Juvenile Idiopathic Arthritis
Juvenile Idiopathic Arthritis  |
| Topics on Continuous Training |
C. Millán Longo*, S. Murias Loza**
*Pediatric Rheumatology Unit. La Paz University Hospital. Madrid. **Clinical Management Area of Pediatrics. Central de Asturias University Hospital. Oviedo
| Abstract
Juvenile idiopathic arthritis (JIA) is the most common chronic rheumatic disease in childhood. It is defined as arthritis of unknown origin, lasting for at least 6 weeks and with onset before 16 years of age. The current classification identifies seven different categories. The most common clinical forms are oligoarthritis and rheumatoid factor-negative polyarthritis. The diagnosis of JIA requires the exclusion of other diseases. An accurate clinical history and a full physical examination are needed. The main complications are skeletal sequelae and anterior uveitis. Macrophage activation syndrome is the most severe complication of systemic JIA. Treatment should be individualized according to the subtype of JIA. Biologic drugs have significantly improved the prognosis of this disease. |
| Resumen
La artritis idiopática juvenil (AIJ) es la enfermedad reumática crónica más frecuente en la infancia y se define como la artritis de etiología desconocida que comienza antes de los 16 años y que persiste durante, al menos, 6 semanas. Se identifican siete categorías, cada una de ellas con unos rasgos clínicos diferenciadores. Las formas clínicas más frecuentes son la oligoartritis y la poliartritis factor reumatoide negativo. El diagnóstico es de exclusión y se basa en una anamnesis y exploración física correctas. Las principales complicaciones son las secuelas esqueléticas y la uveítis anterior. El síndrome de activación macrofágica es la complicación más grave de la categoría sistémica de AIJ. El tratamiento debe ser individualizado en función del subtipo de AIJ. Los fármacos biológicos han mejorado significativamente el pronóstico de esta enfermedad. |
Key words: Juvenile Idiopathic Arthritis; Uveitis; Biological Agents.
Palabras clave: Artritis idiopática juvenil; Uveítis; Fármacos biológicos.
Pediatr Integral 2022; XXVI (3): 141 – 150
OBJECTIVES
• To understand the main etiological aspects and pathophysiologic mechanisms of the disease.
• To delve into the current classification and get to know the clinical and immunological characteristics of each category, as well as the differentiating features of each one of them.
• To carry out an adequate diagnostic approach to a patient with suspected inflammatory arthritis in the pediatric age.
• To recognize the principles of therapeutic management in a patient with juvenile idiopathic arthritis, as well as the generalities of the drugs used today.
• To be aware of the complications associated with the disease and the importance of follow-up and preventive activities, in order to improve the prognosis and quality of life of the patient.
|
|
|


Juvenile idiopathic arthritis
Introduction
Juvenile idiopathic arthritis is the most frequent chronic rheumatic disease in childhood and groups a heterogeneous set of diseases.
Juvenile idiopathic arthritis (JIA) is the most common chronic rheumatic disease in children and is defined as arthritis of unknown etiology that begins before the age of 16 years-old and persists for at least 6 weeks. The term JIA is an “umbrella” term that encompasses a heterogeneous group of diseases, both from a clinical, pathophysiological and immunological point of view(1-3).
Since the first description of the disease by George Frederic Still in 1897(4), and thanks to the knowledge that has been progressively generated on the disease, it is known that JIA differs from chronic arthritis in adults and it can, therefore, be considered as a different entity, with the exception of the polyarticular forms of rheumatoid factor (RF) positive and enthesitis-related arthritis (ERA), similar to rheumatoid arthritis and ankylosing spondylitis, respectively(1).
The term juvenile idiopathic arthritis is the one coined since its proposal by the International League of Associations for Rheumatology (ILAR) in 1995(5), replacing the previously used “juvenile rheumatoid arthritis” of the American College of Rheumatology (ACR)(6) and “juvenile chronic arthritis” of the European League Against Rheumatism (EULAR)(7).
Classification
The current classification identifies seven categories of JIA.
The ILAR classification, subsequently revised and accepted in Edmonton in 2001(8), is currently in force (Table I) and is used for both clinical and research purposes. This classification identifies seven categories of JIA, mutually exclusive, according to clinical and immunological criteria during the first 6 months of disease. It is worth mentioning that this classification has been under review since 2019(9) and modifications are expected soon.

Epidemiology
The population frequency of JIA is variable and the most frequent subtypes are oligoarthritis and RF negative polyarthritis.
The incidence and prevalence of JIA are variable, due to environmental, genetic and social differences between the different populations, with a significant underdiagnosis of the disease in developing countries(2).
Globally, an incidence is estimated between 1.6-23/100,000 children under 16 years of age and a prevalence between 3.8-400/100,000 children under 16 years of age(10). In general, it is more frequent in girls than in boys (2:1), with exceptions, and the age of presentation is variable(11). Among the different categories of JIA, the most frequent are oligoarthritis and RF negative polyarthritis, while psoriatic arthritis and RF positive polyarthritis are the least frequent(1). Table II summarizes the epidemiological characteristics of the different categories.
Etiopathogenesis(1,2)
A multifactorial etiology of JIA is proposed. The systemic category is the one that differs pathophysiologically most from the others as it is an autoinflammatory disease.
The etiopathogenic mechanisms of JIA are not fully understood. The implication of genetic and environmental factors in the deregulation of the immune system is considered, although the variety of clinical forms suggests the presence of different underlying pathogenic mechanisms.
Regarding the genetic basis of JIA, the RF negative oligoarthritis and polyarthritis categories have been the most studied, finding an association with the HLA-DRB1*08, HLA-DRB*11 and HLA-DRB*13 alleles. Positive RF polyarthritis, as well as adult rheumatoid arthritis, have been associated with the HLA-DRB1*04 allele. As for the other categories, enthesitis-related arthritis has been associated with the HLA-B27 allele, and psoriatic arthritis with the IL-23 receptor gene (IL-23R). The genetics of systemic JIA is less well known, although there seems to be an association with the HLA-DRB1*11 allele.
The JIA category that differs pathophysiologically most from the others is systemic. In this, it is the primary or innate immunity that is abnormal, unlike the other forms of JIA, in which the secondary or adaptive immunity is responsible. Thus, systemic JIA is considered an autoinflammatory disease and is mediated by proinflammatory cytokines and there is no production of autoantibodies, while the other forms of JIA are autoimmune diseases.
Regarding the mechanisms of the inflammatory response, both mediators and perpetuators of the same, it is worth noting the involvement of dendritic cells, NK cells and macrophages, as well as CD4+ and CD8+ T lymphocytes, B lymphocytes and plasma cells. All of them accumulate and infiltrate the synovial tissue, where proinflammatory cytokines act, such as tumor necrosis factor (TNF), interferon-γ (IFN-γ) and IL-17, among others. B lymphocytes are also the producers of autoantibodies that play a fundamental role in various forms of JIA: RF and antinuclear antibodies (ANA). It is important to note that the presence of ANA is increasingly considered a differentiating criterion for the different forms of JIA.
Clinical manifestations
Arthritis is the common manifestation of most clinical forms of JIA. Each category presents differentiating clinical characteristics.
Arthritis is the common symptom of most subtypes of JIA(12) and manifests as swelling and/or joint limitation, which is more evident in the last degrees of mobility and in extension. It can present with local increase in temperature, although without erythema of the adjacent skin, and pain is variable, generally of mild or moderate intensity. Stiffness in the morning and after rest is one of the most characteristic clinical manifestations, it can condition gait disturbance or limp and improves with physical activity and throughout the day.
In addition to arthritis, tenosynovitis, enthesitis and dactylitis are other joint manifestations of JIA(1):
• Tenosynovitis: inflammation of the tendon sheaths (Fig. 1). It is frequent and is usually accompanied by arthritis of the adjacent joints. The most frequent locations are the extensor apparatus of the dorsum of the hand and dorsum of the foot and the posterior tibial and peroneal tendons.
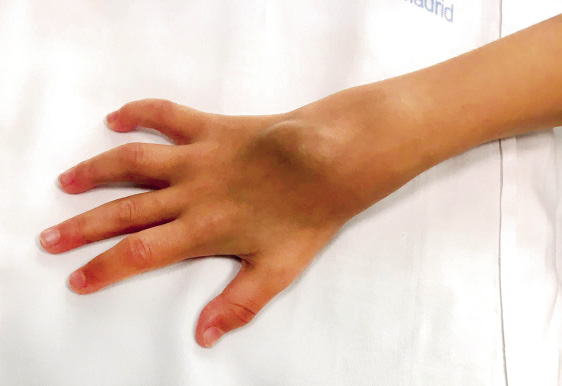
Figure 1. Carpal arthritis with extensor sheath tenosynovitis. Arthritis of the second and fourth proximal interphalangeal and metacarpophalangeal joints is also observed.
• Enthesitis: inflammation of the entheses, that is, the place of insertion of ligaments, tendons and fasciae in the bone. The most frequent locations of enthesitis in JIA are: the insertion of the patellar ligament in the patella and tibial tuberosity, the insertion of the Achilles tendon in the calcaneus (Fig. 2) and the insertion of the plantar fascia in the calcaneus, base of the fifth metatarsal and heads of the first through fifth metatarsals.
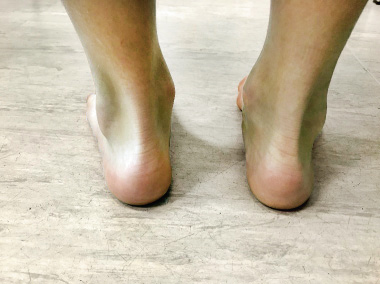
Figure 2. Right Achilles enthesitis.
• Dactylitis: uniform swelling of an entire finger, secondary to inflammation of the tendinous apparatus and periarticular soft tissues, resulting in the characteristic “sausage finger” appearance. Occasionally, the finger presents a fusiform swelling, more pronounced at the base of the finger and the proximal interphalangeal joint.
Systemic symptoms such as fever, asthenia, weight loss and anorexia are common in systemic JIA and may be present in the polyarticular form(12).
Given the variability that exists between the different clinical forms of JIA, the characteristic features of each of them will be presented separately below(1-3).
Oligoarthritis
It is the most common subtype of JIA and usually occurs in girls under 6 years of age. It is characterized by asymmetric arthritis of large joints, with main involvement of the lower limbs. The most commonly involved joints are: the knee and the ankle, but monoarticular presentation is not uncommon at onset. It is called persistent oligoarthritis if after the first 6 months it affects a maximum of four joints, and extended oligoarthritis if the number of affected joints rises to five or more after the same period of time, decreasing the probability of entering long-term remission.
Acute phase reactants (APR) are usually normal or slightly elevated, and up to 70% of patients have positive ANAs at low titers. Approximately one third of patients develop chronic anterior uveitis (CAU), which is typically asymptomatic, hence, regular ophthalmological check-ups are recommended(13) (Table III) for early diagnosis and treatment and thus avoid secondary complications (synechia, band keratopathy, cataracts, macular edema and glaucoma).

RF negative polyarthritis
It is a heterogeneous category in which two main clinical forms are identified:
1. Early-onset asymmetric arthritis before 6 years of age and predominantly in girls, ANA positive and with increased risk of CAU. It is equivalent to oligoarticular JIA, but with a greater number of affected joints.
2. Symmetrical arthritis of large and small joints beginning at school age, without predominance of sex, negative ANA and with elevated APR. It is the subtype equivalent to seronegative rheumatoid arthritis in adults.
In addition, in a small group of patients, a “dry synovitis” can be identified, a form of polyarthritis with little joint swelling, but associated with significant stiffness and limitation, joint contractures and deformities and with a worse response to treatment.
Patients who associate greater systemic inflammation, with asthenia, elevated APR and anemia, frequently present growth retardation, which tends to normalize after the start of treatment and control of the disease.
RF positive polyarthritis
It represents less than 5% of patients with JIA. It typically presents in adolescent girls and is characterized by symmetrical polyarthritis of small joints, especially in the hands, elevated APR, and the presence of anti-citrullinated peptide antibodies. The clinical presentation and prognosis are similar to adult rheumatoid arthritis and, in the same way, it can evolve into a destructive arthritis.
Enthesitis-related arthritis
It mainly affects males of school age or adolescents, and 50-90% are HLA-B27 positive. It is usually an asymmetric and oligoarticular arthritis, especially of the lower limbs. Hip and tarsal involvement is characteristic, uncommon in other forms of JIA, so arthritis in these locations should guide us towards this diagnosis. Unlike ankylosing spondylitis in adults, arthritis in children is usually initially peripheral and axial involvement in the form of sacroiliitis usually develops later in the course of the disease.
Enthesitis is a diagnostic criterion for ERA and the distinctive clinical manifestation compared to other subtypes of JIA. Given that the most affected entheses are the insertions of the patellar ligament, the Achilles tendon and the plantar fascia, a differential diagnosis should be made with osteochondrosis of these locations, that is, Osgood-Schlatter disease (tibial tuberosity), the syndrome Sinding-Larsen-Johansson (inferior pole of the patella) and Sever’s disease (calcaneus).
ERA category is also associated with anterior uveitis, although in this case it is acute and symptomatic, with red eye, pain and photophobia, it occurs in outbreaks and is associated with fewer ocular complications than CAU.
Psoriasic arthritis
It is a heterogeneous and not well-defined category in which two different groups of patients are distinguished:
1. JIA-like oligoarticular presentation, with early-onset asymmetric oligoarthritis, positive ANA and associated with CAU.
2. Presentation similar to ERA and adult psoriasic arthritis, with enthesitis, axial involvement and positive HLA-B27. These patients are usually excluded from this category of JIA due to the ILAR classification exclusion criteria and are usually categorized as undifferentiated arthritis.
A typical feature of psoriatic JIA is the presence of dactylitis, as well as involvement of the distal interphalangeal joints.
Systemic
It is the subtype that differs most from the rest, since, as previously mentioned, it is an autoinflammatory disease and not an autoimmune one. It affects males and females equally and can manifest at any age.
The arthritis is usually symmetrical and polyarticular, although it is not always present initially, which can make diagnosis difficult. Fever typically presents in evening peaks of high fever, associated with: affectation of the general state, myalgias, abdominal pain and a rash which is typically salmon-pink, macular, located on the trunk and root of limbs and evanescent. Other accompanying manifestations are: serositis, adenopathies and hepato-splenomegaly, as in other autoinflammatory processes. In the blood analysis the following can be observed: anemia, leukocytosis with neutrophilia, thrombocytosis and elevation of APR, ferritin, transaminases and D-dimer. Autoantibodies are not detected. 50% of cases evolve to a persistent polyarticular form, difficult to control and with a worse prognosis.
It is important to note that, in the new classification proposal of 2019(9), which is still under study, arthritis is no longer a mandatory criterion for diagnosis, so we could diagnose a patient with systemic JIA in the absence of it (Table IV).

Macrophage activation syndrome (MAS) is the most serious complication of systemic JIA(14). It occurs in 10-15% of patients, although in almost half of the cases it can present subclinically, and can develop both at the beginning of the disease and during its subsequent evolution. Mortality is high (8-20%), so it must be detected and treated early. Clinically, it is characterized by a rapid worsening of the patient secondary to a state of hyperinflammation (“cytokine storm”) with: continuous fever, hepato-splenomegaly and variable involvement of the central nervous system, associated with characteristic laboratory abnormalities. Table V shows its diagnostic criteria.

Table VI summarizes the main complications of JIA.

Diagnosis
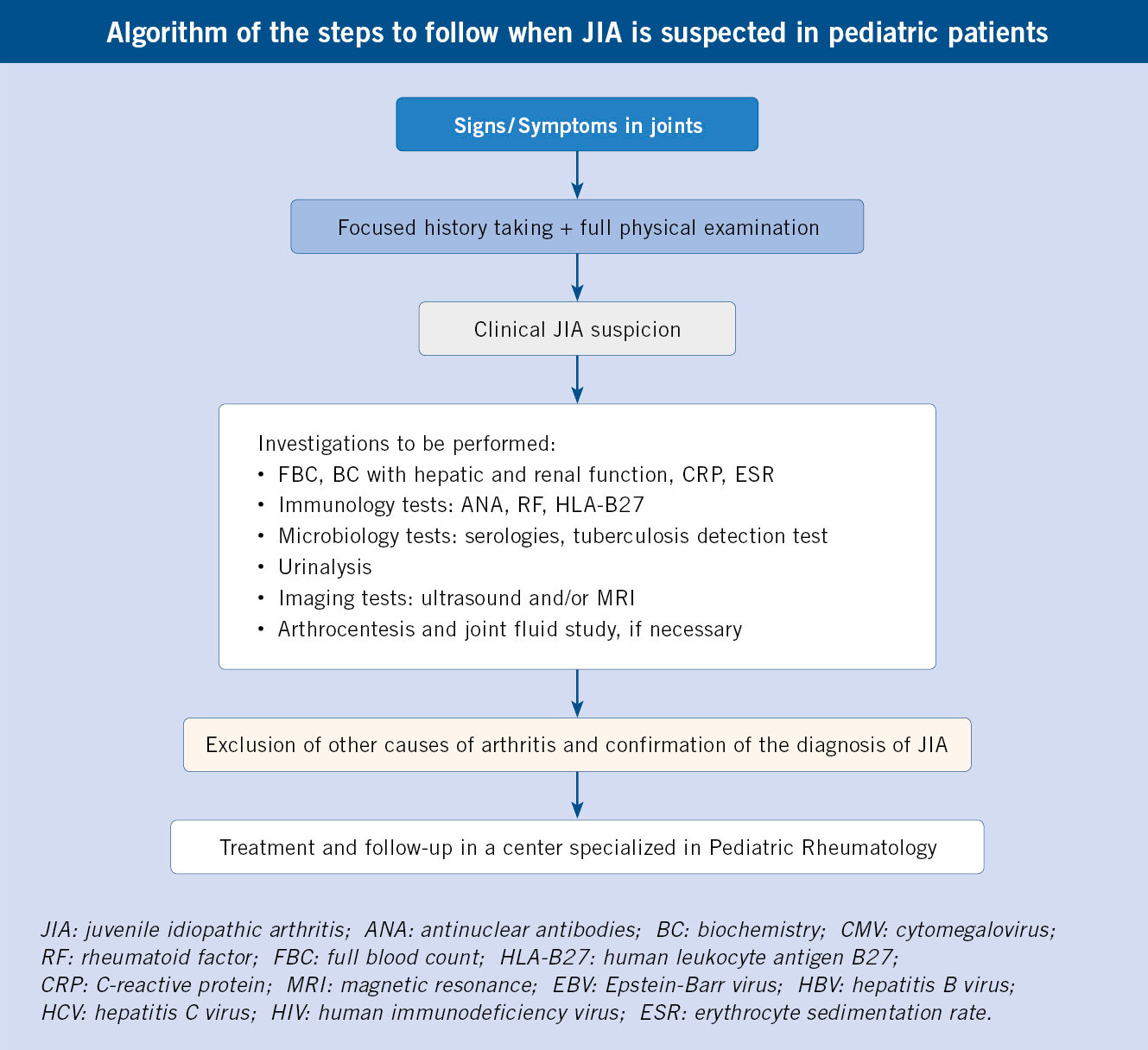
An exhaustive history taking and a full physical examination are essential, as well as the performance of specific complementary tests according to the diagnostic suspicion.
JIA is a diagnosis of exclusion(3), hence, it is necessary to rule out other causes of arthritis(3,15-17). Important issues regarding the history, physical examination, and additional tests when diagnosing JIA are detailed below.
In the clinical history it is necessary to inquire about the following aspects:
• Duration of symptoms: the diagnosis of JIA requires a minimum period of 6 weeks of arthritis.
• Characteristics of the pain: inflammatory rhythm (stiffness in the morning and after rest, with improvement after physical activity) versus mechanical rhythm and predominance of stiffness versus pain in other pathologies (important for the differential diagnosis of infectious arthritis).
• Characterization of joint involvement: number of affected joints, location (symmetric or asymmetric, perypheral or axial).
• Extra-articular symptoms: systemic (fever, affected general condition), cutaneous (exanthema, skin lesions; Fig. 3), gastrointestinal (abdominal pain, altered bowel movements) or ocular (pain, red eye, photophobia, decreased visual acuity).

Figure 3. Psoriasis.
• Family history of arthritis, psoriasis, inflammatory bowel disease, uveitis, or other autoimmune diseases.
Regarding the physical examination, it is necessary to carry out both a general physical examination and a complete musculoskeletal examination:
• General physical exam: rashes, skin lesions, nail pits suggestive of psoriasis, lymphadenopathy, and visceromegaly must be looked for.
• Locomotor system: the examination must be carried out in an orderly and systematic manner, beginning by observing the patient’s spontaneous attitude, assessing anti-algic postures and gait. Remember that arthritis is defined as swelling and/or joint limitation due to pain and that deep joints, such as the shoulder, hip and sacroiliac joints, do not show swelling. We can also find: tenosynovitis, dactylitis or enthesitis. In cases of long-standing arthritis, there may be muscle hypotrophies secondary to less use of the affected limb (Fig. 4).
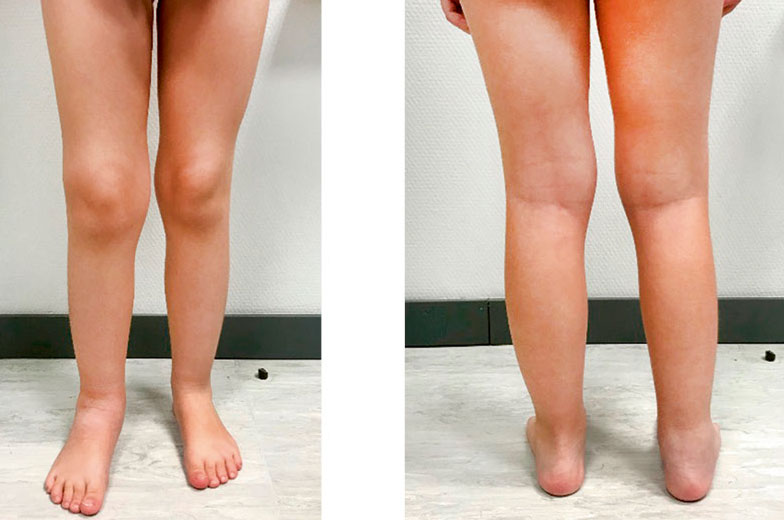
Figure 4. Arthritis of the knee with a flexed attitude (impossibility of full extension), of the right ankle and tarsus, associated with hypotrophy of the right gastrocnemius.
In addition, since sustained inflammation leads to greater vascularization of the area, dysmetria may develop due to the temporary increase in growth rate during the active inflammatory process in favor of the affected limb. pGALS is a tool for assessing the musculoskeletal system aimed at the general pediatrician and that allows the rapid and simple detection of skeletal alterations that can indicate the existence of a rheumatic disease(18).
Complementary tests must be carried out in a targeted manner and based on a compatible history and physical examination:
• Laboratory studies: blood tests (complete blood count, basic biochemistry with liver and kidney function, CRP, ESR, ANA, RF, HLA-B27), serologies of common pathogens (HBV, HCV, HIV, CMV, EBV, parvovirus B19), Mantoux or other tests of screening for tuberculosis and urinalysis.
• Joint ultrasound: it is the most accessible and preferred technique to detect joint effusion or synovial hypertrophy (Fig. 5).
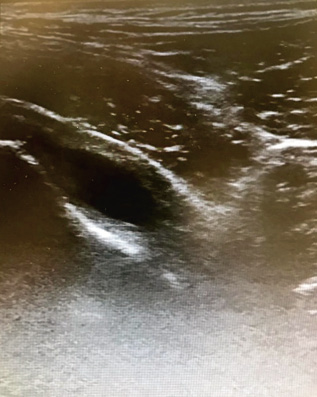
Figure 5. Ultrasound image of hip arthritis.
• X-ray: not commonly used in the pediatric age, although useful to quantify chronic damage (decreased joint space, erosions) and to rule out other processes (neoplasms, fractures and other traumatological processes).
• Magnetic resonance imaging: it is the most sensitive technique to detect joint effusion, study the cartilage and periarticular soft tissues (tendons, ligaments), while ruling out other processes, although it is less frequently used due to: its high cost, difficult access in some centers and the need for sedation in smaller patients. However, it is especially indicated to evaluate the temporomandibular joint (TMJ), the cervical spine and the sacroiliac joints(1).
• Joint fluid study(17): through arthrocentesis, a yellowish, turbid, slightly viscous liquid is obtained (Fig. 6), and with an intermediate cellularity, whose culture is sterile.
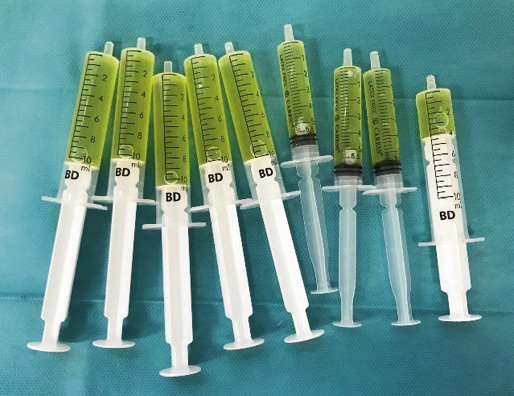
Figure 6. Joint fluid obtained from knee arthrocentesis.
Treatment
The goal of treatment is to achieve short- and long-term clinical remission and, for this, biological drugs have played a key role in recent years.
Treatment should be individualized based on the JIA subtype, presentation, and clinical course of the disease. The main objective is to achieve clinical inactivity, tackling inflammation and controlling symptoms, with the secondary objective of maintaining long-term remission, to achieve normal growth and development, avoid sequelae and enjoy a quality of life similar to that of the general healthy population(19).
The different scientific societies have developed therapeutic guidelines(20,21); in general, proposing a staggered pattern of treatment. However, in recent years, a treat-to-target strategy is usually followed, which consists of quickly seeking control of the disease by using the drugs that are necessary according to the degree of involvement(2,19). To assess activity and prospectively monitor the disease, the most widely used activity index is the Juvenile Arthritis Disease Activity Score (JADAS)(22).
Nonsteroidal anti-inflammatory drugs (NSAIDs)
They are used as symptomatic treatment. They do not modify the course of the disease or control inflammation in the long term.
Glucocorticoids (GC)
Intra-articular GCs are usually used, alone or in combination with other treatments, depending on the number and type of affected joints. Sometimes, the use of oral GC is recommended as bridging therapy until clinical control is achieved in cases of moderate or high activity and depending on the type of involvement. Preferably, their employment should be limited to the shortest possible time.
Disease-modifying antirheumatic drugs (DMARDs)
The most widely used is methotrexate (MTX), a folic acid antagonist, which can be administered both orally and subcutaneously. Its maximum therapeutic effect may take 6-8 weeks. Its main side effects are digestive intolerance and hypertransaminasemia, which is usually mild and transient when the dose is decreased.
Biological agents
They have revolutionized the treatment of JIA since the beginning of the 21st century. In general, they are well tolerated and their safety profiles are favorable. They can be used as monotherapy or in combination with other treatments, usually MTX.
The main biological drugs used are: tumor necrosis factor alpha antagonists (etanercept, adalimumab, infliximab, golimumab), IL-1 antagonists (anakinra, canakinumab), IL-6 antagonist (tocilizumab) and T-lymphocyte antagonist (abatacept).
Janus kinase inhibitors (JAK inhibitors)
They are under investigation as a treatment for JIA, either in monotherapy or associated with other drugs.
Follow-up
Regular follow-up is necessary to monitor clinical activity, detect complications and adjust treatment individually, according to evolution.
JIA is a chronic disease with a variable clinical course that can alternate periods of remission with periods of active inflammation(3). Factors associated with a worse prognosis are: polyarticular presentation, involvement of the carpus, hip or ankle, and the presence of RF. Regarding uveitis associated with JIA, the patients with the highest risk of suffering from this complication are those under 6 years of age with positive ANA and during the first 4 years of the disease(2).
Periodic follow-up is necessary to monitor clinical activity and treatment(3):
• Clinic appointments: are performed every 3-4 months. The objective is to evaluate activity and monitor the appearance of complications: dysmetria after asymmetric arthritis of the lower extremities, requiring the use of lifts in the shorter limb, which is usually partially or totally corrected over time; short stature secondary to persistent inflammation and prolonged use of GC and alterations in mandibular development secondary to TMJ arthritis.
• Analytical tests: they are performed every 3-4 months in patients receiving pharmacological treatment, to monitor full blood count, liver and kidney function, and APR.
• Ophthalmological check-ups(13): in patients with no history of uveitis, the periodicity of check-ups is determined by the subtype of JIA, the age of onset and the duration of disease. In patients with active or previous uveitis, check-ups will be carried out as indicated by the Ophthalmology service depending on the degree of activity, to adjust treatment and ensure good control of uveitis with the least possible toxicity.
Regarding preventive activities and healthy lifestyle habits recommended in patients with JIA(3):
• Diet and supplements: a healthy and balanced diet is recommended, just as in the rest of the child population, since there is no evidence that restrictions or modifications of dietary guidelines provide benefits in patients with JIA. Folic acid supplementation is recommended if they receive treatment with MTX and calcium and vitamin D supplementation if they receive GC at high doses and for a long time.
• Physical activity: limitation of physical exercise is not necessary. In the episodes of active arthritis, pain and stiffness improve with activity and in periods of remission it is equally beneficial and recommended so as to stimulate the recovery of muscle mass.
• Vaccinations(23): live attenuated virus vaccines are contraindicated in patients receiving treatment with GC, immunomodulators or biologicals. On the other hand, the administration of the hepatitis A vaccine is recommended in those receiving MTX or tocilizumab, the quadrivalent ACWY meningococcal vaccine in adolescence, and the meningococcal B and seasonal flu vaccine in all cases.
Role of the Primary Care pediatrician
• The action of the Primary Care pediatrician is essential when it comes to suspecting and recognizing the disease, since early diagnosis and treatment improve the prognosis and reduce the risk of complications and sequelae.
• Upon suspicion of JIA, it is indicated to start symptomatic treatment with NSAIDs at anti-inflammatory doses until the patient is evaluated by a pediatric rheumatologist.
• Early referral to a center specialized in Pediatric Rheumatology is recommended to complete the evaluation and confirm the diagnosis as soon as possible.
• The Primary Care pediatrician, in coordination with the specialist rheumatologist, plays a fundamental role in the follow-up of patients with JIA, for screening and detection of possible complications derived from the disease or from the treatments used.
• The update of the vaccination calendar, as well as the administration of the rest of the vaccines recommended in each case, is key in the patient with JIA. Likewise, it is necessary to be aware that live attenuated virus vaccines are contraindicated in some patients and depending on the treatments received.
• It is necessary to plan and carry out centralized preventive and health promotion activities from Primary Care (healthy diet recommendations, physical exercise) that allow to improve the prognosis and quality of life of patients with JIA.
Conflict of interest
There is no conflict of interest in the elaboration of the manuscript. Declaration of interests: none.
Bibliography
The asterisks show the interest of the article in the opinion of the authors.
1.*** Petty RE, Laxer RM, Lindsley CB, Wedderburn LR, Fuhlbrigge R, Mellins E. Textbook of pediatric rheumatology. 7th ed. Philadelphia: Elsevier; 2016.
2.*** Martini A, Lovell DJ, Albani S, Brunner HI, Hyrich KL, Thompson SD, et al. Juvenile idiopathic arthritis. Nat Rev Dis Primers. 2022; 8: 5.
3.** Murias Loza S, Udaondo Gascón C. Artritis Idiopática Juvenil. In: García JJ, Cruz O, Mintegi S, Moreno JM. M. Cruz Manual de Pediatría. Madrid: Editorial Ergon; 2020. p. 1348-52.
4. Still GF. On a form of chronic joint disease in children. Med Chir Trans. 1897; 80: 47-60.
5. Fink CW. Proposal for the development of classification criteria for idiopathic arthritides of childhood. J Rheumatol. 1995; 22: 1566-9.
6. Brewer EJ, Bass JC, Cassidy JT. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972; 23: 712-9.
7. European League Against Rheumatism: EULAR Bulletin No. 4: Nomenclature and Classification of Arthritis in Children. National Zeitung AG. 1977.
8.*** Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004; 31: 390-2.
9.*** Martini A, Ravelli A, Avcin T, Beresford MW, Burgos-Vargas R, Cuttica R, et al; Pediatric Rheumatology International Trials Organization (PRINTO). Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J Rheumatol. 2019; 46: 190-7.
10. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis. A systematic review. Joint Bone Spine. 2014; 81: 112-7.
11.** Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007; 369: 767-778.
12. Gowdie PJ, Tse SML. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2012; 59: 301-27.
13.*** Cassidy J, Kivlin J, Lindsley C, Nocton J, Section on Rheumatology; Section on Ophtalmology. Ophthalmologic examinations in children with juvenile rheumatoid arthritis. Pediatrics. 2006; 117: 1843-5.
14.*** Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016; 68: 566-76.
15. Prabhu AS, Balan S. Approach to a Child with Monoarthritis. Indian J Pediatr. 2010; 77: 997-1004.
16. Singh S, Mehra S. Approach to Polyarthritis. Indian J Pediatr. 2010; 77: 1005-10.
17. Fernández Fraga P, Murias Loza S. Diagnóstico diferencial de las inflamaciones articulares. Pediatr Integral. 2017; XXI: 154-9.
18.** Foster HE, Jandial S. pGALS – paediatric Gait Arms Legs and Spine: a simple examination of the musculoskeletal system. Pediatr Rheumatol Online J. 2013; 11: 44.
19.** Ravelli A, Consolaro A, Horneff G, Laxer RM, Lovell DJ, Wulffraat NM, et al. Treating juvenile idiopathic arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2018; 77: 819-28.
20. Beukelman T, Patkar NM, Saag KG, Tolleson-Rinehart S, Cron RQ, DeWitt ES, et al. 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res (Hoboken). 2011; 63: 465-82.
21. Ringold S, Angeles-Han ST, Beukelman T, Lovell D, Cuello CA, Becker ML, et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Treatment of Juvenile Idiopathic Arthritis: Therapeutic Approaches for Non-Systemic Polyarthritis, Sacroiliitis, and Enthesitis. Arthritis Care Res (Hoboken). 2019; 71: 717-34.
22. Trincianti C, Van Dijkhuizen EHP, Alongi A, Mazzoni M, Swart JF, Nikishina I, et al; Paediatric Rheumatology International Trials Organisation. Definition and Validation of the American College of Rheumatology 2021 Juvenile Arthritis Disease Activity Score Cutoffs for Disease Activity States in Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2021; 73: 1966-75.
23.** Comité Asesor de Vacunas de la AEP. Vacunación en niños inmunodeprimidos o con tratamiento inmunosupresor. In: Manual de vacunas de la AEP (last update: February 2022).
Recommended bibliography
– Petty RE, Laxer RM,Lindsley CB, Wedderburn LR, Fuhlbrigge R, Mellins E. Textbook of pediatric rheumatology. 7th ed. Philadelphia: Elsevier; 2016.
International reference book of Pediatric Rheumatology. It addresses extensively and in detail, rheumatic diseases in the pediatric age.
– Martini A, Lovell DJ,Albani S, Brunner HI, Hyrich KL, Thompson SD, et al. Juvenile idiopathic arthritis. Nat Rev Dis Primers. 2022; 8:5.
Recent and updated review on JIA, which addresses the different aspects of the disease in an excellent way.
– Petty RE,Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004; 31: 390-2.
Article presenting the currently valid classification of JIA, with the diagnostic criteria for each category.
– Martini A.Ravelli A, Avcin T, Beresford MW, Burgos-Vargas R, Cuttica R, et al; Pediatric Rheumatology International Trials Organization (PRINTO). Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J Rheumatol. 2019; 46: 190-7.
Article on the new classification that has been proposed for JIA and that is currently under study and validation.
– CassidyJ, Kivlin J, Lindsley C, Nocton J, Section on Rheumatology; Section on Ophthalmology. Ophthalmologic examinations in children with juvenile rheumatoid arthritis. Pediatrics. 2006; 117: 1843-5.
Article presenting review recommendations for JIA-associated uveitis.
– Foster HE, Jandial S. pGALS – pediatric Gait Arms Legs and Spine: a simple examination of the musculoskeletal system. Pediatr Rheumatol Online J. 2013; 11:44.
Article that explains the examination of the musculoskeletal system in a systematic and simple way.
| Clinical case |
|
A 5-year 8-month-old boy presented with swelling and pain in both knees, as well as pain in the fingers of 5 weeks’ duration. He reports predominantly morning pain with joint stiffness and limp, which improves throughout the day. He did not refer previous infectious processes or digestive symptoms or skin lesions. Afebrile at all times. Parents report that he has been stagnant in weight lately. The patient continues to perform his daily activities, although sometimes limited by pain. As for his personal history, he is being monitored by a private ophthalmologist. He has no family history of interest. Examination revealed significant swelling of both knees, with a flexed attitude of the right knee and impossibility for full extension, with painful limitation of flexion. He also presented mild swelling of both wrists with pain and limitation of flexion-extension, as well as involvement of the first metacarpophalangeal and second and fourth proximal interphalangeal joints of the right hand. Significant hypotrophy of both quadriceps is identified. Rest of the general physical examination is without any noteworthy findings. Anthropometry at diagnosis: weight at the 21st centile, height at the 65th centile. Joint ultrasound confirmed the presence of joint effusion in both knees and significant synovial hypertrophy, predominantly in the right knee. The blood test shows a slight elevation of APR with: ESR: 42 mm/h (0-20 mm/h); CRP: 15 mg/L (0-5 mg/L); and immunological study with positive ANA: 1/160, and negative RF and HLA-B27.
|