 |
| Temas de FC |
E. Urbaneja Rodríguez
Consulta de Inmunología y Reumatología Pediátricas. Centro Médico La Marquesina, Hospital Recoletas Campo Grande. Valladolid
| Resumen
Las conectivopatías (lupus eritematoso sistémico, dermatomiositis, esclerodermia, síndrome de Sjögren y enfermedad mixta del tejido conectivo) son un grupo heterogéneo de enfermedades autoinmunes de etiopatogenia multifactorial, en las que se combinan factores genéticos y ambientales, desencadenando una alteración del sistema inmune adaptativo. Asocian clínica multisistémica y muy variable, lo que puede complicar su diagnóstico. Su tratamiento debe ser individualizado, en función de la gravedad de las manifestaciones clínicas y, generalmente, está basado en la combinación de corticoides con otros fármacos inmunosupresores. Aunque las conectivopatías son menos frecuentes que en adultos, en edad pediátrica afectan también más al sexo femenino y suelen debutar al final de la primera década de la vida o durante la adolescencia. Todas ellas asocian ciertas peculiaridades pediátricas, que se describen y desarrollan de forma detallada en el siguiente artículo. |
| Abstract
Connective tissue diseases (systemic lupus erythematosus, dermatomyositis, scleroderma, Sjögren’s syndrome and mixed connective tissue disease) are a heterogeneous group of autoimmune diseases with a multifactorial etiopathogenesis in which genetic and environmental factors are combined, triggering an alteration of the adaptive immune system. They associate multisystemic and highly variable symptoms that can complicate their diagnosis. Its treatment must be individualized based on the severity of the clinical manifestations, although it is generally based on the combination of corticosteroids with other immunosuppressive drugs. Although connective tissue diseases are less frequent than in adults, in the pediatric age they mainly affect females and usually manifest at the end of the first decade of life or during adolescence. All of them associate certain pediatric features that are described and developed in detail in the following article. |
Palabras clave: Lupus; Dermatomiositis juvenil; Esclerodermia; Sjögren; Enfermedad mixta del tejido conectivo.
Key words: Lupus; Juvenile dermatomyositis; Scleroderma; Sjogren; Mixed connective tissue disease.
Pediatr Integral 2022; XXVI (3): 163 – 174
OBJETIVOS
• Conocer las características de las principales conectivopatías que debutan en la infancia, haciendo hincapié en sus particularidades pediátricas.
• Realizar una aproximación diagnóstica a las principales conectivopatías.
• Aprender aspectos básicos sobre el tratamiento de estas enfermedades.
• Manejar algunas nociones sobre su seguimiento que se puedan aplicar desde cualquier consulta de Pediatría (detección precoz de brotes, seguimiento de efectos secundarios del tratamiento y la propia enfermedad, disminución del riesgo de infecciones y de factores de riesgo cardiovascular, vacunación, etc.).
Lupus y otras conectivopatías en la infancia
Introducción
Las conectivopatías son enfermedades autoinmunes poco frecuentes en la infancia, debidas a una alteración en la inmunidad adquirida, con predominio de afectación en el sexo femenino y que suelen debutar a finales de la primera década de la vida y en la adolescencia.
Las conectivopatías constituyen un grupo heterogéneo de enfermedades autoinmunes debidas a una alteración en la inmunidad adquirida. Todas ellas son más frecuentes en el sexo femenino y, cuando presentan su debut en edad pediátrica, tienden a aparecer a finales de la primera década de la vida y durante la segunda década, sobre todo en la adolescencia(1).
Presentan una etiopatogenia desconocida y multifactorial, con implicación de factores ambientales unidos a una predisposición genética. Se caracterizan por una afectación multisistémica que suele cursar en brotes de actividad y periodos de remisión.
Aunque en general son raras en la infancia, todas ellas presentan ciertas peculiaridades cuando afectan a niños y adolescentes, asociando una gran morbilidad y, en muchas ocasiones, una demora diagnóstica, ya que presentan un amplio y difícil diagnóstico diferencial, sobre todo, con: procesos infecciosos, neoplasias y vasculitis. El diagnóstico de confirmación generalmente se establece por la combinación de síntomas sugestivos de cada enfermedad sumados a criterios analíticos.
El objetivo del tratamiento de estas patologías es conseguir la remisión/inactividad clínica para prevenir el daño secundario y las complicaciones asociadas(2). No podemos olvidar la interferencia de estas enfermedades en la adaptación escolar, así como los aspectos psicosociales derivados de las mismas. Por todo ello, las conectivopatías que debutan en niños y adolescentes precisan, para su correcto manejo, de un abordaje multidisciplinar en el que el pediatra de Atención Primaria tiene un papel clave.
Procederemos, a continuación, a resumir las características fundamentales y peculiaridades pediátricas de las principales conectivopatías que pueden aparecer en la infancia: lupus eritematoso sistémico (la más frecuente), dermatomiositis juvenil, esclerodermia (tanto su forma localizada como sistémica), síndrome de Sjögren y enfermedad mixta del tejido conectivo.
Lupus eritematoso sistémico (LES)
Generalidades y peculiaridades pediátricas
El LES pediátrico supone un 15-20% del total de pacientes con LES y suele debutar en la adolescencia. Se caracteriza por presentar en la infancia un grado mayor de actividad de la enfermedad y por manifestaciones clínicas más graves que en los adultos, sobre todo a nivel renal y neuropsiquiátrico, que confieren un peor pronóstico.
El lupus eritematoso sistémico (LES) es la conectivopatía mejor conocida y el prototipo de enfermedad autoinmune crónica. El LES pediátrico representa aproximadamente un 15-20% del total de pacientes con LES, con una incidencia en España, según los registros nacionales, de 0,36-0,9 casos/100.000 niños/año y con un pico de incidencia a los 12 años(3), siendo muy poco frecuente su debut en <5 años y presentando estos pacientes manifestaciones clínicas atípicas(4). Es más frecuente en raza asiática, afroamericanos y latinos. Los pacientes pediátricos suelen tener las mismas manifestaciones clínicas que los adultos, aunque presentan un mayor grado de actividad de la enfermedad y manifestaciones clínicas más graves, sobre todo a nivel renal y neuropsiquiátrico, que condicionan una mayor morbimortalidad.
En esta enfermedad se produce una falta de tolerancia a antígenos intrínsecos, con formación de autoanticuerpos e inmunocomplejos y su consecuente depósito en los tejidos provocando daño. La presencia de estos autoanticuerpos es una de las características más destacadas de esta patología, estando los mismos en los niños con LES más presentes en el diagnóstico en comparación con los adultos. Además, se unen una gran cantidad de factores genéticos y epigenéticos que en el LES pediátrico adquieren una mayor importancia respecto a los adultos, encontrándose en hasta un 7-8% del LES pediátrico mutaciones monogénicas causantes de la enfermedad(4).
Clínica
Las manifestaciones clínicas más frecuentes en el LES pediátrico son los síntomas constitucionales, la artritis y el exantema malar. A largo plazo es la afectación renal la que marca el pronóstico de la enfermedad.
Aunque las manifestaciones clínicas pueden ser muy variables y similares a los adultos, los síntomas constitucionales (febrícula o fiebre, astenia, hiporexia, pérdida de peso), la artritis (de pequeñas y grandes articulaciones, simétrica, no erosiva) y el rash o exantema malar (eritema con forma de mariposa en mejillas) son los síntomas iniciales más frecuentes en el LES pediátrico (Fig. 1).

Figura 1. Rash o exantema malar en alas de mariposa, típico de lupus eritematoso sistémico.
Durante la evolución de la enfermedad se puede afectar casi cualquier órgano (afectación renal, neuropsiquiátrica, hematológica, cardiopulmonar, digestiva, mucocutánea, etc.).
La afectación renal, agrupada bajo el término de nefritis lúpica, aparece en el 50-80% de los pacientes con LES pediátrico, en forma de: hematuria, proteinuria-síndrome nefrótico, hipertensión arterial o deterioro de la función renal (Fig. 2).

Figura 2. Hematuria macroscópica como debut clínico en paciente con lupus eritematoso sistémico.
En ocasiones, la afectación renal puede no correlacionarse bien con los síntomas clínicos, por lo que será necesaria una biopsia renal siempre que exista afectación a dicho nivel para clasificar histológicamente el grado de nefritis lúpica, según la clasificación propuesta en 2003, por la International Society of Nephrology/Renal Pathology Society (ISN/RPS) (Tabla I)(5).

La forma difusa (clase IV) es la más frecuente y grave. Según la reciente clasificación diagnóstica de LES, establecida en 2019, por EULAR/ACR, la afectación renal se define por la aparición de proteinuria >0,5 g/24 h o por una biopsia renal con clases II, III, IV o V.
La afectación neuropsiquiátrica puede aparecer en forma de: cefalea, convulsiones, enfermedad vascular cerebral, meningitis aséptica, neuropatía, trastornos del movimiento, cambios del estado de ánimo o psicosis.
A nivel hematológico, la leucopenia, linfopenia y trombopenia son las manifestaciones más frecuentes. Puede aparecer anemia en hasta un 50% de los casos, generalmente debida al trastorno inflamatorio crónico, aunque también puede asociarse a hemólisis con test de Coombs positivo. Existe además un estado de hipercoagulabilidad con un aumento del riesgo de trombosis.
A nivel cardiopulmonar, la pleuropericarditis es la manifestación más frecuente en niños.
A nivel gastrointestinal destacan, sobre todo, la aparición de dolor abdominal y diarrea, aunque pueden asociarse: hepatoesplenomegalia, alteraciones esofágicas, pancreatitis y hepatitis autoinmune.
A nivel mucocutáneo, además del típico exantema malar, pueden aparecer: fotosensibilidad, lesiones vasculíticas en cara, manos y pies (Fig. 3), úlceras orales y nasales, alopecia y fenómeno de Raynaud. Las lesiones cutáneas subagudas y crónicas son raras en la infancia.
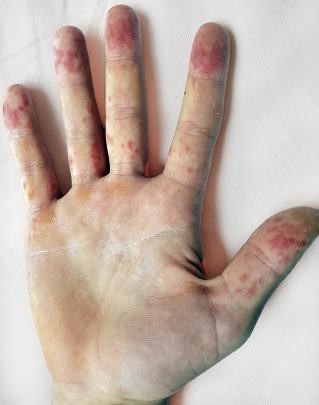
Figura 3. Lesiones de vasculitis en manos en adolescente con lupus eritematoso sistémico.
En casos de gran actividad de la enfermedad, se puede desencadenar un síndrome de activación macrofágico (SAM) que, en comparación con el SAM asociado a artritis idiopática juvenil sistémica, presenta con más frecuencia afectación del SNC e hiponatremia(2).
Diagnóstico
No existen criterios diagnósticos específicos para el LES pediátrico. Se utilizan los criterios de clasificación de adultos (suma de criterios clínicos + analíticos). Las pruebas de laboratorio sirven también para monitorizar la actividad de la enfermedad.
Siempre que se sospeche la enfermedad, se deberá solicitar una analítica con: hemograma, bioquímica completa con función hepática y renal, reactantes de fase aguda, estudio de coagulación que incluya anticuerpos antifosfolípido, estudio de orina y estudio inmunológico, con ANA y anticuerpos específicos (anti-DNAds, anti-Ro, anti-La, anti-Sm y anti-RNP), complemento (C3 y C4) e inmunoglobulinas, así como las pruebas específicas que se precisen según el órgano afectado (ECG, radiografía de tórax, ecografía torácica y/o abdominal, biopsia renal, etc.).
Los niveles de anti-DNAds y los valores de complemento (C3 y C4) sirven para monitorizar la evolución de la enfermedad, siendo estos últimos bajos en periodos de actividad de la misma.
Aunque se está trabajando en ello, actualmente no se dispone de criterios diagnósticos específicos en Pediatría, utilizándose los criterios de clasificación ya conocidos en adultos (Tabla II).

Los más usados históricamente fueron los establecidos en 1982 por el American College of Rheumatology (ACR), que se revisaron en 1997, necesitando cumplir, al menos, 4 de los 11 criterios en cualquier momento desde el inicio de la enfermedad para el diagnóstico de LES(6). En 2012 se publicaron unos nuevos del Systemic Lupus International Collaborating Clinics (SLICC), en los que hay que cumplir para el diagnóstico 4 criterios, de los cuales, al menos, 1 debe ser clínico y otro inmunológico(7). Más recientemente, en 2019 se han propuesto unos nuevos criterios diagnósticos por la European League Against Rheumatism (EULAR) en colaboración con ACR, que presentan una mayor sensibilidad y especificidad respecto a los de 1997 y 2012. La mayor modificación de esta última clasificación es la necesidad de la presencia de ANA positivos a títulos≥1/80 como requisito indispensable (lo que implica la exclusión de pacientes con ANA persistentemente negativos) y se ha incluido la fiebre inexplicada como criterio clínico constitucional. En esta última clasificación, unos ANA positivos ≥1/80 con una puntuación mayor o igual a 10 y un criterio clínico apoyan el diagnóstico de LES(8). Aunque ya hay varias publicaciones sobre la aplicación de estos últimos en el LES pediátrico, se necesitan más estudios para valorar los criterios EULAR/ACR 2019 en la infancia, sobre todo, teniendo en cuenta que los niños con LES de menor edad pueden tener ANA negativos.
Tratamiento
El tratamiento del LES pediátrico debe ser individualizado y está basado en la combinación de medidas generales y terapia farmacológica, que varía en función de la gravedad de las manifestaciones clínicas que predominan en cada paciente.
Entre las medidas generales destaca la importancia de limitar la exposición solar, recomendando protección diaria. Es fundamental incidir también en la realización de una dieta variada con ingesta adecuada de calcio, ejercicio físico regular y una correcta inmunización según el calendario vacunal vigente. Los adolescentes con LES deben evitar el tabaco, ya que puede empeorar la enfermedad y disminuir la eficacia de algunas medicaciones.
El tratamiento farmacológico de la enfermedad varía en función de la extensión y gravedad de la misma y está basado en las recomendaciones aplicadas a pacientes adultos(1,4,9). Los pilares fundamentales y la primera línea de tratamiento para casi todos los pacientes con LES son los corticoides y la hidroxicloroquina. Los corticoides ejercen un potente efecto antiinflamatorio e inmunosupresor, mediante la reducción no selectiva en la expresión de citoquinas proinflamatorias. La dosis de corticoides se ajustará en función de la gravedad de las manifestaciones clínicas. Generalmente, se administra prednisona por vía oral, pero en casos graves, se utilizan bolos intravenosos de metilprednisolona. Siempre habrá que recordar que si se realiza un tratamiento prolongado con corticoides en la infancia, se deberán iniciar suplementos de calcio y vitamina D para la profilaxis de osteoporosis y realizar controles seriados de peso y tensión arterial. La hidroxicloroquina es un fármaco antipalúdico oral que resulta muy eficaz para el control de las manifestaciones cutáneas y articulares, reduce la frecuencia de brotes, retrasa la aparición de nuevos síntomas y previene el riesgo trombótico. Se recomienda una exploración oftalmológica anual durante el tratamiento con hidroxicloroquina, ya que puede provocar toxicidad retiniana(9).
En casos moderados se añadirá al tratamiento de primera línea fármacos inmunosupresores ahorradores de corticoides como: metotrexato (sobre todo en casos de afectación cutánea y articular), azatioprina (afectación cutánea, articular y, particularmente, en nefritis lúpica durante el embarazo), micofenolato mofetilo (sobre todo en nefritis lúpica, aunque también en enfermedad cutánea resistente y serositis) o ciclofosfamida (manifestaciones orgánicas graves y potencialmente mortales a nivel renal, cardiopulmonar y neuropsiquiátrico).
En casos refractarios a la terapia anteriormente descrita, pueden utilizarse fármacos biológicos contra las células B, como rituximab (anticuerpo monoclonal quimérico anti-CD20) o belimumab (anticuerpo monoclonal humano que inhibe al estimulador del linfocito B, BLYS). Los inhibidores de las JAK-Kinasas se encuentran en fase de ensayo clínico en niños. En un futuro, la terapia que involucra a células madre mesenquimales podría formar parte del arsenal terapéutico en los pacientes con LES.
En casos de nefritis lúpica, la utilización de diferentes combinaciones de fármacos (corticoterapia + inmunosupresores) ha mejorado mucho su pronóstico(10). En general, en fase de inducción suele utilizarse ciclofosfamida en pulsos o micofenolato mofetilo oral. El micofenolato tiene una eficacia similar a la ciclofosfamida y presenta un perfil de toxicidad más bajo. Posteriormente, como terapia de mantenimiento, se usan micofenolato o azatioprina orales. Es importante recordar que durante el embarazo no pueden usarse ni metotrexato ni micofenolato, por sus efectos teratogénicos, y que en mujeres de edad fértil no se debe usar ciclofosfamida por sus efectos gonadotóxicos.
Dermatomiositis juvenil (DMJ)
Generalidades y peculiaridades pediátricas
La DMJ es una enfermedad autoinmune que se caracteriza por la inflamación crónica de piel y músculo estriado, que suele debutar en edad escolar. A diferencia de los adultos, los niños con DMJ no tienen un mayor riesgo de neoplasia, pero asocian con mayor frecuencia: calcinosis, lipodistrofia y afectación cutánea severa.
La dermatomiositis juvenil (DMJ) es la miopatía inflamatoria idiopática más frecuente en la infancia y una enfermedad autoinmune que afecta a piel y músculo estriado, produciendo inflamación y necrosis del tejido muscular, que suele debutar en edad escolar con una edad media de aparición de 7 años. Presenta una incidencia de 2-4 casos/1.000.000 niños/año(1). Al igual que en otras enfermedades autoinmunes, su etiopatogenia se relaciona con una interacción de factores ambientales (infecciones, luz ultravioleta, fármacos) en un individuo genéticamente predispuesto. A diferencia de la dermatomiositis que aparece en adultos, los niños afectos no tienen un mayor riesgo de neoplasia. Además, de forma característica, los pacientes pediátricos asocian más frecuentemente: calcinosis, lipodistrofia y enfermedad cutánea grave y presentan una mejor respuesta al tratamiento y menor mortalidad que los adultos(11).
Clínica
La asociación de manifestaciones cutáneas características y pérdida de fuerza muscular proximal y simétrica a nivel de cintura escapular y pelviana es muy sugestiva de DMJ.
Las manifestaciones cutáneas son específicas y patognomónicas de esta enfermedad y se observan en el 80% de pacientes: eritema en heliotropo (exantema violáceo en párpados superiores que puede acompañarse de edema palpebral) y pápulas de Gottron (lesiones rosadas que se asientan en prominencias óseas, sobre todo, a nivel de articulaciones metacarpofalángicas e interfalángicas, aunque también pueden aparecer en codos y rodillas) (Figs. 4-6).
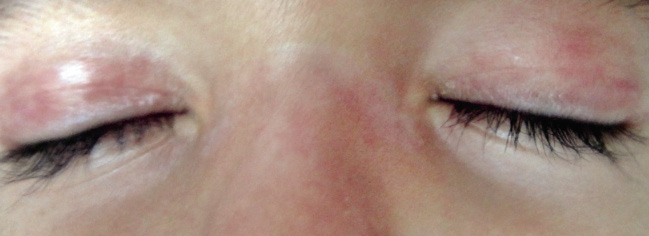
Figura 4. Eritema en heliotropo en paciente con dermatomiositis juvenil.
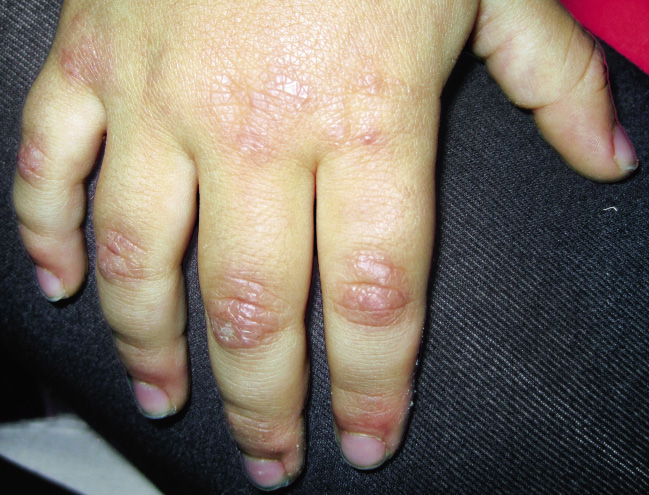
Figura 5. Pápulas de Gottron en articulaciones metacarpo-falángicas e interfalángicas de manos.

Figura 6. Pápulas de Gottron en codos en paciente con dermatomiositis juvenil.
Las alteraciones en los capilares periungueales también son frecuentes en la DMJ. Durante la evolución de la enfermedad pueden aparecer complicaciones cutáneas como: calcinosis (que se relaciona con enfermedad grave y prolongada e inicio tardío del tratamiento), úlceras y lipodistrofia.
La afectación muscular se manifiesta en forma de debilidad proximal y generalmente simétrica, que afecta a cintura escapular (dificultad para levantar los brazos) y pelviana (dificultad para subir y bajar escaleras). El signo de Gowers (el niño trepa sobre sí mismo para levantarse del suelo) es positivo. En casos graves se pueden afectar la musculatura faríngea y respiratoria, asociando dificultad para la deglución, disfagia, disfonía y voz nasal. Existen casos de afectación cutánea exclusiva sin asociación de clínica muscular, denominándose formas amiopáticas.
La afectación gastrointestinal constituye una de las complicaciones más graves de la enfermedad y es debida a una vasculitis intestinal. Se caracteriza por dolor abdominal y existe riesgo de sangrado o perforación.
La afectación cardiopulmonar es menos frecuente que en adultos y puede manifestarse en forma de: cardiomegalia, pericarditis, miocarditis y enfermedad pulmonar intersticial.
Diagnóstico
El diagnóstico de sospecha de DMJ está basado en la suma de criterios clínicos (afectación cutánea característica + debilidad muscular proximal) y analíticos (elevación de enzimas musculares). En ocasiones, se necesita una biopsia muscular para confirmar la enfermedad.
La DMJ se caracteriza analíticamente por una elevación de enzimas musculares (CPK, GOT, LDH y aldolasa). Los ANA pueden ser positivos. También pueden encontrarse anticuerpos específicos de miositis (anti-p155, anti-NXP2, anti-MDA5, anti-SRP, etc.) que están asociados con distintos subgrupos de la enfermedad, con diferente fenotipo, y anticuerpos asociados a miositis (anti-Pm-Scl, anti-U1-RNP, anti-Ro, anti-La, anti-Sm, etc), que aparecen en pacientes con clínica de solapamiento. El electromiograma muestra un patrón miopático con denervación, pero no es específico de la enfermedad. La confirmación diagnóstica se realiza mediante biopsia muscular. En los últimos años, la resonancia magnética se ha incorporado como nueva técnica diagnóstica no invasiva y ayuda a identificar fácilmente las zonas con inflamación muscular, permitiendo estudiar amplias áreas musculares. Se recomienda, además, un estudio cardiológico (ECG y ecocardiograma) y pulmonar (radiografía de tórax y función pulmonar) al inicio del cuadro clínico y durante su evolución si se precisa.
La fuerza muscular debe ser evaluada al diagnóstico y de forma periódica mediante escalas de valoración validadas en población pediátrica, como CMAS o MMT-8.
Clásicamente, para el diagnóstico de DMJ se utilizan los criterios de Bohan y Peter, descritos en 1975 (Tabla III)(12).

En 2017 se han publicado unas nuevas recomendaciones del Single Hub and Access point for paediatric Rheumatology in Europe (SHARE) para el diagnóstico y manejo del paciente con DMJ(13).
Tratamiento
El tratamiento de la DMJ está basado en la corticoterapia y en el uso de metotrexato. En casos resistentes a la terapia habitual, se pueden usar inmunoglobulinas o fármacos biológicos.
El tratamiento de la DMJ está basado en el uso de corticoides a altas dosis (orales y/o endovenosos), con descenso posterior hasta mantener dosis bajas durante un mínimo de 12 meses, junto con metotrexato oral o subcutáneo como ahorrador de corticoides. En pacientes con intolerancia al metotrexato, se recomienda cambiar a micofenolato mofetilo o ciclosporina A. En pacientes con enfermedad grave se puede añadir ciclofosfamida.
En casos refractarios, el uso de inmunoglobulinas intravenosas de forma periódica se ha mostrado eficaz. Como último escalón terapéutico, se pueden utilizar fármacos anti-TNF o rituximab. Es importante incidir en la importancia de la protección solar y administrar suplementos de calcio y vitamina D. La fisioterapia y el ejercicio físico aeróbico controlado son de gran importancia para la rehabilitación y la prevención de complicaciones en estos pacientes.
Esclerodermia localizada (EL)
Generalidades y peculiaridades pediátricas
La EL es la forma más frecuente de esclerodermia en la infancia, afecta exclusivamente a la piel y suele debutar en edad escolar. En las formas pediátricas se describe una afectación más profunda y prolongada respecto a los adultos.
La esclerodermia localizada (EL), también llamada morfea, es una conectivopatía autoinmune que se caracteriza por una fibrosis o exceso de colágeno en la piel, afectando de forma exclusiva a piel y tejidos adyacentes. Es la forma de esclerodermia más frecuente en la infancia, con una incidencia de 0,34-2,7 casos/100.000 niños/año y es de 6 a 10 veces más frecuente que la esclerosis sistémica. Suele debutar entre los 6-9 años, incluso puede estar presente desde el nacimiento. Aunque su etiopatogenia es desconocida, se habla de una combinación de factores genéticos y ambientales que da lugar a una inflamación y aumento en la producción del colágeno de la piel.
Existen diferentes subtipos de la enfermedad en función de la profundidad y el patrón de afectación. Su aparición en edad pediátrica asocia una mayor frecuencia de formas lineales, con afectación más profunda y mayor duración de la enfermedad respecto a los casos descritos en adultos(1,14).
Clínica
La EL se caracteriza por un endurecimiento cutáneo progresivo sin asociación de síntomas sistémicos. El subtipo más frecuente en la infancia es la forma lineal.
La EL se caracteriza, inicialmente, por un edema cutáneo unilateral con bordes eritemato-violáceos que, posteriormente, asocia engrosamiento e induración de la piel y tejido subcutáneo. En casos avanzados pueden aparecer: atrofia cutánea, cambios de pigmentación, contracturas y dismetrías.
En un 25% de pacientes aparecen manifestaciones extracutáneas, en forma principalmente de artralgias o artritis, aunque también pueden asociarse: mialgias, miositis, escoliosis y hemiatrofia. Cuando se afectan cara y cuero cabelludo, se denomina “coup de sabre” y puede asociar: alopecia, clínica ocular (uveítis, afectación palpebral) y neurológica (cefalea, convulsiones, alteraciones del comportamiento).
Las diferentes formas de EL se clasifican en 5 subtipos (Tabla IV)(15).

El subtipo de EL más frecuente en la infancia es la forma lineal, afectando fundamentalmente a extremidades, hueso frontal o tronco (Fig. 7).

Figura 7. Afectación en tronco de forma lineal de esclerodermia localizada.
Las manifestaciones gastrointestinales, cardíacas y renales son muy poco frecuentes en las formas localizadas. La progresión a forma sistémica es excepcional(14).
Diagnóstico
El diagnóstico de EL está basado en la clínica y se confirma mediante biopsia cutánea. No existen hallazgos de laboratorio característicos de esta enfermedad.
No hay alteraciones analíticas características, aunque los ANA y el factor reumatoide pueden ser positivos. La biopsia cutánea se caracteriza por un infiltrado inflamatorio con predominio linfocitario a nivel perivascular y perianexial. La dermis puede estar edematosa con alteraciones en el colágeno.
Tratamiento
No existe tratamiento específico para la EL. En casos leves, se administra tratamiento tópico y, en casos moderados-graves, tratamiento inmunosupresor sistémico.
No existen guías estandarizadas para su tratamiento en Pediatría, lo que traduce una gran variabilidad terapéutica. En casos leves, se puede usar tratamiento tópico con corticoides, inhibidores de la calcineurina (tacrolimus, pimecrolimus) o imiquimod. La fototerapia no suele emplearse en la infancia por sus potenciales efectos secundarios. En casos moderados-graves, se recomienda tratamiento inmunosupresor sistémico, siendo la combinación de metotrexato oral o subcutáneo unido a corticoides orales la pauta más eficaz descrita en numerosas publicaciones. Como alternativa pueden usarse micofenolano mofetilo o fármacos biológicos(1).
Esclerosis sistémica (ES)
Generalidades y peculiaridades pediátricas
La ES es una conectivopatía excepcional en la infancia, con afectación multisistémica además de cutánea, que suele aparecer en edad escolar y que asocia menor gravedad que en pacientes adultos.
La esclerosis sistémica (ES) es una enfermedad autoinmune crónica, excepcional en la infancia, con una incidencia de 0,27-1 caso/1.000.000 niños/año y una edad media de aparición de 7-9 años. Se estima que solo el 10% de la ES debuta antes de los 16 años. Se caracteriza por afectación de piel y órganos internos, en general, con menor participación sistémica que en el adulto en el momento del diagnóstico y con menor mortalidad asociada(1,14).
Clínica
El fenómeno de Raynaud es la primera manifestación clínica de la enfermedad en la mayoría de casos. Además de la induración cutánea, aparecen síntomas: músculo-esqueléticos, digestivos, respiratorios y/o renales.
La enfermedad se inicia con fenómeno de Raynaud (Fig. 8) y alteraciones capilaroscópicas que asocian edema e induración progresiva de dedos (puffy fingers).

Figura 8. Fenómeno de Raynaud complicado con ulceración digital como primera manifestación de esclerosis sistémica en adolescente.
El patrón capilaroscópico se caracteriza por pérdida de capilares y existencia de megacapilares, con destrucción de la arquitectura normal de la circulación del lecho ungueal. La piel está dura y seca, con áreas de alteración en la pigmentación. Existe además una disminución de la apertura oral con aumento de pliegues periorales, telangiectasias y calcinosis.
A nivel músculo-esquelético, suele asociar artralgias y artritis (sobre todo en manos).
A nivel digestivo, puede asociar: microstomía (disminución de apertura oral) y microquilia (disminución de los labios), alteraciones del gusto, caída de piezas dentales, reflujo gastroesofágico y alteraciones en la motilidad digestiva.
A nivel respiratorio, asocia alteración de la función pulmonar, enfermedad intersticial e hipertensión pulmonar.
Diagnóstico
Existen unos criterios de clasificación provisionales para la ES juvenil, basados en criterios clínicos y de laboratorio.
El hemograma y los reactantes de fase aguda suelen ser normales. Los ANA pueden ser positivos y es característica la positividad de los anticuerpos antitopoisomerasa (anti-Scl 70) y anticentrómero. Se deben realizar radiografía de tórax y estudio de función pulmonar, ECG y ecocardiograma.
Hay publicados unos criterios de clasificación provisionales para la ES juvenil elaborados por PRES/ACR/EULAR en 2007, más restrictivos que los aplicados en adultos (Tabla V).
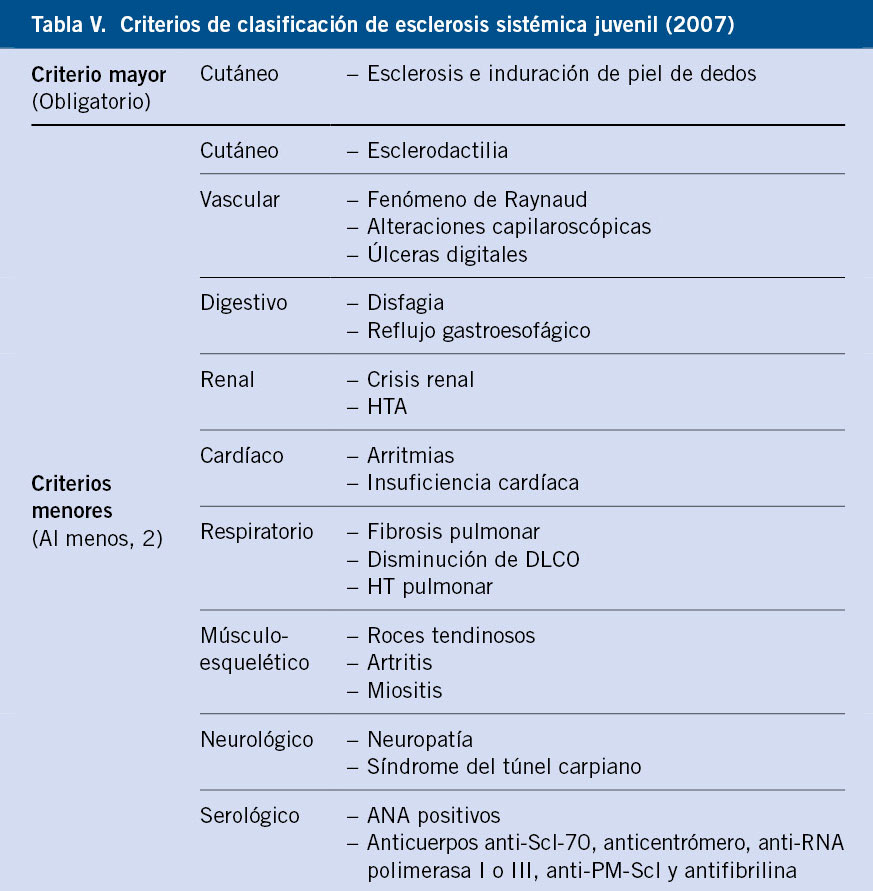
Cuando se cumple el criterio mayor de forma obligatoria y, al menos, 2 de los criterios menores presentan una sensibilidad del 90% y especificidad del 96% para el diagnóstico de ES juvenil(16).
Tratamiento
El tratamiento de la ES es sintomático y varía en función de las manifestaciones clínicas asociadas y su gravedad.
El tratamiento de la ES es sintomático y varía enormemente en función de la gravedad de la clínica asociada(14). El fenómeno de Raynaud precisa de medidas físicas (protección contra el frío, evitación de traumatismos y adecuada hidratación de la piel) y tratamiento con antagonistas del calcio (nifedipino). En casos refractarios se usan: antagonistas del receptor de angiotensina (losartan), inhibidores de la endotelina (bosentan), inhibidores de la fosfodiesterasa (sildenafilo) o prostaglandinas (iloprost). Para el reflujo gastroesofágico se usan los inhibidores de la bomba de protones. La terapia inmunosupresora sistémica con metotrexato, ciclofosfamida o fármacos biológicos es la base del tratamiento para el resto de manifestaciones clínicas. La fisioterapia también es un pilar fundamental en estos enfermos.
Síndrome de Sjögren (SS)
El SS es una enfermedad autoinmune crónica que afecta al sistema glandular exocrino en forma de sequedad generalizada. Es poco frecuente su debut en edad pediátrica y se presenta en la infancia generalmente en forma de parotiditis recurrente.
Generalidades y peculiaridades pediátricas
El síndrome de Sjögren (SS) es una enfermedad autoinmune crónica debida a la infiltración linfocitaria de las glándulas exocrinas, produciendo pérdida de su función y sequedad generalizada, que suele debutar en edad adulta y es infrecuente en Pediatría. Se desconoce su incidencia real en niños debido a los pocos casos descritos. La edad media de aparición de la enfermedad son los 10-12 años(17). A diferencia de lo que ocurre en adultos, en la infancia el síntoma inicial más frecuente es la parotiditis recurrente, lo que hace su diagnóstico mucho más complicado(2).
Clínica
En niños, el SS se inicia como parotiditis recurrente. En adultos, el SS se inicia con sequedad bucal y ocular, síntomas que se presentan raramente en la infancia.
La parotiditis recurrente es el síntoma inicial en la mayoría de casos de SS pediátrico, aunque en Pediatría ante un caso de parotiditis recurrente siempre se deberán excluir primero otras causas como: infecciones (sobre todo, víricas), tumores, malformaciones anatómicas, litiasis glandular y parotiditis recurrente juvenil.
La sequedad bucal (xerostomía) y ocular (xeroftalmia) se instauran con tiempo de evolución de la enfermedad, por lo que son raras en la infancia, apareciendo solo en un 8% de los casos con debut en edad pediátrica(2). La xerostomía asocia dificultad para masticar, disfagia y una necesidad constante de beber agua. La lengua está seca, roja, depapilada, con queilitis angular y los labios están secos con fisuras (rágades). Es frecuente la asociación de caries y periodontitis. La xeroftalmia se manifiesta como sensación de cuerpo extraño o de “arenilla”, que asocia picor, fotofobia y disminución de agudeza visual. Se puede producir una irritación crónica con destrucción del epitelio conjuntival en forma de queratoconjuntivitis seca, que aumenta la susceptibilidad a infecciones oculares. La xeroftalmia se pone de manifiesto con el test de Schirmer y la tinción con rosa de Bengala.
Además, el SS puede asociar: síndrome constitucional, artralgias o artritis, citopenias, lesiones vasculíticas, afectación pleuropulmonar, renal y neurológica, siendo las artralgias y las adenopatías las formas más frecuentes de afectación extraglandular en la infancia. A diferencia de lo que ocurre en adultos, el linfoma de células B es una complicación poco frecuente en edad pediátrica(2).
Diagnóstico
No existen criterios de clasificación específicos para el SS en Pediatría. Por tanto, el diagnóstico debe estar centrado en una clínica compatible, en la detección de autoanticuerpos y en la demostración de lesiones estructurales en el parénquima glandular mediante ecografía.
No existen criterios de clasificación específicos para el SS infantil y los criterios aplicados en adultos son poco sensibles, porque no incluyen manifestaciones tan frecuentes en la infancia como la parotiditis recurrente, por lo que no deben aplicarse.
Los ANA pueden ser positivos en más del 80% de casos. Los anticuerpos anti-Ro y anti-La son positivos en el 30% y 70% de los casos, respectivamente, y su positividad se acompaña de más manifestaciones extraglandulares, de más alteraciones analíticas y del desarrollo de bloqueo cardíaco congénito en hijos de mujeres con anticuerpos anti-Ro. Algunos pacientes pueden presentar también positividad del factor reumatoide e hipergammaglobulinemia(17).
La ecografía permite evaluar la estructura y vascularización del parénquima glandular submandibular y parotídeo. La presencia de lesiones ovaladas hipoecóicas, la alteración de la estructura del parénquima glandular y el aumento de vascularización detectado mediante Doppler son hallazgos muy sugestivos de SS(17,18).
En casos de difícil diagnóstico, una biopsia de glándulas salivares menores puede confirmar la enfermedad(1). Se observa un infiltrado linfocitario difuso y en focos, con desestructuración de la arquitectura glandular(18).
Tratamiento
El tratamiento del SS en edad pediátrica es sintomático y se basa en la combinación de AINES y corticoides en fase aguda e hidroxicloroquina como terapia de mantenimiento.
No existe ninguna terapia que modifique la evolución del SS. El tratamiento es puramente sintomático y está basado en una buena hidratación y en la combinación de AINES y corticoterapia en fase aguda. El tratamiento crónico con hidroxicloroquina consigue una disminución en la frecuencia de los episodios de parotiditis. En casos refractarios o con afectación extraglandular, se pueden usar otros inmunosupresores(18).
Enfermedad mixta del tejido conectivo (EMTC)
Generalidades y peculiaridades pediátricas
La EMTC se caracteriza por solapar la clínica de dos o más conectivopatías y por presentar anticuerpos anti-RNP positivos como marcador serológico. Es muy poco frecuente su debut en la infancia.
La enfermedad mixta del tejido conectivo (EMTC) es una enfermedad autoinmune sistémica con incidencia desconocida en la infancia, que se caracteriza por el solapamiento de características clínicas de: LES, esclerosis sistémica, dermatomiositis juvenil y artritis idiopática juvenil. Suele presentarse en edad adulta, siendo excepcional su aparición en niños, ya que solo un 23% de los pacientes con EMTC inician la enfermedad en la infancia, con una edad media de aparición de 11 años(2,19,20). La hipertensión pulmonar que asocia la enfermedad en adultos es rara y mucho menos severa en la infancia.
Clínica
Las manifestaciones clínicas más frecuentes de la EMTC en la infancia son el fenómeno de Raynaud inicialmente, seguido de: artritis, miositis, esclerodactilia, edema de manos y dedos y enfermedad pulmonar intersticial.
La EMTC suele iniciarse con fenómeno de Raynaud, que precede al resto de manifestaciones clínicas en meses o años. Progresivamente, se asocian manifestaciones: articulares (poliartritis de manos), musculares (debilidad muscular, miositis), cutáneas (eritema malar, fotosensibilidad, esclerodactilia, edema de manos y dedos) y cardiopulmonares (pericarditis, pleuritis, fibrosis intersticial e hipertensión pulmonar). En edad pediátrica, la alteración de la función pulmonar es frecuente, incluso en ausencia de síntomas respiratorios, aunque la presencia de hipertensión pulmonar asociada es mucho menor que en adultos(20).
Diagnóstico
La EMTC debe sospecharse ante un paciente con síntomas sistémicos de diferentes conectivopatías solapados entre sí que presente anticuerpos anti-RNP positivos, ya que no existen criterios de clasificación específicos en Pediatría.
No existen criterios de clasificación específicos en Pediatría, por lo que el diagnóstico se basa en la aplicación de alguno de los diferentes criterios de clasificación propuestos para adultos. A nivel analítico, pueden aparecer: citopenias, hipocomplementemia, elevación de enzimas musculares, ANA y factor reumatoide positivos. La EMTC se asocia a la aparición de títulos elevados de anticuerpos anti-RNP, aunque su presencia no es patognomónica de esta patología e incluso pueden llegar a negativizarse en edad adulta en casos de remisión de la enfermedad(20).
Tratamiento
No existe tratamiento específico para la EMTC. Se usan los fármacos ya explicados para el resto de conectivopatías, ajustados en función de la gravedad de la enfermedad.
El tratamiento de la EMTC es similar al ya explicado para el resto de conectivopatías y se ajustará en función de la gravedad de los síntomas. La mayoría de los niños responden bien a dosis bajas de corticoides, AINES e hidroxicloroquina(1,20). Si se asocia artritis o miositis se añadirá metotrexato. En casos de afectación orgánica grave, se escalará el tratamiento inmunosupresor.
Funciones del pediatra de Atención Primaria
• Pensar en la posibilidad de conectivopatía ante un paciente pediátrico, generalmente de sexo femenino y edad escolar/adolescente que presente síntomas sistémicos no relacionados con causa infecciosa ni tumoral.
• Integrar todos los aspectos clínicos, diagnósticos y terapéuticos de estas enfermedades, siendo el pilar fundamental dentro del seguimiento multidisciplinar que necesitan estos pacientes.
• Incidir en la educación sobre estas enfermedades en el propio paciente y sus familiares, indicando la importancia del buen cumplimiento terapéutico, la protección solar, el ejercicio físico y la prevención tanto de infecciones como de factores de riesgo cardiovascular.
• Revisar periódicamente el calendario vacunal de estos enfermos para conseguir una correcta vacunación (prestando especial atención a la vacunación frente a gripe, neumococo, varicela, triple vírica y SARS-CoV-2).
• Controlar los efectos adversos de los tratamientos utilizados en estos pacientes (controles analíticos periódicos, control del crecimiento, etc.).
• Identificar de forma precoz los brotes o recaídas de estas enfermedades, para lograr un ajuste precoz de su tratamiento y evitar secuelas.
Conflicto de intereses
No hay conflicto de interés en la elaboración del manuscrito. Declaración de intereses: ninguno.
Bibliografía
Los asteriscos reflejan el interés del artículo según la autora.
1.*** Clemente Garulo D. Conectivopatías. Pediatr Integral. 2017; XXI: 207-18.
2.** Tarvin SE, O’Neil KM. Systemic Lupus Erythematosus, Sjögren Syndrome and Mixed Connective Tissue Disease in Children and Adolescents. Pediatr Clin of North Am. 2018; 65: 711-37.
3. Torrente-Segarra V, Salman Monte TC, Rúa-Figueroa I, Sánchez-Alonso F, López-Longo FJ, Galindo- Izquierdo M, et al. Juvenile- and adult-onset systemic lupus erythematosus: a comparative study in a large cohort from the Spanish Society of Rheumatology Lupus Registry (RELESSER). Clin Exp Rheumatol. 2017; 35: 1047-55.
4. Charras A, Smith E, Hedrich CM. Systemic Lupus Erythematosus in children and young people. Curr Rheumatol Rep. 2021; 23: 20.
5.** Weening JJ, D´Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erithematosus revisited. Kidney Int. 2004; 65: 521-30.
6.** Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997; 40: 1725.
7.** Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and Validation of Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheum. 2012; 64: 2677-86.
8.** Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019; 71: 1400-12.
9. Durcan L, O´Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet. 2019; 393: 2332-43.
10.*** Boteanu A. Lupus eritematoso sistémico pediátrico. Protoc diagn ter pediatr. 2020; 2: 115-28.
11.*** Iglesias Jiménez E. Dermatomiositis juvenil. Protoc diagn ter pediatr. 2020; 2: 155-62.
12.** Bohan A, Peter JB. Polymiositis and dermatomyositis: Parts 1 and 2. N Engl J Med. 1975; 292: 344-47.
13. Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman B, et al. Consensus-based recommendation for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017; 76: 329-40.
14.** Suzanne C Li. Scleroderma in Children and Adolescents: Localized Scleroderma and Systemic Sclerosis. Pediatr Clin North Am. 2018; 65: 757-81.
15. Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006; 18: 606-13.
16.** Zulian F, Woo P, Athreya BH, Laxer RM, Medsger TA Jr, Lehman TJ, et al. The Pediatric Rheumatology European Society/American College of Rheumatology/European League Against Rheumatism provisional classification criteria for juvenile systemic sclerosis. Arthritis Rheum. 2007; 57: 203-12.
17. Wright TB. Updates in childhood Sjogren´s syndrome. Curr Opin Pediatr. 2021.
18.*** Nieto González JC, Monteaguado Sáez I, Serrano Benavente B. Síndrome de Sjögren. Protoc diagn ter pediatr. 2020; 2: 187-94.
19. Berard RA, Laxer RM. Pediatric Mixed Connective Tissue Disease. Curr Rheumatol Rep. 2016; 18: 28.
20.*** Bethencourt Baute JJ, Expósito Pérez L, Bustabad Reyes S. Enfermedad mixta del tejido conectivo. Protoc diagn ter pediatr. 2020; 2: 195-200.
Bibliografía recomendada
– Clemente Garulo D. Conectivopatías. Pediatr Integral. 2017; XXI: 207-18.
Excelente número monográfico en español sobre patología reumatológica en la infancia.
– Tarvin SE, O’Neil KM. Systemic Lupus Erythematosus, Sjögren Syndrome and Mixed Connective Tissue Disease in Children and Adolescents. Pediatr Clin of North Am. 2018; 65: 711-37.
Artículo en inglés que resume de forma muy completa y didáctica las características pediátricas de estas 3 enfermedades.
– Weening JJ, D´Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erithematosus revisited. Kidney Int. 2004; 65: 521-30.
Artículo de referencia en inglés para la clasificación internacional de la nefritis lúpica.
– Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997; 40: 1725.
Artículo de referencia en inglés para la clasificación de LES.
– Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and Validation of Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheum. 2012; 64: 2677-86.
Artículo de referencia en inglés para la clasificación de LES.
– Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019; 71: 1400-12.
Artículo de referencia en inglés para la clasificación de LES más reciente.
– Boteanu A. Lupus eritematoso sistémico pediátrico. Protoc diagn ter pediatr. 2020; 2: 115-28.
Excelente revisión en español de esta enfermedad, abordando los aspectos epidemiológicos, clínicos, diagnósticos y terapéuticos más destacados.
– Iglesias Jiménez E. Dermatomiositis juvenil. Protoc diagn ter pediatr. 2020; 2: 155-62.
Excelente y completa revisión en español sobre los aspectos más destacados de esta enfermedad.
– Bohan A, Peter JB. Polymiositis and dermatomyositis: Parts 1 and 2. N Engl J Med. 1975; 292: 344-47.
Artículo en inglés de referencia en esta enfermedad, que aborda sus aspectos más destacados y resume sus criterios diagnósticos.
– Suzanne C Li. Scleroderma in Children and Adolescents: Localized Scleroderma and Systemic Sclerosis. Pediatr Clin North Am. 2018; 65: 757-81.
Excelente revisión de inglés sobre la esclerodermia en Pediatría.
– Zulian F, Woo P, Athreya BH, Laxer RM, Medsger TA Jr, Lehman TJ, et al. The Pediatric Rheumatology European Society/American College of Rheumatology/European League Against Rheumatism provisional classification criteria for juvenile systemic sclerosis. Arthritis Rheum. 2007; 57: 203-12.
Artículo en inglés que hace referencia a los criterios de clasificación de la esclerosis sistémica en Pediatría, señalando las peculiaridades de esta enfermedad en la infancia.
– Nieto González JC, Monteaguado Sáez I, Serrano Benavente B. Síndrome de Sjögren. Protoc diagn ter pediatr. 2020; 2: 187-94.
Excelente revisión en español sobre los aspectos más destacados de esta enfermedad.
– Bethencourt Baute JJ, Expósito Pérez L, Bustabad Reyes S. Enfermedad mixta del tejido conectivo. Protoc diagn ter pediatr. 2020; 2: 195-200.
Excelente revisión en español de los aspectos más destacados de esta enfermedad.
| Caso clínico |
|
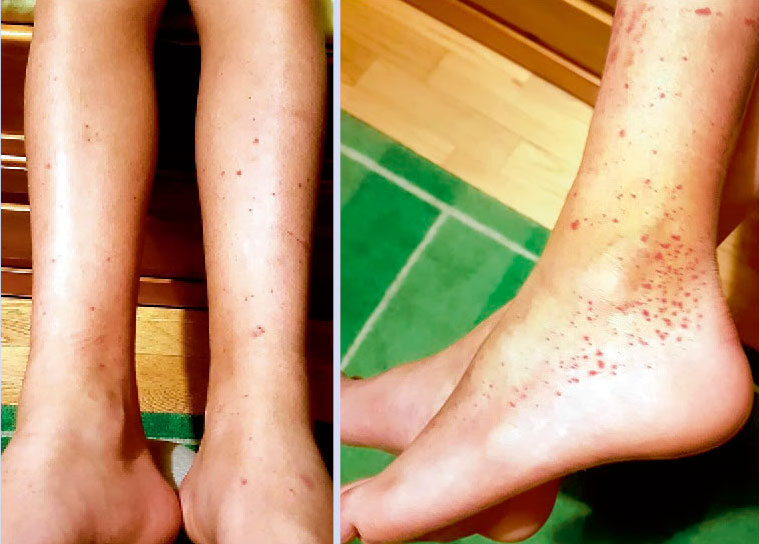
Varón de 13 años derivado a la consulta de Reumatología Pediátrica desde Atención Primaria para estudio de cuadro clínico con: astenia, aftas, artralgias y lesiones purpúricas en miembros inferiores de un mes de evolución (Fig. 9).
Figura 9. Lesiones purpúricas en miembros inferiores sugerentes de vasculitis. Anamnesis Antecedentes familiares: madre con hipotiroidismo autoinmune. Prima con enfermedad celíaca. Sin otros antecedentes reumáticos ni autoinmunes. Antecedentes personales: embarazo, parto y periodo neonatal normales. Lactancia materna 6 meses. Beikost sin incidencias. Vacunas al día. Sin alergias. Desarrollo psicomotor normal. Enfermedad actual: desde hace 1 mes está mucho más cansado, come peor por la aparición de numerosas úlceras orales y han salido lesiones eritemato-violáceas en miembros inferiores, asociando artralgias con rigidez matutina hasta media mañana. No ha notado inflamación articular. No se ha tomado la temperatura. Sin otros síntomas asociados. Infección respiratoria de vías altas hace dos meses, que fue tratada de forma sintomática. Exploración física Temperatura 38,3ºC. Ojeroso, leve palidez cutánea asociada. Lesiones purpúricas no confluentes en miembros inferiores que no blanquean a la presión. Auscultación cardio-pulmonar: normal. Abdomen: hepatoesplenomegalia. Sin adenopatías. Úlceras en mucosa oral. Locomotor: Sin articulaciones limitadas, dolorosas ni tumefactas. No puntos dolorosos. Sin dactilitis. Sin entesitis. Maniobras sacroilíacas negativas. Fuerza conservada. Marcha normal. Exploraciones complementarias • Analítica sangre: Hb: 11 g/dl con VCM 86 fL; y test de Coombs positivo. Leucocitos: 4.420/mm3 con linfocitos: 824/mm3 y neutrófilos: 3.430/mm3. Plaquetas: 95.000/mm3. Bioquímica: normal (incluyendo perfil hepático y renal). PCR: 25 mg/l. VSG: 60 mm. ANA positivos 1/1250 con anti-DNAds positivos y anti-RNP positivos, factor reumatoide positivo, C3: 85 mg/dl, C4: 4,2 mg/dl. Estudio de coagulación: normal. • Analítica de orina: 50-100 hematíes/campo. Resto normal. • Microbiología: mantoux negativo. Serologías VHB, VHC y VIH negativas. Frotis faríngeo negativo para virus y bacterias.
|


 Juvenile Idiopathic Arthritis
Juvenile Idiopathic Arthritis