 97% de los casos, la afectación renal aparece en los 6 primeros meses. El 1,6% de los pacientes que tuvieron una alteración renal puntual acaban presentando afectación renal permanente y en los que tuvieron nefritis o síndrome nefrótico, puede ocurrir hasta en el 19,5%(6).
En un metaanálisis con 2.400 pacientes con VIgA/PSHN, se identificaron factores de riesgo para desarrollar afectación renal(6) (Tabla III).

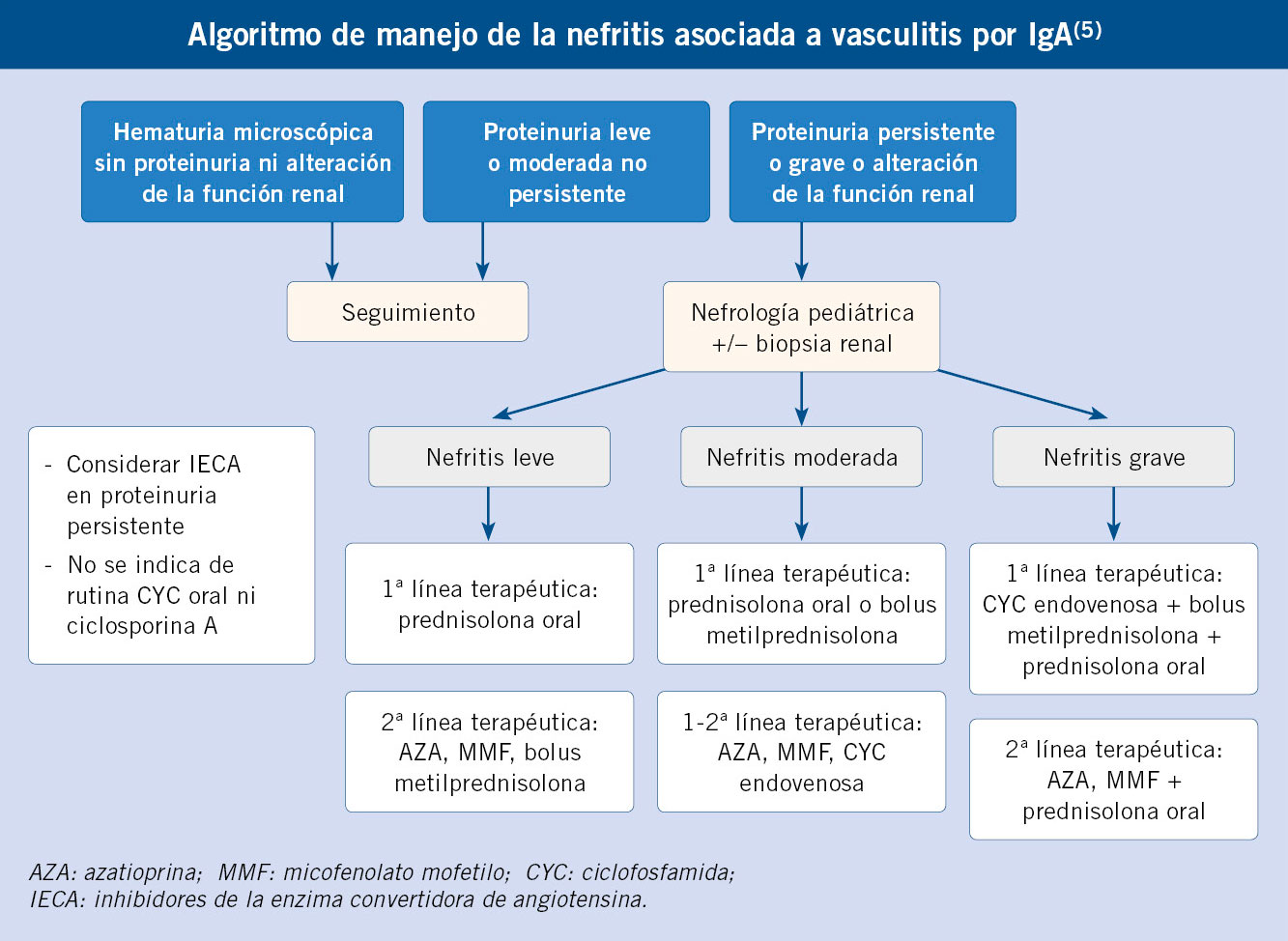
Las guías SHARE publicadas en 2019, para el diagnóstico y tratamiento de la VIgA, recomiendan realizar un control de la tensión arterial y sedimento urinario durante los primeros 6 meses. En aquellos pacientes con proteinuria moderada o afectación del filtrado glomerular, se recomienda valoración y seguimiento por una unidad de Nefrología Pediátrica(5).
Manifestaciones urológicas
Las manifestaciones aparecen, sobre todo en varones, en forma de: escroto agudo, epididimitis, orquitis y complicaciones del cordón espermático (hematoma y edema). La eco-doppler permite establecer un diagnóstico, aunque puede ser necesaria la exploración quirúrgica para descartar torsión testicular. Rara vez, puede existir una obstrucción ureteral o ureteritis secundaria, típicamente aparecen al mes o a los dos meses, en ocasiones, asociada a nefritis(1).
Manifestaciones neurológicas
La afectación del sistema nervioso es poco frecuente (<1%), pudiendo presentarse en forma de: cefaleas con cambios emocionales (irritabilidad, apatía…), convulsiones, síndrome de Guillain-Barré, neuropatía periférica, ataxia, etc.(6).
Manifestaciones pulmonares
La afectación pulmonar es rara. Se han descrito casos aislados de hemorragia difusa alveolar, neumonía intersticial y fibrosis intersticial(1).
Diagnóstico VIgA
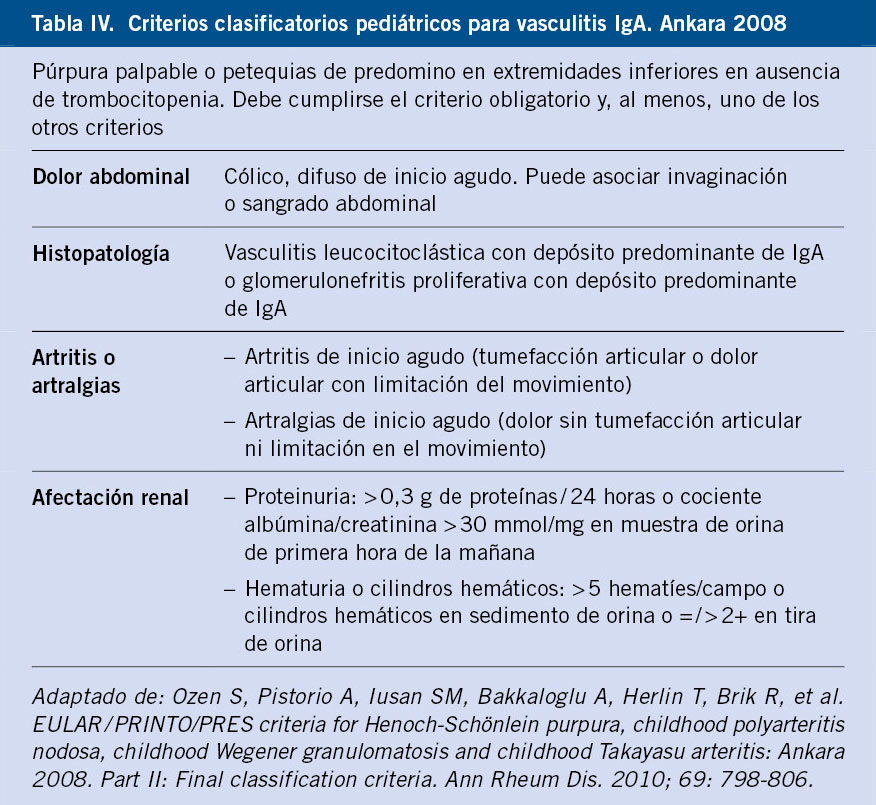
El diagnóstico de la VIgA/PSH es clínico. A todo paciente con VIgA se le debe valorar la función renal al diagnóstico. La característica típica de la nefritis por VIgA en la biopsia renal es el depósito mesangial difuso de IgA en la inmunofluorescencia.
El diagnóstico fundamentalmente es clínico. En el consenso de Ankara 2008, se establecieron los criterios clasificatorios para VIgA en edad pediátrica por parte de EULAR/PRINTO/PRES, no se trata de criterios diagnósticos (Tabla IV).
No existen pruebas complementarias específicas para llevar a cabo el diagnóstico, pero sí nos ayudan en el diagnóstico diferencial y en conocer el grado de afectación. En el ámbito del laboratorio, en la fase aguda, podemos encontrar hallazgos inespecíficos (anemia, leucocitosis con neutrofilia…) y ligera elevación de VSG y PCR. El estudio de autoinmunidad (ANA, ANCA y FR) suele ser negativo, con niveles de complemento normales. En torno a un tercio de los casos pueden presentar elevación de IgA sérica, no siendo diagnóstica de la patología. En todo niño con sospecha de vasculitis por IgA, se debe estudiar la función renal; para ello, se realizará una evaluación del filtrado glomerular, sedimento urinario a primera hora de la mañana (hematuria, proteinuria, Índice Proteína/Creatinina -IProt/Cre-) y evaluación de la tensión arterial(5-6).
Las pruebas de imagen como la ecografía abdominal, se recomiendan en aquellos casos de dolor abdominal severo o sangrado abdominal, para descartar invaginación intestinal y perforación secundaria a isquemia de la pared intestinal.
La biopsia cutánea será necesaria para el diagnóstico solo en aquellos casos con una presentación atípica. Las guías de práctica clínica SHARE de 2019 recomiendan realizar biopsia cutánea de las lesiones agudas de <=48 horas de aparición, en aquellos casos con rash atípico (lesiones extensas o distribución difusa) para descartar otros diagnósticos(5).
Desde el punto de vista histológico, el hallazgo típico es una reacción leucocitoclástica con depósito predominante de IgA en la pared de pequeños vasos, con una especificidad del 100%. El depósito de IgA es lo que permite diferenciarla de la vasculitis leucocitoclástica; sin embargo, la ausencia de depósitos de IgA no excluye el diagnóstico de VIgA(5-6).
La biopsia renal se lleva a cabo en casos de duda diagnóstica o con afectación renal grave, existiendo una buena correlación entre la gravedad y los hallazgos histopatológicos. En las guías SHARE 2019, se establecieron las indicaciones de biopsia renal y se definió la clasificación de severidad desde el punto de vista anatomopatológico para la nefritis por VIgA(5) (Tablas V y VI).

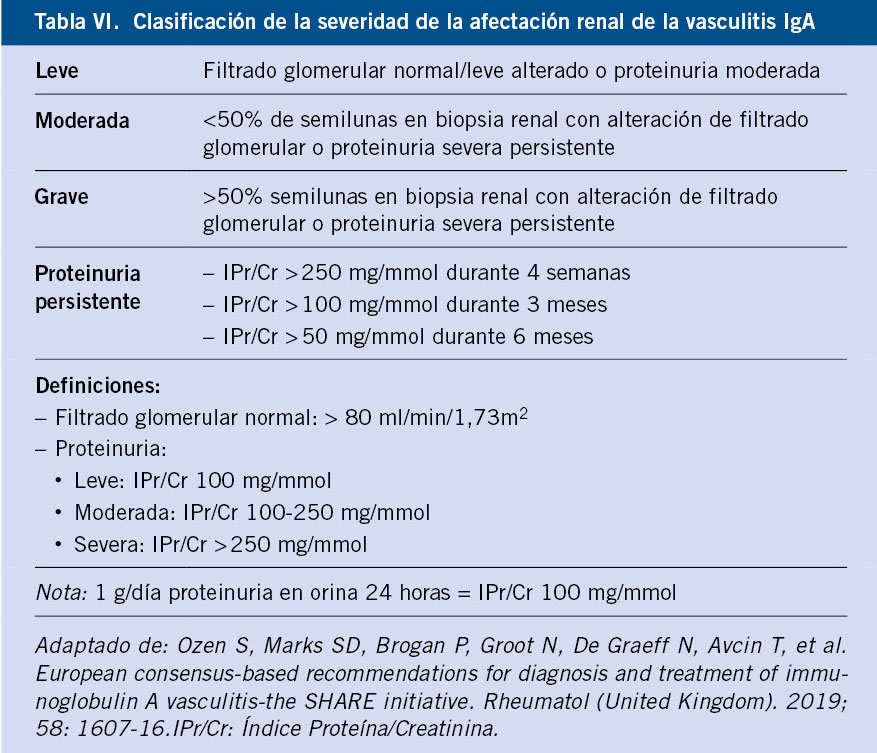
En la microscopía óptica, en la nefritis por IgA, se puede observar desde una proliferación mesangial leve hasta una glomerulonefritis con formación de semilunas grave. El depósito mesangial difuso de IgA en la inmunofluorescencia es la característica típica de la nefritis por VIgA, pudiéndose acompañar de depósitos de C3 (75%)(5-6).
Diagnóstico diferencial
El diagnóstico diferencial se debe realizar, sobre todo en pacientes con presentación atípica. Entre los posibles diagnósticos a tener en cuenta estarían otras vasculitis como: vasculitis ANCA positivo, patología autoinmune como PTI (púrpura trombopénica idiopática), conectivopatías como lupus, patología autoinflamatoria como fiebre mediterránea familiar (FMF), o procesos infecciosos como parvovirus B19, sepsis, etc.(6).
Existe una entidad con manifestaciones clínicas similares, el edema agudo hemorrágico del lactante, que afecta predominantemente a lactantes de 4 a 24 meses, con curso autolimitado en 1-3 semanas, siendo excepcional la afectación visceral como ocurre en la VIgA.
Tratamiento
El tratamiento principal es conservador. El uso de corticoides está indicado en afectación gastrointestinal severa, orquitis, vasculitis del sistema nervioso central, nefritis o hemorragia pulmonar. El uso de corticoides no previene de la afectación renal.
La mayoría de los casos presentan una resolución espontánea; por ello, el principal manejo es conservador, con medidas de soporte como: reposo, analgesia e hidratación(5-7).
En función de las recomendaciones de tratamiento elaboradas por el grupo SHARE para la VIgA, el manejo de las artralgias/artritis y el dolor abdominal leve se puede realizar con analgesia convencional con paracetamol o AINES, evitando el uso de los últimos si existe compromiso renal. No se ha demostrado que el uso de AINES aumente el riesgo de sangrado abdominal, pero sí están contraindicados en caso de sangrado digestivo activo(5).
El uso de glucocorticoides está limitado a una serie de indicaciones: afectación gastrointestinal severa, orquitis, vasculitis del sistema nervioso central, nefritis o hemorragia pulmonar; en estos casos, se puede realizar una inducción con bolus de metilprednisolona a dosis de 10-30 mg/kg/día (máximo 1 g) durante 3 días e incluso podría ser necesario el uso de otros inmunosupresores o plasmaféresis(5).
En los pacientes con dolor abdominal intenso o sangrado rectal, afectación cutánea severa o artralgias intensas, se puede considerar el uso de corticoterapia, ya que ha demostrado reducir la intensidad y duración del dolor, además de prevenir la intervención quirúrgica. La dosis habitual es de 1-2 mg/kg/día de prednisolona (máximo 60 mg) durante 1-2 semanas en pauta descendente. En los pacientes con afectación abdominal y ante el posible síndrome de malabsorción secundario al edema de la pared intestinal, se recomienda su uso vía endovenosa. El uso de corticoides no previene la afectación renal ni las recurrencias(5).
En los casos con curso refractario, en los que se han utilizado corticoides con adecuada respuesta, se puede añadir un ahorrador de corticoides (colchicina, dapsona, micofenolato mofetilo, azatioprina, inmunoglobulinas, ciclosporina…). Existen publicaciones de casos con afectación cutánea severa o recurrente tratados con colchicina con resolución de las lesiones(5).
Las recomendaciones para el tratamiento de la nefritis por VIgA siguen siendo controvertidas ante la falta de evidencia científica. En las recientes recomendaciones del grupo SHARE, se establece como recomendación general, el tratamiento con inhibidores de la enzima convertidora de la angiotensina (IECA) o antagonistas del receptor de la angiotensina II (ARA II), para prevenir o limitar la lesión glomerular secundaria en niños con VIgA, que presentan compromiso renal con proteinuria persistente (>3 meses de duración), independientemente de si reciben prednisolona u otro inmunosupresor(5-8).
En la nefritis leve, el tratamiento de primera línea es prednisolona oral. En la nefritis moderada, el tratamiento de primera línea es prednisolona oral o metilprednisolona en pulsos endovenosos. En la nefritis grave, se recomienda el uso de ciclofosfamida endovenosa con metilprednisolona en pulsos endovenosos o prednisolona oral, como tratamiento de primera línea.
Se puede usar azatioprina (AZA) o micofenolato mofetilo (MMF) como tratamiento de segunda línea en pacientes con nefritis, según los hallazgos histopatológicos hallados en la biopsia, además del tratamiento de mantenimiento con corticoides en casos de nefritis grave(5-8).
Pronóstico
El pronóstico general es bueno. A nivel renal, el 95% de los pacientes con afectación alcanzan una resolución completa.
El pronóstico general es excelente, al ser una patología autolimitada a 2-4 semanas. Un tercio de los pacientes pueden presentar recurrencias de las lesiones cutáneas o dolor abdominal dentro de las primeras 6 semanas. La morbilidad temprana de la patología depende en gran medida de la afectación gastrointestinal, sin embargo, la morbimortalidad a largo plazo estará determinada por la afectación renal(1-6).
A nivel renal, el pronóstico es bueno, con una resolución completa en cerca del 95% en aquellos pacientes que presentan hematuria y proteinuria. En un 2-15% de los casos, puede producirse un daño renal, con alteración del filtrado glomerular o hipertensión arterial, y en menos del 1% un fallo renal.
Se consideran factores de mal pronóstico renal: debut por encima de los 8 años, afectación abdominal, púrpura persistente, síndrome nefrítico/nefrótico, disminución de la actividad factor XIII, hipertensión arterial, fallo renal al inicio de los síntomas y presencia en la biopsia renal de esclerosis glomerular/glomerulonefritis con más del 50% de semilunas/atrofia y fibrosis tubulointersticial(1-6).
Enfermedad de Kawasaki
Introducción
La EK es una vasculitis sistémica aguda autolimitada que puede generar complicaciones potencialmente graves.
La enfermedad de Kawasaki (EK) es una vasculitis sistémica aguda autolimitada, que afecta a vasos de pequeño y mediano calibre y puede llegar a producir complicaciones potencialmente graves. La clínica característica es: fiebre, conjuntivitis bilateral no supurativa, eritema labial y oral, exantema, adenopatía látero-cervical y cambios en extremidades (descamación ungueal y edema en dorso de manos y pies)(9-10).
Epidemiología
Es la segunda vasculitis más frecuente en Pediatría y supone la causa más común de enfermedad cardíaca adquirida en la infancia en nuestro medio.
La EK es la segunda vasculitis más frecuente en la infancia tras la VIgA y supone la causa más común de enfermedad cardíaca adquirida en nuestro medio. El 25% de los casos que no reciben tratamiento presentarán afectación coronaria, disminuyendo al 4% en los que reciben inmunoglobulinas(9).
La incidencia ha ido en aumento en los últimos años. La incidencia en Europa se sitúa entorno los 5,4-15 casos/100.000 niños menores de 5 años/año. En un estudio realizado en Cataluña, la incidencia fue de 8 casos/100.000 niños menores de 5 años/año. En países asiáticos, la incidencia es mayor, siendo en Japón de 309 casos/100.000 niños menores de 5 años/año. El 77% de los casos ocurren en menores de 5 años, con un pico de incidencia entre los 18-24 meses, es poco frecuente en menores de 3 meses o mayores de 5 años; en estos grupos etarios, el riesgo de aneurisma coronario es mayor. Hay un predomino en el sexo masculino de 1,5:1. El mayor pico de incidencia ocurre en primavera e invierno. En Japón, el riesgo de recurrencia es del 3%(11).
La tasa de mortalidad no se conoce en nuestro medio; sin embargo, la mayor mortalidad se da entre el 15º y el 45º día desde el inicio de la fiebre, coincidiendo con el pico máximo de hipercoagulabilidad por trombocitosis e inflamación coronaria(10).
Etiología
La etiología es desconocida. La teoría más aceptada es la existencia de un desencadenante infeccioso transportado hasta las vías respiratorias en personas genéticamente predispuestas.
Tras décadas de investigación aún no se conoce la etiología de la enfermedad. La teoría más aceptada es la existencia de un desencadenante infeccioso inhalado que infectaría las células epiteliales bronquiales ciliadas en personas genéticamente predispuestas. Debido a la estacionalidad de la enfermedad, hay estudios en curso que orientan a un transporte del agente desencadenante por vientos de la troposfera. Se ha considerado la posibilidad de la existencia de un superantígeno(9-10).
La alta incidencia en población asiática sugiere que la genética juega un papel importante en la enfermedad. Estudios del genoma humano han permitido identificar marcadores genéticos de susceptibilidad, severidad y refractariedad al tratamiento. En alguno de estos estudios se han identificado genes del sistema HLA como: HLA B5, B44, Bw51, DR3 y DRB3*0301, que se asocian a enfermedad de Kawasaki en caucásicos. Además, se han identificado tres genes (ITPKC, ORAI1 y SLC8A1) relacionados con la vía de señalización del calcio, que activan la vía de la calcineurina y la traslocación del factor nuclear de células T activadas (NFAT), generando la transcripción de genes proinflamatorios (IL1B y TNF-alfa). Este hallazgo es importante, ya que tiene implicaciones terapéuticas, pudiendo utilizar la ciclosporina y el tacrólimus como tratamientos para inhibir la vía de la calcineurina. El gen de la caspasa 3, en el cromosoma 4, se ha asociado a resistencia al tratamiento con inmunoglobulinas y afectación coronaria. El gen FAM167A-BLK, que se había descrito asociado a enfermedades, como el lupus y la artritis reumatoide, también se ha asociado a un mayor riesgo de presentar EK(9-11).
En la EK participan tanto el sistema inmunológico innato como el adaptativo. La activación del sistema innato genera un gran número de neutrófilos circulantes, leucinas, IL1 e IL6, y factor de necrosis tumoral. La naturaleza autolimitada y no recurrente de la enfermedad sugiere la creación de células T y B de memoria que protegen ante futuros eventos(9).
Clínica
Las manifestaciones típicas no aparecen de forma secuencial, existiendo manifestaciones transitorias; de ahí la importancia de una historia clínica minuciosa con un alto índice de sospecha.
El inicio suele ser agudo, pudiendo estar precedido por síntomas respiratorios de vías altas o gastrointestinales. Las manifestaciones no aparecen secuencialmente, existiendo manifestaciones transitorias, de ahí la importancia de realizar una historia clínica completa. Los signos y síntomas clínicos en los pacientes no tratados desaparecen en una media de 12 días. De forma clásica, se diferencia tres fases evolutivas: un periodo febril agudo, de unos 10 días de duración; un periodo subagudo, de 2-4 semanas; y una fase de convalecencia(11-12).
Fiebre
Es el síntoma guía, suele ser persistente con escasa respuesta a antitérmicos, alcanzando picos de hasta 40ºC. La duración media sin tratamiento es de 1-3 semanas y en los tratados con inmunoglobulinas suele ceder a las 36 horas post-infusión. En los casos que cede espontáneamente a los 7 días, no se puede descartar el diagnóstico(9).
Manifestaciones oculares
La conjuntivitis ocurre en el 85% de los pacientes y suele aparecer al inicio de la fiebre. Se manifiesta como inyección conjuntival bilateral, predominantemente bulbar no exudativa que resuelve sin secuelas. Otra manifestación menos frecuente es la uveítis, sobre todo en niños de 2 años, entre el 5º-8º día de la enfermedad(11).
Manifestaciones de la mucosa oral
Los labios eritematosos, secos, fisurados con sangrado y la lengua aframbuesada son signos típicos. Además, podemos encontrar eritema orofaríngeo. No suelen aparecer vesículas ni exudado(9).
Manifestaciones cutáneas
El exantema se inicia durante los primeros 5 días desde el inicio de la fiebre. La erupción suele comenzar en el tronco, extendiéndose a extremidades, pudiendo confluir, sobre todo a nivel genital, donde produce una descamación temprana característica. La forma más típica de presentación es como exantema macular, urticarial, morbiliforme o lesiones en diana, no apareciendo habitualmente petequias, ampollas o vesículas(9-11).
En la fase aguda es característico un eritema palmo-plantar y edema, en ocasiones, doloroso en dorso de manos y pies. La descamación ungueal se inicia a nivel periungueal a los 10-15 días tras el inicio de la fiebre y entorno a las 6 semanas pueden aparecer unos surcos lineales transversales en las uñas (líneas de Beau)(9-11).
Linfadenopatía
La linfadenopatía cervical anterior unilateral de más de 1,5 cm de diámetro no es la característica más típica, presentándose en menos del 50% de los casos. Inicialmente, puede confundirse con una adenitis cervical bacteriana(11).
Manifestaciones cardíacas
La afectación puede ocurrir a cualquier nivel, manifestándose como: miocarditis, pericarditis, insuficiencia mitral (25%) o dilatación coronaria. En los primeros 10 días, no suelen detectarse los aneurismas, observándose inicialmente una hiperrefringencia de la pared en la ecocardiografía, apareciendo a partir de la 4-6ª semana. Los pacientes con aneurismas rara vez presentan síntomas, salvo isquemia miocárdica o rotura, debido al rápido crecimiento en la fase aguda(10).
Algunos pacientes en la fase aguda pueden presentarse con shock cardiogénico (5%). Estos pacientes tienen más riesgo de desarrollar afectación coronaria, disfunción miocárdica y refractariedad al tratamiento con inmunoglobulinas(10).
En estudios recientes histopatológicos, se han podido identificar tres procesos patológicos a nivel coronario(10-11):
1. Arteritis necrosante por infiltración de neutrófilos activados: afecta a la adventicia arterial, generando el aneurisma. Autolimitado con cese a las 2 semanas.
2. Vasculitis subaguda/crónica: infiltración de linfocitos, células plasmáticas, eosinófilos y macrófagos. Inicio a las 2 semanas, persistiendo durante meses.
3. Proliferación miofibroblástica luminal: es la causa de estenosis arterial. Inicio en las 2 primeras semanas hasta meses.
Otras manifestaciones
Otros manifestaciones son: artritis/artralgias de grandes articulaciones, dolor abdominal, vómitos y diarreas. A nivel respiratorio pueden presentar derrame pleural y, menos frecuentemente, infiltrado intersticial peribronquial. En lactantes es característica una marcada irritabilidad secundaria a meningitis aséptica. Otras manifestaciones neurológicas menos frecuentes son: sordera neurosensorial reversible, parálisis facial periférica unilateral y uveítis. Una de las complicaciones graves y poco frecuente al debut es el síndrome de activación macrofágica(10-11).
Diagnóstico
El diagnóstico de la enfermedad de Kawasaki fundamentalmente es clínico, apoyándose en unos criterios diagnósticos bien definidos para la enfermedad.
El diagnóstico de la EK se realiza a través de criterios diagnósticos clínicos (Tabla VII).

El diagnóstico de EK incompleto debe considerarse en niños con fiebre y alguno de los criterios clínicos con hallazgos analíticos y/o ecocardiograma compatibles con EK, una vez se han descartado otros diagnósticos(9).
En el ámbito del laboratorio, hay marcadores que apoyan el diagnóstico como: elevación de reactantes de fase aguda (PCR, VSG, PCT, leucocitosis con neutrofilia, NT-proBNP); hiponatremia, hipoalbuminemia, transaminasitis y piuria estéril, así como la trombocitosis a partir del décimo día(10). Se recomienda a todos los pacientes una valoración cardiológica con electrocardiograma y ecocardiograma. Además, dentro del estudio diagnóstico inicial se recomienda valoración con ecografía abdominal y valoración oftalmológica(10).
Diagnóstico diferencial
En la actualidad, a raíz de la pandemia por SARS-COV-2, el síndrome multisistémico inflamatorio (MIS-C) se ha convertido en uno de los principales diagnósticos diferenciales, debido a su similitud en la presentación clínica.
El diagnóstico diferencial debe realizarse con: procesos infecciosos bacterianos o virales (adenovirus, parvovirus B19, CMV, VEB, herpes virus…); enfermedades mediadas por toxinas (escarlatina, shock tóxico); otras patologías reumáticas (artritis idiopática juvenil de inicio sistémico, PAN cutánea, lupus eritematoso sistémico); o reacciones inmunitarias (síndrome Steven Johnson, enfermedad del suero)(12).
En los últimos dos años, a raíz de la pandemia por SARS-COV-2, el síndrome multisistémico inflamatorio (MIS-C), secundario a infección por COVID-19, se ha convertido en uno de los principales diagnósticos diferenciales, al compartir muchas de las características clínicas con la EK. Godfred-Cato et al., han identificado tres formas clínicas de presentación, denominando a una de ellas como fenotipo Kawasaki like(13-14).
Existen diferencias que nos van a permitir diferenciar ambas entidades, una de ellas es el antecedente de infección y/o contacto estrecho confirmado por SARS-COV-2 positivo en las 4-6 semanas previas en los casos de MIS-C. Además, los pacientes con MIS-C presentan más clínica digestiva y neurológica, así como una mayor incidencia de miocarditis y disfunción cardíaca. Desde el punto de vista analítico, destaca una elevación marcada de la ferritina junto citopenias, siendo características la linfopenia y plaquetopenia(13).
Tratamiento
El tratamiento de elección son las inmunoglobulinas, cuyo uso dentro de los 10 primeros días de la enfermedad reduce el riesgo de afectación coronaria por debajo del 5%.
El objetivo del tratamiento es disminuir la inflamación, evitando el daño arterial coronario y previniendo las posibles trombosis asociadas(9).
El tratamiento de elección son las inmunoglobulinas (IVIG), cuyo mecanismo de acción se desconoce. El tratamiento está indicado dentro de los primeros 10 días desde el inicio de la fiebre y pasado el 10º día en pacientes con persistencia de la clínica, elevación de parámetros inflamatorios analíticos o afectación ecocardiográfica sugestiva de EK. El tratamiento con IVIG en los 10 primeros días disminuye el riesgo de aneurismas coronarios por debajo del 5%.
El tratamiento de primera línea en la fase aguda son las IVIG en infusión única a dosis de 2 g/kg junto aspirina (AAS) a dosis antiinflamatorias (30-50 mg/kg/día cada 6 horas). Una vez que el paciente permanezca afebril durante 48-72 horas, se disminuirá a dosis antiagregantes, de 3-5 mg/kg/día en dosis única, manteniéndola durante 6-8 semanas hasta conseguir la normalización de los parámetros analíticos y el ecocardiograma. El tratamiento adyuvante con aspirina no reduce la frecuencia de aneurismas coronarios, pero sí disminuye el riesgo trombótico.(10-15).
En el 10-20% de los casos, la fiebre persiste pasadas 36 horas postratamiento con IVIG, considerándose refractarios a tratamiento; en estos casos existen diferentes opciones terapéuticas, siendo la más recomendada por los expertos una segunda dosis de IVIG y adicionar pulsos de metilprednisolona 30 mg/kg/día (máximo 1 g) durante 3 días, continuando con prednisona oral 1 mg/kg/día en pauta descendente en los siguientes 21 días(9,10,15).
Otras opciones terapéuticas de segunda línea en caso de refractariedad y con eficacia probada en reducir la inflamación son los fármacos biológicos, como Infliximab IV 6 mg/kg (1 o 2 dosis separadas por una semana). Otros tratamientos utilizados en pacientes refractarios son anakinra y ciclosporina(12,15,16).
En los pacientes con factores de riesgo para EK grave y desarrollo de afectación coronaria, se recomienda de entrada el uso de corticoterapia, ya que disminuye la inflamación, disminuyendo la clínica y el riesgo de daño(9-17) (Tabla VIII).

Diferentes metaanálisis y el estudio RAISE concluyen que el uso combinado de IVIG y corticoides en el tratamiento de la EK grave reduce el riesgo de presentar aneurismas coronarios sin aumentar efectos secundarios(9-17).
El tratamiento preventivo de trombosis en caso de aneurismas se hará según el tamaño y se mantendrá hasta la normalización de plaquetas y reactantes de fase aguda. Si el aneurisma persiste, el tratamiento antiagregante y/o anticoagulante será indefinido(9-12).
• Aneurisma pequeño (Z score ≥ 2,5 a < 5): dosis bajas de AAS (3-5 mg/kg/día).
• Aneurismas de tamaño moderado
(Z score ≥ 5 a < 10 y diámetro máximo < 8 mm): AAS (3-5 mg/kg/día) + tienopiridina (clopidogrel 0,2-1,0 mg/kg/día).
• Aneurismas gigantes (Z score ≥ 10 o diámetro máximo > 8 mm) o de rápido crecimiento: AAS (3-5 mg/kg/día) + warfarina.
Los pacientes con EK deberán retrasar la vacunación de virus vivos durante 11 meses tras la última dosis de IVIG, ya que puede disminuir la respuesta vacunal.
Pronóstico
La EK incompleta supone un factor de riesgo de afectación coronaria. Debido a su infradiagnóstico, muchos de los infartos en la 3ª y 4ª década de la vida se atribuyen a este diagnóstico.
Las manifestaciones cardiológicas suponen la principal causa de morbimortalidad, tanto en fase aguda como a largo plazo. El pronóstico de los pacientes sin aneurismas coronarios o dilatación a las 6 semanas de enfermedad es bueno, pero en aquellos en los que persiste, se deberá realizar un seguimiento cardiovascular hasta la edad adulta. El 50-70% de los aneurismas regresan en los siguientes 2 años. Cerca del 20% de los menores de 6 meses tratados dentro de los primeros 10 días presentarán aneurismas coronarios frente al 5% en mayores de 6 meses(11).
En un reciente metaanálisis, donde se incluyeron 20 estudios, se concluyó que la EK incompleta supone un factor de riesgo para el desarrollo de afectación coronaria(12-15). Diferentes estudios sugieren una alta prevalencia de eventos cardíacos en pacientes jóvenes relacionados con EK. En EE.UU., el 5% de los infartos agudos de miocardio (IAM) en menores de 40 años ocurren en pacientes con historia conocida o sospecha de EK en la infancia, siendo en Japón del 9% los casos de IAM y muerte súbita(10).
Otras vasculitis
Vasculitis ANCA
Poliangeítis con granulomatosis (PAG)
Anteriormente denominada granulomatosis de Wegener, es una vasculitis granulomatosa que afecta a vasos de pequeño calibre, principalmente a nivel renal y de vías respiratorias altas y bajas. Los criterios clasificatorios son: inflamación granulomatosa en la pared arterial o el área perivascular, afectación de vía aérea superior, afectación laríngea-traqueo-bronquial, afectación pulmonar, ANCA positivos (MPO o PR3) y afectación renal en forma de proteinuria, hematuria o glomerulonefritis necrotizante(18).
Poliangeítis granulomatosa eosinofílica (PGE)
Previamente denominada síndrome de Churg-Strauss, es una vasculitis necrotizante de pequeño vaso con un intenso infiltrado eosinofílico. La localización más frecuentemente es el tracto respiratorio y a nivel cardiovascular. Clínicamente, se caracteriza por presentarse como: asma severa, rinitis alérgica y lesiones cutáneas(18).
Poliangeítis microscópica (PAM)
Es otra vasculitis asociada a positividad ANCA, especialmente a anti-MPO. Afecta a vasos de pequeño calibre, principalmente a nivel pulmonar y renal, siendo la glomerulonefritis necrotizante su afectación más característica(18).
Vasculitis mediano vaso
La PAN es una vasculitis de mediano vaso. Su diagnóstico definitivo es una combinación de clínica compatible (alteraciones cutáneas, mialgias, hipertensión arterial, neuropatía periférica y afectación renal) junto a una anatomía patológica con necrosis en los vasos y/o la existencia de alteraciones en la angiografía, como: aneurismas, estenosis u oclusión de vasos. Se desconoce su etiopatogenia, aunque existen factores que predisponen a la patología, como se ha demostrado la asociación de PAN con mutaciones en el gen MEFV. En los últimos años, se ha identificado que el déficit de ADA2 (adenosina desaminasa 2), causado por una alteración en el gen CERC1, presenta en algunos pacientes un cuadro similar a una PAN (v. más adelante)(16).
Vasculitis gran vaso
La arteritis de Takayasu es una vasculitis granulomatosa que afecta predominantemente a la aorta y sus ramas primarias (subclavias, carótidas, arco aórtico, aorta abdominal, pulmonares…). Esta vasculitis predomina en países asiáticos y en mujeres entre la 2ª y 3ª década de la vida. Los criterios de clasificación son alteraciones angiográficas de la aorta o sus ramas principales, mostrando aneurismas/dilatación (criterio obligatorio), más 1 de los siguientes 5 criterios: déficit de pulso o claudicación, discrepancia en tensión arterial, soplos, hipertensión arterial y elevación de reactantes de fase aguda(2-16).
Vasculitis monogénicas
Mención especial por su importancia en el diagnóstico diferencial, a pesar de ser muy poco frecuentes, merecen un grupo de enfermedades autoinflamatorias monogénicas que cursan con afectación predominantemente vascular y entre las que cabe destacar las que se citan a continuación.
Síndrome DADA2
La deficiencia de la adenosina desaminasa tipo 2 está causada por mutaciones en el gen CECR1, con un patrón de herencia autosómico recesivo. Supone uno de los principales diagnósticos diferenciales de la panarteritis nodosa, con la que comparte similitudes clínicas y anatomopatológicas. Se han descrito varios fenotipos de presentación. El fenotipo clásico se manifiesta como: fiebre recurrente, lesiones cutáneas en forma de livedo racimosa y ulceraciones digitales, afectación neurológica en forma de infartos cerebrales (lacunares) o neuropatía periférica. Desde el punto de vista histológico, es una vasculitis necrosante sistémica que afecta a vasos de mediano calibre. Analíticamente, podemos encontrar: elevación de reactantes de fase aguda inespecíficos (PCR, VSG, leucocitosis) y, en algunos casos, se observan citopenias e hipogammglobulinemia de IgM. El diagnóstico se lleva a cabo mediante la cuantificación de la actividad enzimática de la enzima adenosina desaminasa, encontrándose disminuida en los pacientes afectos. El diagnóstico definitivo es la confirmación genética de mutaciones en el gen CECR1. El tratamiento que ejerce un buen control sobre la enfermedad son los fármacos biológicos anti TNF (etanercept, infliximab, adalimumab). En las formas clínicas con inmunodeficiencia o aplasia importante, el tratamiento es el trasplante hematopoyético(19).
Síndrome de SAVI
El síndrome de SAVI (o STING-Associated Vasculopathy with Onset in Infancy) es una enfermedad autoinflamatoria causada por mutaciones en el gen TMEM173, que regula la proteína STING encargada de regular la producción de interferón B (IFN-B). En los pacientes afectos hay una producción elevada de IFN-B. El patrón de herencia es autosómico dominante. Clínicamente, se manifiesta desde los primeros meses de vida por: fiebre recurrente, lesiones cutáneas violáceas acrales, telangiectasias, infartos e ulceraciones digitales distales y de pabellones auditivos y cartílago nasal, así como livedo reticular. Es característica la enfermedad pulmonar intersticial. Histopatológicamente, se observa una vasculopatía inflamatoria de pequeño vaso con afectación exclusiva de capilares. Los pacientes con síndrome de SAVI presentan una firma elevada de los genes que codifican el IFN, siendo útil como biomarcador para el diagnóstico y monitorización de la enfermedad. El diagnóstico se realiza confirmando las mutaciones genéticas en el gen TMEM173. Dentro de las opciones de tratamiento, los fármacos que parecen más eficaces son los inhibidores de las JAK (baricitinib, tofacitinib y ruxolitinib), ya que bloquean la vía de señalización del IFN(19).
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito. Declaración de intereses: ninguno.
Función del pediatra de Atención Primaria
• Diagnóstico diferencial inicial con otros cuadros clínicos sugestivos de vasculitis.
• En la VIgA, el manejo inicial es conservador, reservando los corticoides para casos graves de afectación gastrointestinal, orquitis, afectación SNC, nefritis o hemorragia pulmonar. El uso de corticoides no previene la afectación renal.
• El pediatra de Atención Primaria es clave en el control periódico ambulatorio. Se recomienda monitorizar la función renal, al menos, durante los primeros 6 meses tras el diagnóstico (toma de tensión arterial y cribado proteinuria/hematuria) para detectar de forma precoz la nefritis secundaria por VIgA.
• Es importante tener un alto índice de sospecha diagnóstica en aquellos pacientes con cuadros de fiebre sin foco, para realizar un diagnóstico precoz de la enfermedad de Kawasaki. Destacar la importancia del inicio del tratamiento dentro de los 10 primeros días de la enfermedad, para disminuir el riesgo de afectación coronaria. Igualmente es importante el seguimiento cardiológico posterior.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Camacho MS, Lirola MJ. Púrpura de Schönlein-Henoch, enfermedad de Kawasaki y otras vasculitis. Pediatr Integral. 2017; XXI: 183-95.
2. Ruperto N, Ozen S, Pistorio A, Dolezalova P, Brogan P, Cabral DA, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: Overall methodology and clinical characterisation. Ann Rheum Dis. 2010; 69: 790-7.
3. Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010; 69: 798-806.
4. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013; 65: 1-11.
5.** Ozen S, Marks SD, Brogan P, Groot N, De Graeff N, Avcin T, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatol (United Kingdom). 2019; 58: 1607-16.
6. Brogan P, Nott KA. Immune Complex Small-Vessel Vasculitis: IgA Vasculitis (Henoch-Schönlein) and Hypersensitivity Vasculitis. Petty, Laxer, Lindsley, Wedderburn, Mellins, Fuhlbrigge. Pediatric Rheumatology. Eighth edition. Philadelphia. Elsevier. 2021; 33: 456-66.
7. Eleftheriou D, Brogan PA. Therapeutic advances in the treatment of vasculitis. Pediatr Rheumatol [Internet]. 2016; 14: 1-9. Disponible en: http://dx.doi.org/10.1186/s12969-016-0082-8.
8.** Nicoara O, Twombley K. Immunoglobulin A Nephropathy and Immunoglobulin A Vasculitis. Pediatr Clin North Am. 2019; 66: 101-10.
9.*** McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American Heart Association. Circulation. 2017; 135: 927-99.
10.** Tascón AB, Malfaz FC, Sombrero HR, Fernández-Cooke E, Sánchez-Manubens J, Pérez-Lescure Picarzo J. Consenso nacional sobre diagnóstico, tratamiento y seguimiento cardiológico de la enfermedad de Kawasaki. An Pediatría. 2019; 90: 135-6.
11. Son MBF, Burns JC, Newburger JW. Kawasaki Disease. Petty,Laxer, Lindsley, Wedderburn, Mellins, Fuhlbrigge. Pediatric Rheumatology. Eighth edition. Philadelphia. Elsevier. 2021; 35: 475-83.
12. Sánchez-Manubens J. Enfermedad de Kawasaki. Protoc diagn ter pediatr. 2020; 2: 213-24.
13. Esteve-Sole A, Anton J, Pino-Ramírez RM, Sánchez-Manubens J, Fumadó V, Fortuny C, et al. Similarities and differences between the immunopathogenesis of COVID-19-related pediatric multisystem inflammatory syndrome and Kawasaki disease. J Clin Invest. 2021; 131: 1-10.
14.** Henderson LA, Canna SW, Friedman KG, Gorelik M, Lapidus SK, Bassiri H, et al. American College of Rheumatology Clinical Guidance for Multisystem Inflammatory Syndrome in Children Associated With SARS-CoV-2 and Hyperinflammation in Pediatric COVID-19: Version 2. Arthritis and Rheumatology. 2021; 73: 13-29.
15. De Graeff N, Groot N, Ozen S, Eleftheriou D, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of Kawasaki disease – the SHARE initiative. Rheumatology. 2018; 58: 672-82.
16.*** De Graeff N, Groot N, Brogan P, Ozen S, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of rare paediatric vasculitides-the SHARE initiative. Rheumatol (United Kingdom). 2019; 58: 656-71.
17. Kobayashi T, Saji T, Otani T, Takeuchi K, Nakamura T, Arakawa H, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. 2012; 379: 1613-20.
18. David A. Cabral, Kimberly A. Morishita. Antineutrophil Cytoplasmic Antibody Associated Vasculitis. Petty,Laxer, Lindsley, Wedderburn, Mellins, Fuhlbrigge. Pediatric Rheumatology. Eighth edition. Philadelphia. Elsevier. 2021; 36: 484-97.
19. Hernández-Rodríguez J, Prieto-González S, Espígol-Frigolé G, Cid MC. Nuevos síndromes autoinflamatorios con vasculopatía inflamatoria (síndrome de DADA2 y síndrome de SAVI). José Hernández-Rodríguez, Juan Ignacio Aróstegui, Jordi Yagüe. Avances en enfermedades autoinflamatorias. Marge Books; 2015. p. 155-69.
Bibliografía recomendada
– Ruperto N, Ozen S, Pistorio A, Dolezalova P, Brogan P, Cabral DA, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: Overall methodology and clinical characterisation. Ann Rheum Dis. 2010; 69: 790-7.
Artículo de revisión donde se establecen los criterios clasificatorios validados para varias vasculitis en población pediátrica. Ankara 2008.
– Ozen S, Marks SD, Brogan P, Groot N, De Graeff N, Avcin T, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatol (United Kingdom). 2019; 58: 1607-16.
Últimas recomendaciones Europeas elaboradas por el grupo SHARE para el diagnóstico y tratamiento de la vasculitis por IgA. Indicaciones de cuándo realizar biopsia renal y manejo de la nefritis por IgA.
– McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American Heart Association. Circulation. 2017; 135: 927-99.
Recomendaciones bien detalladas y estructuradas de la American Heart Association sobre el manejo de la enfermedad de Kawasaki.
– Tascón AB, Malfaz FC, Sombrero HR, Fernández-Cooke E, Sánchez-Manubens J, Pérez-Lescure Picarzo J. Consenso nacional sobre diagnóstico, tratamiento y seguimiento cardiológico de la enfermedad de Kawasaki. An Pediatría. 2019; 90: 135-6.
Consenso nacional completo y práctico con algoritmos diagnósticos y de tratamiento sobre el manejo de la enfermedad de Kawasaki.
– De Graeff N, Groot N, Brogan P, Ozen S, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of rare paediatric vasculitides-the SHARE initiative. Rheumatol (United Kingdom). 2019; 58: 656-71.
Últimas recomendaciones europeas elaboradas por el grupo SHARE para el diagnóstico y tratamiento de la vasculitis en la edad pediátrica.
| Caso clínico |
|
Varón de 4 años que consulta en Urgencias por presentar, desde hace 48 horas, lesiones eritematosas en extremidades inferiores que han ido progresando, afectando a brazos, con edema en manos y pies doloroso, asocia leve dolor abdominal. Afebril, antecedente hace 1 semana de proceso respiratorio de vías altas. Exploración Hemodinámicamente estable con TA 109/73 mmHg, lesiones purpúricas palpables en extremidades inferiores y superiores, tronco y cara, confluentes a nivel de nalgas. Edema de manos y pies doloroso al tacto, dolor abdominal a la palpación profunda sin signos de irritación peritoneal, sin masas ni megalias. Pruebas complementarias • Analítica: – Hemograma: Hb: 12,9 g/dl; Hto: 36,6%; plaquetas: 424.000mm3; leucocitos: 9.700 mm3. – Bioquímica: creatinina: 0,43 mg/dl; urea: 31 mg/dl; PCR: 10,9 mg/L; proteínas: 60 g/L; albúmina: 38 g/L. • Bioquímica urinaria: sin hematuria; Índice Pr/Cr: 0,18. Evolución Se ingresa para control del dolor, iniciando a las 24 horas aumento en la intensidad del dolor abdominal y cuadro de vómitos asociado. Se realiza ecografía abdominal con escaso líquido libre en pelvis y escaso edema de asas intestinales y hemoglobina en heces positiva.
|

 Juvenile Idiopathic Arthritis
Juvenile Idiopathic Arthritis