 |
| Historia de la Medicina y la Pediatría |
M. Zafra Anta*, V.M. García Nieto**
*Servicio de Pediatría del Hospital Universitario de Fuenlabrada, Madrid. Miembro del Grupo de Historia de la Pediatría de la AEP. **Coordinador del Grupo de Historia de la Pediatría de la AEP. Director de Canarias Pediátrica
Pediatr Integral 2022; XXVI (2): 116.e1 – 116.e6
Enfermedades pediátricas que han pasado a la historia (8). Síndrome de Reye
Generalidades
El síndrome de Reye (SR) es una enfermedad rara y potencialmente mortal, que se define como una encefalopatía aguda no inflamatoria, rápidamente progresiva, con disfunción hepática anictérica y afectación sistémica, que suele comenzar varios días después de una enfermedad viral, incluso aparentemente recuperada(1,2). Es casi exclusiva de menores de 18 años de edad. En general, se produce en pacientes sin enfermedades previas conocidas, aparentemente sanos. Se caracteriza por la aparición de vómitos persistentes, que se suceden súbitamente de letargia o confusión, y puede evolucionar rápidamente a convulsiones y coma.
El SR es muy poco frecuente actualmente. Se describió en 1963, y su incidencia creció en las décadas de los 60 y 70 hasta situarse en 1 caso por 100.000 niños. Desde los años 80 ha descendido hasta casi desaparecer; su incidencia actual es de 0,79/1.000.000 de niños; esto es, 2 casos al año en todo EE.UU.(1,3-5).
Se desconoce la etiología exacta del SR. Muchos casos se producen poco después de una infección por gripe o por varicela. La administración durante esos procesos de ácido acetilsalicílico (AAS) o aspirina® aumenta el riesgo hasta 20-35 veces. La hipótesis patogénica que prevalece en los últimos años, considera el síndrome como el resultado de una respuesta inusual a la infección viral precedente, que está determinada por factores genéticos del huésped, modificados por algunos agentes exógenos desencadenantes, entre estos, el ácido acetilsalicílico(6).
Descripción del síndrome de Reye, apunte histórico
El síndrome de Reye se describió en Australia(7). Antes de la década de 1950 no existía como brotes epidémicos o no se tenía constancia médica de ello.
Ralph Douglas Kennet Reye (1912-1977), un anatomopatólogo australiano (Fig. 1), publicó en la revista Lancet, en 1963, con Graeme Morgan y Jim Baral, una serie de 21 casos de “encefalopatía y degeneración grasa de las vísceras” que habían ingresado en el Royal Alexandra Hospital for Children de Sídney (Australia), entre marzo de 1951 y marzo de 1962: Encephalopaty and fatty degeneration of the viscera. A disease entity in childhood(7).

Figura 1. El profesor Ralph Douglas K. Reye. Fotografía del archivo de la Universidad de Sídney. Disponible en:
https://www.sydney.edu.au/medicine/museum/mwmuseum/index.php/Reye,_Ralph_Douglas_Kenneth.
La edad que tenían los pacientes era desde los 5 meses hasta los ocho años y medio. El estudio describe que los dos primeros casos fueron interpretados como encefalitis o sepsis, y solo se hizo una investigación retrospectiva en colaboración con el Departamento de Salud local a partir de que se agregaran otros 5 casos. El cuadro clínico que presentaban los niños era similar, en el ingreso tenían estupor o coma, que había sido precedido por vómitos persistentes de varias horas de duración. Existía además el antecedente de 1-3 días antes, incluso hasta 3 semanas, de una enfermedad menor con poca afectación, con rinorrea, tos, otalgia o exantema. Al ingreso se objetivaba: polipnea e hipotonía o hipertonía muscular, hepatomegalia, hipoglucemia, líquido cefalorraquídeo sin alteraciones, excepto hipoglucorraquia y elevación de transaminasas. El estado de coma se profundizaba y aparecía opistótonos o convulsiones difíciles de tratar. De los 21 pacientes, 17 fallecieron en pocas horas, los otros cuatro sobrevivieron sin secuelas. El perfil clínico era tan llamativo y el empeoramiento tan brusco, tan catastrófico, que los médicos de Urgencias lo identificaban rápidamente. El estudio necrópsico encontró: aumento de tamaño de tejido cerebral, cardiaco y renal, pero especialmente una degeneración grasa en el hígado, difusa y microvesicular.
Como observación, el diccionario de la Real Academia Nacional de Medicina señala que la pronunciación original aproximada es /rái/, pero entre hispanohablantes se oye también /réye/. El propio Douglas K. Reye refería que su apellido se pronunciaba como eye (“ojo” en inglés).
Tras los primeros casos, en el Royal Alexandra, se realizó una búsqueda de posible ingesta o inhalación de tóxicos retrospectivamente por personal enviado por las autoridades sanitarias locales, con una visita a los padres de niños afectados. El cuadro recordaba a la enfermedad de los “vómitos de Jamaica”, que se produce por efecto de la hipoglicina A, concretamente por ingestión de frutas inmaduras de Ackee. Pero tenía diferencias. Se encontró en un caso la presencia en orina de una pteridina (son cofactores de enzimas, como ocurre en la fenilcetonuria y otras enfermedades metabólicas).
Reye y cols., propusieron a Lancet la publicación de esta serie, porque estaban convencidos de que formaban un grupo diferente respecto de aquellos niños en los que los cambios grasos, especialmente en el hígado, eran una manifestación secundaria de una variedad de enfermedades. Esperaban que la experiencia de otros pudiera ayudar a sugerir una respuesta a los problemas de etiología, prevención y tratamiento.
Se recibieron varias cartas a Lancet y, posteriormente, también publicaciones en otras revistas científicas, señalando casos de “encefalopatía y degeneración grasa de vísceras” en muchos otros países: Inglaterra, Escocia, Sudáfrica, Checoslovaquia, Nueva Zelanda, EE.UU. Canadá(5,8). Desde 1968, en medios científicos, este cuadro se nominó como “Síndrome de Reye”.
Definición actual del síndrome de Reye
Según ORPHANET(2), el portal web de enfermedades raras y medicamentos huérfanos, resultado de un consorcio de 40 países, el SR se codifica actualmente como CIE-10: G93.7 y se define como: “Enfermedad sistémica poco frecuente caracterizada por vómitos persistentes que se presentan junto a confusión, letargia, desorientación, hiperreflexia, hiperventilación y taquicardia, con rápida progresión a convulsiones y encefalopatía no inflamatoria que evoluciona a coma y fallecimiento. Por lo general, se desarrolla entre las 12 horas y 3 semanas después de la recuperación de una enfermedad viral, como infecciones del tracto respiratorio superior o gastroenteritis. El síndrome está asociado a hepatomegalia, esteatosis hepática aguda, degeneración del hígado graso y múltiples alteraciones en las determinaciones analíticas”.
El Centro de Control de Enfermedades (CDC) de los EE.UU. definió el SR(3) con los siguientes criterios operativos: “encefalopatía aguda no inflamatoria que se documenta clínicamente por: a) alteración de la conciencia (el CDC estableció también niveles de coma); b) un estudio de líquido cefalorraquídeo que contenga menos o igual a 8 leucocitos/mm3 o una muestra histológica que demuestre edema cerebral sin inflamación perivascular o meníngea (si están disponibles); c) hepatopatía documentada por una biopsia de hígado o una autopsia considerada diagnóstica del SR o bien un aumento de tres veces o más en los niveles de la transaminasa glutámico-oxalacética sérica (SGOT), la transaminasa glutámico-pirúvica sérica (SGPT) o el amoníaco sérico; y d) que no haya otra explicación para las anomalías cerebrales y hepáticas”.
Algunas publicaciones incluyen en la definición: cuadro “consecutivo a una infección por un virus de la gripe o de la varicela en concomitancia con la ingesta de ácido acetil salicílico”, como la edición actual del diccionario de la Real Academia Nacional de Medicina. En los textos reconocidos de Pediatría y Medicina Interna, en sus últimas ediciones, apenas se citan 4 líneas, y en relación con salicilatos. En el Nelson, Tratado de Pediatría (Nelson Textbook of Pediatrics), en su 21 ed., se cita en el apartado de hepatopatías mitocondriales secundarias: “está precipitado en un individuo genéticamente susceptible por la interacción de una infección vírica (gripe, varicela) y el empleo de salicilatos…”
Donde más se cita el SR en la bibliografía pediátrica actual, es para tomar precauciones en la indicación de uso de salicilatos, como en la enfermedad de Kawasaki(9).
La revisión actual de UpToDate(9) la incluye dentro de encefalopatías tóxico-metabólicas.
Como podemos observar, no hay unanimidad en la definición del SR en la literatura médica. Todavía hay debate en la etiología, al menos, en señalar a la aspirina® como el principal factor de riesgo.
La aspirina® como etiología del síndrome de Reye: de una sospecha a una aceptación “sin dudas”
El SR tenía, en la mayoría de ocasiones, un curso clínico bifásico, de forma que antes de la aparición de vómitos y letargia había un antecedente de infección por patógenos virales, fundamentalmente la gripe A y B, así como la varicela. Con mucha menor frecuencia, se encuentra asociación con: coxsackie, parainfluenza, Epstein-Barr, citomegalovirus, adenovirus, hepatitis e incluso con patógenos bacterianos: Chamydia (Bordetella pertussis, Mycoplasma y Sighela)(10).
En la década de los 60 y 70, se buscó la posibilidad de una causa infecciosa vírica y una asociación tóxica o farmacológica. Desde la descripción de los primeros casos, se incluía la toma de AAS como posible factor relacionado(10), por cuanto se asemejaba la clínica a la de la intoxicación por AAS. La intoxicación por salicilatos puede producir una encefalopatía que se manifiesta como: letargo, convulsiones y coma, acompañada de depresión respiratoria y colapso cardiovascular. Estos signos suelen ir precedidos de hiperpnea, vómitos, inquietud y delirio(1).
Diversos estudios epidemiológicos encontraron relación entre el uso de salicilatos y el desarrollo de SR(5). Las principales publicaciones que señalaron a la aspirina® fueron en EE.UU. Hay que citar las siguientes:
• Estudio de Arizona(12), un estudio piloto de casos-control, en diciembre de 1978, publicado en 1980. Con 7 niños diagnosticados de SR durante 4 días y 16 controles. Ante sus resultados, se organizaron dos estudios en los inviernos siguientes.
• Estudio de Michigan 1, realizado en el invierno de 1979-1980. De casos-control. Con 25 niños con SR frente a 46 controles. El Michigan 2, invierno 1980-81, con 12 casos y 13 controles(5,13).
El Ohio study(14), el más determinante, tomó 97 niños de 225 con diagnóstico de SR, de diciembre 1978 a marzo de 1980, frente a 110 controles. Más del 80% de los niños diagnosticados de SR habían tomado aspirina® en las 3 semanas anteriores, como antitérmico o analgésico, sobre todo en relación con gripe o varicela. Este estudio de cuestionario retrospectivo fue criticado por defectos metodológicos. Era un producto de venta sin receta, muy difundido en los hogares. Hasta 1962, la FDA tuvo poca autoridad para los medicamentos de venta libre. A pesar de las deficiencias y de los resultados no confirmatorios definitivos, la publicación fue determinante para incluir avisos en las cajas de aspirina® primero, luego para la prohibición del uso en menores de 12 años, especialmente fuera de las farmacias y, finalmente, para la prohibición de la venta de aspirina® infantil en los EE.UU. Todo en los años 80.
La Fundación Síndrome de Reye (https://www.reyessyndrome.org/) fue promovida por los padres de una niña de 5 años fallecida en 1973 por SR. Se proporcionó financiación para la investigación y, posteriormente, también para la promoción pública de la información sobre el riesgo de la aspirina®. A consecuencia de esta difusión en los medios de comunicación, se empezó a dejar de consumir para la fiebre en niños y adolescentes. Dick Van Dyke, un actor norteamericano de gran prestigio en esas décadas, fue portavoz de la organización, después de que su nieta Jessica muriera a los 13 años a causa del síndrome en 1987. Jessica Van Dyke murió tras una varicela con fiebre, en la que tomó un total de 4 comprimidos de aspirina®(15). En este artículo de prensa de Doyle, se enfatiza mucho en los intereses de la industria –los lobbies de la industria de la aspirina– y la necesidad de establecer un aviso en la etiqueta de los fármacos.
Algunos autores, como Orlowski(5), señalan que los estudios epidemiológicos iniciales tuvieron una metodología imperfecta, con defectos en varios aspectos: diagnóstico no uniforme de los casos, con o sin biopsia, y sin realizarse confirmación del diagnóstico virológico de los controles. Eran estudios con preguntas retrospectivas, que pueden encontrar asociación temporal, pero difícilmente asociación causal. Por ejemplo, se encontró asociación de las fenotiazinas con el SR; pero si se consideraba el diagnóstico desde la fase de inicio de letargia (las fenotiazinas son antieméticos y antipsicóticos usados entonces). En el estudio de Ohio, la hipótesis de partida era encontrar la infección desencadenante, pero se modificó a posteriori la hipótesis para incluir a la aspirina®, esto se considera un defecto metodológico grave. Después, ya no se consideraría ético hacer estudios, por ejemplo de cohortes sin incluir la recomendación de evitar el uso de AAS.
Las publicaciones señalan como prueba de la relación causal, que el SR bajó de incidencia en relación temporal con las recomendaciones de Salud pública en EE.UU. de etiquetado y de restricción del uso de aspirina®(3) (Fig. 2).
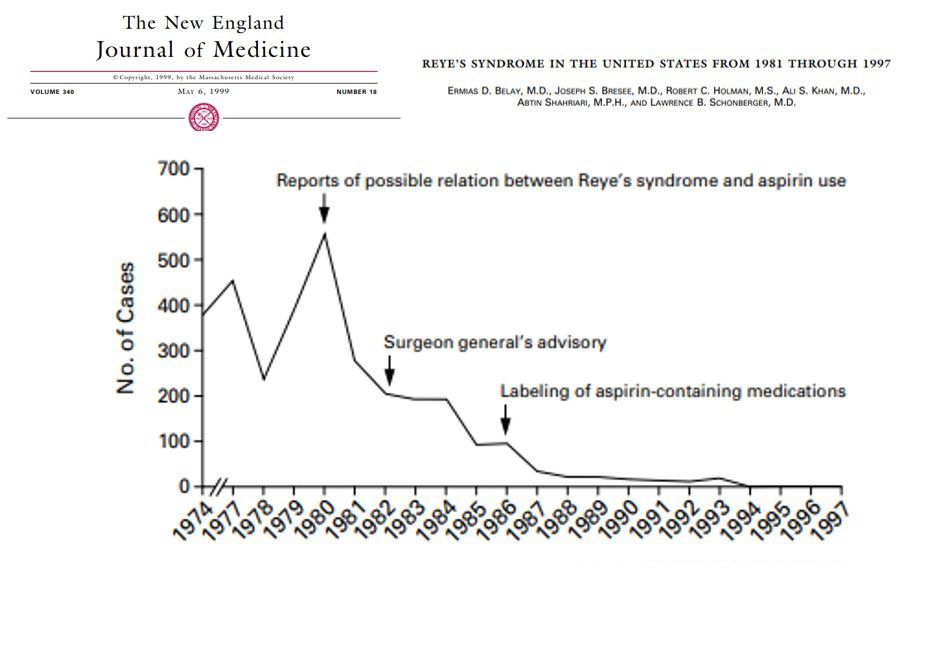
Figura 2. Casos comunicados de síndrome de Reye en EE.UU. de 1981 a 1997. De Belay et al.(3)
Casos comunicados de síndrome de Reye, en EE.UU., desde 1981 a 1997, mostrando: los anuncios públicos de riesgo epidemiológico de la aspirina, el comunicado del Cirujano General (ministro de sanidad) y Etiquetado de las medicaciones con aspirina(3).
La vigilancia nacional del SR en EE.UU. comenzó en 1973. El CDC informaba de más de 500 casos al año en 1979-80, y menos de 100 casos a partir de 1986. En esa época, por cálculo de publicaciones había unos 12 casos al año en España y unos 50 en Inglaterra. Actualmente, no es una enfermedad de declaración obligatoria.
De hecho, la incidencia empezó a disminuir antes de dichas recomendaciones oficiales. Se postula que quizá fue la propia población estadounidense la que dejó de consumir aspirina infantil®.
Estos estudios y la respuesta de la opinión pública, conducida por los medios de comunicación y las gestiones de la Fundación Síndrome de Reye de EE.UU., se han transformado para muchos autores en un gran éxito de Salud Pública(13). “Esta historia de éxito fue resultado de la colaboración entre los grupos biomédicos que realizaron los estudios críticos y un público comprometido con la prevención de una peligrosa enfermedad en los niños”.
Bennett(4) en su revisión, comenta que la identificación de las reacciones adversas graves a los medicamentos, asociadas a fármacos de uso común, puede eludir su detección durante años. El síndrome de Reye (SR), la fibrosis sistémica nefrogénica y la aplasia pura de células rojas, entre los pacientes con enfermedad renal crónica, fueron reconocidos como asociados a aspirina®, gadodiamida y eritropoyetina, respectivamente. Se precisaron unos 81, 13 y 17 años tras la introducción del fármaco en la práctica y la identificación de una relación causal.
Por el contrario, Orlowsky(5) y cols., señalan estudios de los años 80, que citan un porcentaje bajo de pacientes con SR, que había tomado AAS (Sudáfrica, Tailandia, Japón, Irlanda, España, Alemania Occidental, Hong Kong, etc.), si bien eran series retrospectivas. En Australia se tomaba poca aspirina infantil para la fiebre desde los años 50. Algunos estudios, como el de Orlowski en 1987, no consiguieron establecer relación causal.
Orlowsky(5) dice que la desaparición del SR probablemente se pueda relacionar con la mejora en la capacidad diagnóstica de errores innatos del metabolismo que simulan SR clínica, bioquímica y anatomopatológicamente. Una serie de errores innatos del metabolismo predisponen al SR o son responsables de algunos casos de SR. Entre ellos se encuentran: la deficiencia de acil-coenzima A deshidrogenasa de cadena media y otros trastornos de la oxidación de ácidos grasos, así como trastornos del ciclo de la urea(16). La microscopía electrónica pronto señaló el daño mitocondrial en el SR.
Algunos autores consideran que gran parte de estos defectos enzimáticos específicos son trastornos hereditarios, que suelen hacerse evidentes antes de los tres años de edad. En estos casos predominarían: episodios recurrentes, presencia de la enfermedad en los hermanos, hipoglucemia frecuente, aumento de tamaño del corazón y debilidad muscular(3,6).
Solo se han notificado casos de síndrome de Reye en raras ocasiones, en asociación con el tratamiento con salicilatos para la enfermedad de Kawasaki o en el tratamiento con antiinflamatorios no esteroideos para la artritis idiopática juvenil u otras enfermedades del tejido conectivo. Se alude a que dosis bajas de AAS no se relacionan con SR.
Síndrome de Reye en España, primeras noticias y publicaciones
Respecto al impacto en España en los medios de comunicación, hemos realizado una búsqueda en hemeroteca de prensa, ABC y La Vanguardia (Fig. 3).

Figura 3. Noticias en prensa de divulgación en España: ABC, La Vanguardia, años 1985-86.
Encontramos una primera noticia en 1973, que se refiere a una sesión clínica en Sevilla, en el hospital Virgen del Rocío, de Navarro, Abajo, Giráldez y Guerao (ABC, Sevilla, 1-2-1973, p. 42). Posteriormente, no encontramos noticias hasta 1986, donde la prensa se hace eco de la prohibición en Reino Unido del uso de aspirina® en menores de 12 años (La Vanguardia, 12 de junio 1986, p. 32) y de la recomendación de evitar el uso en ese país (La Vanguardia, 1 de agosto de 1986, p. 18). Esto solo en páginas interiores. En ABC, se cita la enfermedad de Reye como una de las 10 causas más importantes de fallecimientos en niños (ABC, 5-10-1986, p. 52). Los titulares señalaban además: “no existen pruebas claras que demuestren su relación (de la ingesta de aspirina®) con el síndrome de Reye” (ABC, 12-6-1986, p. 51). No fue sino hasta el 27 de junio de 2003, en que la Agencia Española del Medicamento prohibió su uso en menores de 16 años (circular 10/93), en la que revisaba la autorización de comercialización de las especialidades farmacéuticas con AAS/salicilatos.
Publicaciones españolas
Las primeras publicaciones de casos de SR en revistas médicas nos constan en 1972 y 1973. Vidal, Ruiz, Carbonell, Delgado(17-20). Resultan muy interesantes de leer las publicaciones de 1977 Álvarez Baleriola(21) y Ull Laita(22), que además revisan la anatomía patológica (Fig. 4).
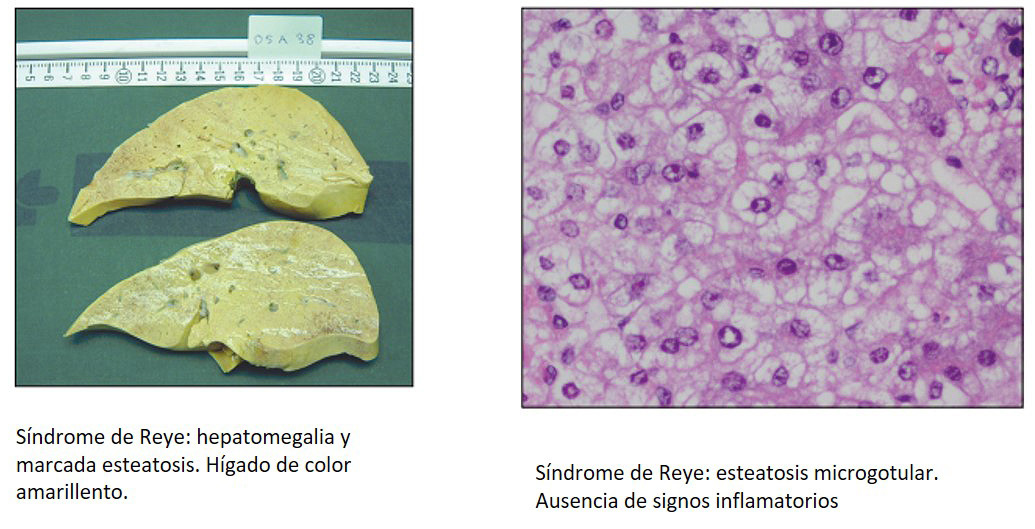
Figura 4. Anatomía patológica del síndrome de Reye. Tomado de referencia Ortiz(27).
Desde 1976 aparecen casos en Anales Españoles de Pediatría:
• En 1976, con edema cerebral unilateral. En 1977, de síndrome de West secundario a un síndrome de Reye.
• De gran interés resulta la casuística de un centro, de Gutiérrez Benjumea(23) y, especialmente, de Palomeque(24), estudio cooperativo, es el estudio más citado internacionalmente. Por último, Baldellou(25) en una editorial-revisión narrativa de 2003, que parece poner casi un punto y final.
Baldellou y Martínez Pardo(25,26), en la primera década del siglo XXI señalan, como sin género de dudas, que la mayoría de los diagnósticos de SR eran enfermedades metabólicas. El ácido acetil salicílico (aspirina) se metaboliza como salicilil-CoA a través de la beta-oxidación mitocondrial de ácidos grasos de cadena media y corta, y el ácido valproico lo hace como valproil-CoA por la misma vía. Estos medicamentos han estado relacionados con casos de síndrome de Reye, en los que además es posible que exista una alteración del metabolismo intermediario sutil, que se manifiesta solo cuando hay sobrecarga de un medicamento capaz de inundar el sistema deficiente. Los virus de la familia herpes, incluida varicela, y el influenza B (también A) en su replicación intracelular, tienen capacidad de alterar la síntesis de enzimas y el transporte intramitocondrial, afectándose la beta-oxidación de ácidos grasos libres.
Breve resumen de la actualidad del síndrome de Reye(1,10)
• Fisiopatología del síndrome de Reye. No se conoce aún con precisión. Se produce una lesión mitocondrial en el contexto de una infección viral. La aspirina®, especialmente si se emplean dosis antipiréticas o antiinflamatorias, puede causar o perpetuar el daño mitocondrial, provocando la inhibición del metabolismo de los ácidos grasos. Las alteraciones neurológicas probablemente sean el resultado de los niveles elevados de amonio, resultado de la disfunción mitocondrial hepática. Hay edema astrocitario, edema cerebral difuso, aumento de la presión intracraneal y lesión neuronal. También se producen otros efectos de alteraciones metabólicas. La presión intracraneal y el nivel de amonio en sangre pueden ser normales en los primeros momentos, incluso con letargia, pero luego se elevan. El daño es multisistémico.
• Diagnóstico diferencial. Enfermedades que presentan un cuadro clínico/patológico que se asemeja al síndrome de Reye.
- Enfermedad metabólica: acidurias orgánicas, trastornos en la fosforilación oxidativa, defectos en el ciclo de la urea (carbamoil-fosfato-sintetasa, ornitina transcarbamilasa), defectos en el metabolismo de la oxidación de ácidos grasos, deficiencias en acil-CoA deshidrogenasa, deficiencia de carnitina sistémica, deficiencia hepática de carnitina palmitoil transferasa, deficiencia de 3-OH-3metilglutaril-CoA y fructosemia.
- Infecciones e intoxicaciones del sistema nervioso central: meningitis, encefalitis y encefalopatía tóxica.
- Shock hemorrágico con encefalopatía.
- Ingesta de fármacos, drogas o tóxicos: salicilato y valproato.
• Tratamiento del SR, fundamentalmente es de soporte, en el entorno de una UCI pediátrica, requiere procedimientos y técnicas para mantener la estabilidad hemodinámica, la función respiratoria y monitorizar la presión intracraneal. Se deben corregir la hipoglucemia y la deshidratación, evitando la sobrecarga de líquidos, corregir la acidosis y la hiperamoniemia(16), como con el empleo descrito de fenilacetato-benzoato de sodio o polisulfato de sodio, incluso hemodiálisis, si los niveles son superiores a 500 mg/dl); las alteraciones de la coagulación; la hipertensión intracraneal; las convulsiones; y las complicaciones (neumonías por aspiración, arritmias cardiacas, pancreatitis, hemorragia gastrointestinal, insuficiencia renal y sepsis).
Epílogo
Antes de los años 60-70 del pasado siglo XX, la aspirina® y otros salicilatos eran los principales agentes antipiréticos y antiinflamatorios utilizados, incluidos adultos, adolescentes, bebés y niños. En esa época, además, era frecuente la sobredosis accidental o intencionada. Los entonces datos crecientes de asociación epidemiológica entre el tratamiento AAS para enfermedades víricas y el desarrollo del síndrome de Reye, llevaron a la autoridad sanitaria de EE.UU. a emitir avisos, limitaciones y, finalmente, prohibir el uso de aspirina infantil®. Lo propio ocurrió en otros países; en algunos se tardó más de 10 años en tener esta precaución.
El síndrome de Reye casi ha desaparecido. La aspirina® ha sido sustituida por otros antipiréticos. También por el gran avance en el estudio, diagnóstico y tratamiento de las enfermedades metabólicas, que representan muchos casos de SR o similares, especialmente en menores de 3 años de edad. Desgraciadamente, los antiinflamatorios no esteroideos y el paracetamol no están exentos de efectos adversos.
Aunque parece que durante mucho tiempo seguirá habiendo ciertas controversias. Hay preguntas epidemiológicas sin resolver aún, como: por qué prácticamente fueron inexistentes los casos con presentación familiar (que tienen genética, entorno y medicación similar); cómo no se tenía noticia de brotes epidémicos antes de los años 50, y el AAS se utilizaba desde principios de siglo XX; o el significado de la aparición de casos también en edades escolar y adolescente-adulto joven.
En los últimos 30-50 años, han sido espectaculares los avances en el conocimiento de las enfermedades metabólicas, a todos los niveles: molecular, genético, anatomía patológica, etc. También se han producido progresos en el conocimiento de los efectos de la aspirina a múltiples niveles, los positivos y también los adversos. La historia de la farmacología y de la medicina del siglo XX, en parte, es la historia de la aspirina.
Bibliografía
1. Chiriboga CA, Patterson MC. Acude toxic-metabolic encephalopathy in children. UpToDate.con/contents/. 2022.
2. Orphanet. Síndrome de Reye. Disponible en: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=3096. Acceso el 23 enero de 2022.
3. Belay ED, Bresee JS, Holman RC, Khan AS, Shahriari A, Schonberger LB. Reye’s syndrome in the United States from 1981 through 1997. N Engl J Med. 1999; 340: 1377-82.
4. Bennett CL, Karen M. Starko KM, Thomsen HS, Sartor O, Macdougall LC, et al. Linking drugs to obscure illnesses: Lessons from Pure red cell aplasia, Nephrogenic systemic fibrosis, and Reye’s syndrome. A report from the Southern Network on Adverse Reactions (SONAR). J Gen Intern Med. 2012; 27: 1697-1703.
5. Orlowski JP, Hanhan UA, Fiallos MR. Is aspirin a cause of Reye’s síndrome? A case against. Drug Saf. 2002, 25: 225-31.
6. Ferretti S, Gatto A, Curatola A, Pansini V, Graglia B, Chiaretti A. Atypical Reye syndrome: three cases of a problem that pediatricians should consider and remember. Acta Biomed. 2021; 92: e2021110.
7. Reye RDK, Morgan G, Basal J. Encephalopaty and fatty degeneration of the viscera. A disease entity in childhood. Lancet. 1963; 2: 749-52.
8. Becroft DMO. Syndrome of encephalopathy and fatty degeneration of viscera in New Zealand children. BMJ. 1966; 2: 135-40.
9. McCrindle BW, Rowley AH, Newburguer JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki Disease: A scientific Statement for Health professionals from the American Heart Association. Circulation. 2017; 135: e927-e999.
10. Chapman J, Arnold JK. Reye Syndrome. StatPearls Publishing LLC. Bookshelf ID: NBK526101PMID: 30252357. Última actualización el 10 de julio de 2021. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK526101/.
11. Giles HM. Encephalopathy and fatty degeneration of the viscera. Lancet. 1965; 1: 1075.
12. Starko KM, Ray CG, Domínguez LB, Stromberg WL, Woodall DF. Reye’s síndrome and salicylate use. Pediatrics. 1980; 66: 859-64.
13. Monto AS. The disappearance of Reye’s syndrome-a public health triumph. N Eng J Med.1999; 340: 1423-4.
14. Halpin TJ, Holtzhauer FJ, Campbell RJ, Hall LJ, Correa-Villaseñor A, Lanese R, et al. Reye’s syndrome and medication use. JAMA. 1982; 248: 687-91.
15. Doyle L. Aspirin and deadly Reye’s Syndrome: warnings can be missed. Hemeroteca: “Los Angeles Times”. 1987.
16. Gil Ortega D, Cocho JA, Merinero B. Protocolos de diagnóstico y tratamiento de los Errores Congénitos del Metabolismo. 2ª edición. Ergon. 2018. Disponible en: http://aepmi.org/mm/file/protocolos-AECOM-2-ed.pdf.
17. Vidal ML, Hortelano JG, Scarpellini A, López Barea F. Síndrome de Reye (a propósito de dos casos). Bol S Ped Madr. 1972; 19: 191-202.
18. Ruiz A, Larrauri J, López Barea F, Vidal ML. Encefalopatía y degeneración grasa de las vísceras (síndrome de Reye). Comunicación de cuatro casos y revisión de la literatura. Med Clín. 1973; 60: 452-58.
19. Carbonell Estrany J, Ruiz Gómez D. Síndrome de Reye. Diálisis. Curación. Bol Soc Val Pediat.1972; 59: 141.
20. Delgado A, Bernaola E, et al. Síndrome de Reye. Estudio clínico y ultramicroscópico. Rev Esp Ped. 1975; 31: 561-80.
21. Álvarez Baleriola I. Síndrome de Reye: estudio morfológico e histoquímico de la esteatosis. Patología. 1977; X: 93-110.
22. Ull Laita M, Gómez Ribas B. Síndrome de Reye: anatomía patológica y revisión patogénica. Patología. 1977; X: 111-20.
23. Gutiérrez Benjumea A, Navarro González J, Cintado Bueno C, Tovaruela Santos A. Síndrome de Reye: revisión de nuestra casuística. Anales de Pediatría. 1979; 12: 511-22.
24. Palomeque A, Domenech P, Martínez-Gutiérrez A. Síndrome de Reye en España, 1980-1984 (Estudio cooperativo, Sección de CIP de la AEP). An Esp Pediatr. 1986; 24: 285-9.
25. Baldellou Vázquez A. Síndrome de Reye. Cuarenta años después. An Pediatr (Barc). 2003; 59: 319-22.
26. Martínez-Pardo M, Sánchez Valverde F. Síndrome de Reye: fracaso mitocondrial hepático agudo con encefalopatía. Etiología metabólica. Protocolos AEP. 1ª ed. Disponible en: https://www.aeped.es/sites/default/files/documentos/11-reye.pdf.
27. Ortiz J, González San Martín F, Muñoz Singi R, Geijo F, Bullón A. Síndrome de Reye en paciente pediátrico. Rev Esp Enf Dig. 2007; 99: 165-6.

 Behavior disorders
Behavior disorders