 |
| Temas de FC |
M.V. Barajas Sánchez, J. Martín Valbuena![]()
Unidad de Neumología Pediátrica. Complejo Asistencial Universitario de Salamanca
| Resumen
Las malformaciones congénitas pulmonares y de la vía aérea engloban un grupo heterogéneo de patologías que afectan al sistema respiratorio. Su origen se encuentra en un incorrecto desarrollo de diferentes estructuras pulmonares durante la embriogénesis. Las manifestaciones clínicas son muy variables, dependiendo de la entidad y del grado de afectación, abarcando desde pacientes completamente asintomáticos hasta casos no compatibles con la vida. El cribado prenatal mediante ecografía permite detectar un gran número de estas patologías que requerirán otras pruebas diagnósticas específicas para una mejor caracterización postnatal. El tratamiento dependerá tanto de la repercusión clínica que implique cada malformación como de posibles riesgos futuros, individualizando cada caso en busca de un balance riesgo-beneficio óptimo. Hay casos que se manejan de manera expectante con diferentes tipos de controles sin llevar a cabo medidas terapéuticas excesivas y otros requerirán intervención urgente. Es importante para cualquier pediatra conocer las diferentes entidades para una detección precoz de complicaciones y llevar a cabo un manejo adecuado. |
| Abstract
Congenital lung and airway malformations include a heterogeneous group of pathologies affecting the respiratory system. Their origin lies in an incorrect development of lung structures during embryogenesis. Clinical features are highly variable, depending on the type of malformation itself and the extent of the affected area, ranging between completely asymptomatic patients to cases incompatible with life. Prenatal ultrasound screening detects many of these pathologies, though specific diagnostic procedures will be required postnatally to achieve a better characterization of the lesions. Treatment will depend on both the clinical impact of each malformation as well as future risks involved in certain pathologies, individualizing each case looking for an optimum risk/benefit balance. Some cases will require expectant management with different assessments without excessive therapeutic measures and others will require urgent intervention. It is important for every pediatrician to know the different entities to achieve an early detection of complications and carry out adequate management. |
Palabras clave: Malformación congénita de la vía aérea pulmonar; Secuestro pulmonar; Enfisema lobar congénito; Hipoplasia pulmonar.
Key words: Congenital malformation of the pulmonary airway; Pulmonary sequestration; Congenital lobar emphysema; Pulmonary hypoplasia.
Pediatr Integral 2026; XXX (1): 7 – 14
OBJETIVOS
• Entender la etiopatogenia general de las malformaciones congénitas pulmonares y de la vía aérea, entendiendo el momento del desarrollo en el que se producen y el lugar de afectación.
• Tener un alto grado de sospecha clínica, reconociendo los síntomas y signos más frecuentes, especialmente en aquellos casos paucisintomáticos y sin diagnóstico prenatal.
• Conocer la utilidad e indicaciones de las principales pruebas diagnósticas disponibles.
• Reconocer las posibles complicaciones, asociaciones con otras patologías y criterios de mal pronóstico dentro de las diferentes patologías que serán clave para el seguimiento.
• Conocer los riesgos y beneficios de las diferentes intervenciones terapéuticas, para adecuar el manejo de los pacientes.
Malformaciones congénitas pulmonares y de la vía aérea
https://doi.org/10.63149/j.pedint.106
Introducción
Las malformaciones pulmonares y de la vía aérea son una entidad rara, aunque con aumento de la incidencia debido al diagnóstico prenatal, con un abordaje comprometido en muchos casos.
Las malformaciones congénitas pulmonares y de la vía aérea son un grupo de anomalías debidas a la alteración del desarrollo durante la embriogénesis del pulmón y de la vía aérea. Los defectos en el proceso de separación traqueal del intestino anterior y del desarrollo de las regiones ramificadas del pulmón son la base de la mayoría de las malformaciones pulmonares congénitas. Es una patología rara, con incidencia estimada de 1 caso cada 2.500 a 8.000 recién nacidos(1), siendo el diagnóstico intrauterino el más frecuente en la actualidad, debido al avance de las técnicas de imagen prenatal, lo que permite un manejo de forma precoz.
El diagnóstico prenatal se realiza con la ecografía, que forma parte del cribado durante el embarazo. Es una prueba sensible para el diagnóstico y muy valiosa en el seguimiento y control evolutivo de estas malformaciones. El diagnóstico prenatal se basa en la presencia de masas de apariencia quística o sólida ocupantes de espacio dentro del tórax fetal o en el tamaño anormal de los pulmones y la consiguiente desviación del corazón de su posición normal de 45º.
La resonancia magnética fetal es una técnica de imagen que apoya a la ecografía en el abordaje de estas malformaciones. Es capaz de localizar y definir con más precisión la lesión pulmonar.
Las principales herramientas en el diagnóstico postnatal son la radiografía de tórax (Rx tórax) y la angiografía por tomografía computarizada (angio-TAC). La radiografía de tórax es la prueba inicial de elección ante la sospecha prenatal de una malformación congénita pulmonar por su carácter no invasivo y su rentabilidad. Sin embargo, nunca se debe descartar una malformación diagnosticada prenatalmente en un recién nacido basándose únicamente en la radiografía de tórax, ya que el 50 % de los pacientes con estas malformaciones puede tener una radiografía normal.
Nos encontramos ante una patología rara, con un aumento de casos en las últimas dos décadas debido al diagnóstico prenatal(2,3). Esto supone un nuevo reto para los pediatras, dado que el manejo postnatal de aquellos recién nacidos asintomáticos es controvertido.
Etiopatogenia
Se han propuesto varios mecanismos fisiopatológicos que alterarían el correcto desarrollo del sistema respiratorio durante la embriogénesis.
Para comprender las causas de estas malformaciones congénitas pulmonares, es preciso recordar la embriogénesis pulmonar. El desarrollo y maduración del aparato respiratorio comienza en torno a la cuarta semana de gestación y finaliza alrededor de los 8 años. Se describen cinco periodos evolutivos en la embriogénesis pulmonar. El periodo embrionario ocurre en las 6 primeras semanas de gestación. A partir del intestino primitivo, se origina un divertículo ventral, que dará origen al desarrollo del tejido epitelial del árbol respiratorio. El periodo pseudoglandular ocurre entre la 6ª-16ª semana de gestación; en este periodo se produce la ramificación dicotómica de los bronquios y termina con la formación de los bronquiolos terminales. Entre la semana de gestación 16 a 26 tiene lugar el periodo canalicular, donde tiene lugar la formación de los acinos y se inicia la irrigación de la vía respiratoria. Al final de esta etapa aparece el surfactante. Los bronquiolos terminales se transforman en bronquiolos respiratorios y se forman los sáculos, lugar en el que se realiza el intercambio gaseoso. Este periodo es conocido como periodo sacular, y tiene lugar entre la semana 28 a 36 de la gestación. El último periodo se denomina periodo alveolar; ocurre desde la semana 36 hasta los 8 años de vida. Durante este periodo, se forman los septos y las estructuras alveolares que seguirán su desarrollo posterior (Fig. 1).
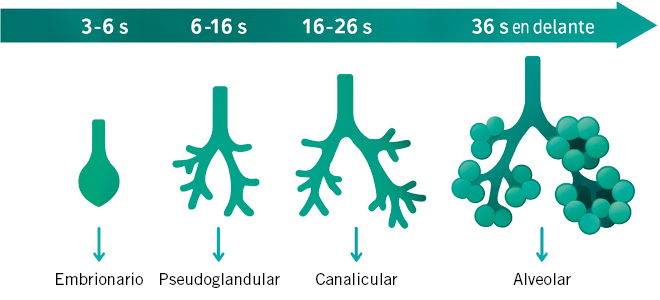
Figura 1. Fases del desarrollo embrionario pulmonar. s: semanas de gestación.
La etiología de las malformaciones congénitas pulmonares es desconocida, pero se sospecha que el desarrollo de las malformaciones pulmonares congénitas puede deberse a una combinación de diferentes factores que interactúan entre sí en diversos periodos del desarrollo pulmonar(4).
Las principales causas propuestas son:
• Defectos en la diferenciación y separación de la porción ventral del intestino anterior primitivo.
• Obstrucción de la vía aérea intraútero.
• Anomalías vasculares.
• Factores genéticos.
De estos factores, el que se propone como origen más probable de estas malformaciones es la obstrucción de la vía aérea intraútero y, dependiendo del nivel de obstrucción y el momento de la gestación en el que ocurra, tendríamos un tipo u otro de malformación(5,6).
Tipos de malformaciones congénitas broncopulmonares
Las malformaciones congénitas pulmonares se pueden clasificar en función de si predomina afectación parenquimatosa, vascular o mixta. Abordaremos aquellas malformaciones más frecuentes con las características clínicas, su diagnóstico y manejo terapéutico de cada una de ellas.
Agenesia y aplasia pulmonar
Supone la ausencia total o parcial de un pulmón con hiperplasia compensadora del contralateral. Su manejo dependerá del desarrollo de infecciones de repetición en el pulmón afecto.
Son anomalías raras, con una incidencia de 1 por cada 15.000 recién nacidos. La agenesia se define, como la ausencia completa de tejido pulmonar, bronquio y arteria pulmonar (tipo I); que se diferencia de la aplasia pulmonar, porque esta presenta un bronquio rudimentario (tipo II). El pulmón contralateral es hiperplásico. Dichas anomalías predominan en el lado izquierdo.
Este defecto congénito se puede presentar de forma aislada o asociado con otras anomalías a nivel esquelético, cardiovascular, gastrointestinal o genitourinario. Puede formar parte de síndromes, como el de VACTERL.
Su pronóstico va a depender de las anomalías asociadas. La afectación bilateral presenta elevada mortalidad al nacimiento.
El diagnóstico cada vez es más frecuente que se realice mediante ecografía prenatal, apoyándose en la resonancia magnética (RM) fetal.
En la radiografía de tórax muestra ausencia de pulmón y desplazamiento del mediastino hacia el lado afecto, con disminución de espacios intercostales y elevación del diafragma. Otras pruebas diagnósticas que apoyan el diagnóstico son la angio-TAC o la angiografía por resonancia magnética (angio-RM).
Las manifestaciones clínicas en los casos de diagnóstico postnatal pueden variar desde pacientes asintomáticos a infecciones pulmonares de repetición con sibilancias que pueden hacer pensar en asma en niños mayores.
No existe un tratamiento curativo específico para esta patología. En la aplasia pulmonar, el esbozo bronquial puede dar lugar a infecciones de repetición, por lo que debe ser extirpado. Siempre deben buscarse y evaluarse la posible corrección de aquellas malformaciones asociadas.
Hipoplasia pulmonar
Consiste en la incorrecta diferenciación pulmonar, muchas veces asociada a otras patologías y con una clínica variable.
Se define como una reducción del volumen pulmonar, con disminución del número de ramificaciones bronquiales o insuficiente diferenciación pulmonar.
Podemos distinguir la hipoplasia pulmonar primaria de causa desconocida y la secundaria a otras patologías, siendo especialmente grave la asociada a hernia diafragmática congénita. La hipertensión pulmonar asociada a la hipoplasia vascular es el principal factor pronóstico en estos pacientes.
Las manifestaciones clínicas de la forma primaria producen síntomas inmediatamente tras el nacimiento, manifestándose como distrés respiratorio grave. Además, la disminución de vascularización origina hipertensión pulmonar. En el caso de hipoplasia secundaria, el espectro clínico varía desde pacientes asintomáticos a infecciones respiratorias de repetición, disnea o sibilantes recurrentes.
El manejo de la hipoplasia pulmonar primaria de los recién nacidos es conservador y depende de la gravedad de las alteraciones. En el caso de la hipoplasia pulmonar secundaria, se basa en la corrección quirúrgica de las malformaciones asociadas (p. ej., hernia diafragmática) tras la estabilización del paciente.
Hiperinsuflación lobar congénita
Atrapamiento aéreo distal a obstrucción bronquial por efecto valvular, con variable repercusión clínica y diagnóstico generalmente postnatal. Si es asintomático, no suele tener complicaciones.
La hiperinsuflación lobar congénita es una patología poco frecuente, con una incidencia estimada de alrededor de 1 por cada 20.000 a 30.000 nacimientos(7,8). Antes conocida como enfisema lobar congénito; en la actualidad, se prefiere el término de hiperinsuflación en lugar de “enfisema”, dado que el tejido pulmonar afecto es sano.
Se caracteriza por una alteración localizada del cartílago en la pared bronquial, ocasionando un efecto valvular y, consecuentemente, una hiperinsuflación y atrapamiento aéreo en uno o varios lóbulos pulmonares. El origen de la anomalía puede ser primario (ausencia de cartílago bronquial, estenosis bronquial o debilidad de la pared bronquial) o secundario a anomalías vasculares (sling de la arteria pulmonar o retorno venoso pulmonar anómalo). En el 50 % de los pacientes no se llega a identificar la causa. Los lóbulos más afectados son el lóbulo superior izquierdo y el lóbulo medio.
En recién nacidos y lactantes puede manifestarse, como un distrés respiratorio grave, infecciones recurrentes, sibilancias, atelectasias debido a la compresión pulmonar o ser un hallazgo incidental(9). Hasta en un 20 % de los casos se asocia a cardiopatías (comunicación intraventricular y persistencia del ductus permeable)(10).
Dado que en el cribado prenatal la ecografía tiene baja sensibilidad, el diagnóstico suele realizarse de manera postnatal. En la Rx tórax podemos encontrar distensión del lóbulo afecto con desplazamiento del mediastino con compresión y atelectasia del pulmón contralateral. Otras pruebas de imagen, como la RM o la TC, pueden definir la lesión y establecer la causa (Fig. 2). La fibrobroncoscopia en esta patología es importante para valorar la anatomía bronquial y descartar causas secundarias de obstrucción bronquial intrínsecas (cuerpo extraño bronquial, tapón de moco, etc.) o extrínsecas (sling vascular, tumoraciones, etc.).

Figura 2. Área hipodensa bien delimitada en lóbulo medio con estructuras vasculares distribuidas de manera periférica. Hallazgos compatibles con enfisema lobar congénito.
Se considera tratamiento quirúrgico en pacientes sintomáticos, con lobectomía del lóbulo afecto. En los casos de pacientes asintomáticos, se opta por un tratamiento conservador, dado que es improbable que presente complicaciones(11).
Malformación congénita de la vía aérea pulmonar
Grupo heterogéneo de malformaciones del árbol traqueobronquial con diferente expresión clínico-radiológica según la localización de la lesión. Alguna de ellas conlleva ciertos riesgos asociados.
La malformación congénita de la vía aérea pulmonar (MCVAP), anteriormente conocida como malformación adenomatoidea quística, es la malformación pulmonar más frecuente, con una incidencia estimada de 1 caso por 8.500 a 35.000 recién nacidos(7,12). El diagnóstico se realiza a través de la ecografía prenatal, en el segundo trimestre, entre la 21-24 semanas de edad gestacional, ofreciendo una imagen hiperecogénica, a veces difícil de diferenciar de otras malformaciones. Las lesiones pueden regresar durante la gestación en un 50 %.
Se caracterizan por un patrón anormal de las vías respiratorias, que tiene lugar durante la morfogénesis de la ramificación pulmonar. Tienen su origen en diferentes niveles del árbol traqueobronquial y en diferentes etapas del desarrollo pulmonar, dando lugar a una lesión pulmonar multiquística de tejido pulmonar no funcionante.
Estas malformaciones ocurren esporádicamente y no están asociadas a factores maternos, como edad, raza o exposiciones. En estudios recientes, se han identificado mutaciones asociadas a los genes FGFR2, DICER1, KRAS y TP53, lo que plantea un posible origen genético en determinados subtipos. Destaca la MCVAP tipo 4, conocida como blastoma pleuropulmonar (BPP) tipo 1, que se asocia con mutación en DICER1 con un alto potencial de malignidad(8).
La nueva clasificación de las MCVAP, realizada en 2002 por Stoker, las reclasifica en 5 tipos, basándose en el lugar donde se produce la lesión, número y tamaño de los quistes y distinciones histopatológicas que son de utilidad clínica (Tabla I).

La presentación clínica de la MCVAP es variable, desde dificultad respiratoria grave en el periodo neonatal en relación con el tamaño de la lesión a pacientes asintomáticos. Según estudios recientes(13), alrededor del 25 % de los pacientes asintomáticos con diagnóstico de MCVAP desarrollan síntomas alrededor de los 6-7 meses de edad. En la etapa de lactante y escolar, las complicaciones más frecuentes son infecciones de repetición (neumonías recurrentes) por sobreinfección de los quistes, tos crónica, neumotórax o hemoptisis.
El diagnóstico de las MCVAP se realiza por técnicas de imagen, ecografía prenatal, Rx tórax, tomografía axial computarizada de alta resolución (TACAR) y RM.
Los hallazgos en la Rx tórax son variables, dependiendo del tipo de MCVAP. Los tipos 1, 2 y 4 se caracterizan por quistes de paredes finas con ocupación interior o contenido aéreo, asociando una masa sólida en los tipos 1 y 4. El tipo 3 se presenta como una masa grande, sólida y homogénea. Además, en la Rx tórax podemos observar desplazamiento mediastínico hacia el lado contralateral, con hipoplasia del pulmón ipsilateral debido al efecto masa. Con esta prueba no es posible delimitar y definir bien las lesiones, por lo que nos apoyamos en técnicas de imagen avanzadas, como la TACAR, que nos permite determinar el tamaño de los quistes, la extensión anatómica e identificar otras alteraciones asociadas. Se debe realizar con contraste para evaluar la vascularización, especialmente si sospechamos asociación con secuestro pulmonar (Fig. 3).

Figura 3. Lesión multiquística en lóbulo inferior izquierdo. Presenta quistes de pared fina, menores de 2 cm, con contenido aéreo en su interior. Irrigación por rama arterial dependiente de aorta descendente, compatible con lesión híbrida MCVAP tipo II con secuestro pulmonar asociado.
Manejo prenatal
Para el manejo prenatal de la MCVAP, es de gran utilidad la monitorización con ecografías seriadas del tamaño de la lesión en relación con el perímetro cefálico, mediante el cálculo de la ratio entre el volumen de la malformación y la circunferencia craneal (RVC). Si la RVC es superior a 1,6, se correlaciona con un alto riesgo de desarrollar hidrops fetalis y muerte fetal. En fetos de menos de 32 semanas con hidrops fetalis asociado a MCVAP estaría indicada, como terapia de primera línea, corticoides prenatales (betametasona). Aunque la respuesta es variable, han demostrado responder mejor los que presentan lesiones microquísticas. En los estudios realizados, se ha observado una supervivencia de un 80 %(14).
En caso de no respuesta o ante la presencia de quistes grandes, se pueden beneficiar de terapias de segunda línea, como la cirugía prenatal con la colocación de shunt toracoamniótico, para reducir la compresión pulmonar(15).
Manejo postnatal
La Rx tórax es una herramienta de primera línea, debido a su carácter no invasivo y rentabilidad. Se debería realizar antes del alta del recién nacido. Sin embargo, nunca debemos descartar una MCVAP prenatal basándonos en esta prueba. Ante una Rx tórax normal, se debe programar, dentro de los primeros 6 meses, un angio-TAC para evaluar la persistencia o desaparición de la lesión detectada en la ecografía prenatal, así como un seguimiento multidisciplinar por neumólogo y cirujano pediátrico(16).
En la Rx tórax y TACAR postnatal se pueden observar uno o varios quistes o masa sólida con microquistes en su interior. Como diagnóstico de elección, solicitaremos la angio-TAC, ante la posibilidad de lesiones híbridas (definidas como combinación de MCVAP y secuestro pulmonar) para poder localizar la arteria aberrante.
Si el recién nacido es sintomático, hay consenso entre la mayoría de los expertos en la indicación de tratamiento quirúrgico de la lesión (lobectomía), siendo la vía de abordaje más frecuente realizada la toracoscopia, por las ventajas que ofrece de seguridad y recuperación más precoz(17,18).
En los pacientes asintomáticos, con lesiones pequeñas y sin factores de riesgo de malignización, continúa la controversia sobre qué actitud es la más adecuada. Los expertos que apoyan la cirugía temprana, recomendada antes del primer año de vida, se basan en el riesgo de infección que dificultaría la cirugía posterior y en la posibilidad de malignización. La cirugía electiva precoz presenta las ventajas de una recuperación rápida, así como un crecimiento compensatorio pulmonar(19).
En los pacientes asintomáticos, en los que se opta por la observación, se apoyan en las siguientes premisas: muchos son asintomáticos y/o son hallazgos casuales; en la posibilidad de regresión de las MCVAP, que algunos estudios han reportado hasta un 20 % de los recién nacidos asintomáticos con lesiones quísticas confirmadas en el TAC durante el periodo neonatal; y en los riesgos quirúrgicos(20).
En estos casos se debe realizar un seguimiento para detectar complicaciones durante el primer año de vida y realizar imagen (TACAR/RM) a los 6 meses y, posteriormente, anual, pero no existe un consenso sobre la estrategia óptima para realizar dichas pruebas de imagen(21); además de valorar el riesgo-beneficio de radiación y coste al realizar TAC anual, ansiedad familiar o la posibilidad de pérdida de seguimiento.
Riesgo de malignización
El riesgo de malignización de las MCVAP se encuentra de manera general entre 1-3 %(22), siendo las de tipo 1 y 4 las que conllevan mayor riesgo, asociándose a carcinoma bronquioalveolar y blastoma pleuropulmonar tipo 1 (BPP), respectivamente. Se han propuesto factores de alto riesgo para el desarrollo de BPP en niños con lesiones quísticas pulmonares asintomáticas. Entre estos factores de riesgo, encontramos la aparición conjunta con neumotórax, quistes pulmonares bilaterales o multifocales, MCVAP tipo 4, síndrome DICER1 o antecedentes familiares de BPP o enfermedades relacionadas.
El síndrome DICER1 es un síndrome con patrón de herencia autosómico dominante con penetrancia disminuida, que predispone al cáncer hereditario y está relacionado con mutaciones en el gen DICER1 (presente en 2/3 de los casos de BPP). Puede asociarse con enfermedad quística renal y poliposis intestinal(7).
Secuestro pulmonar
Tejido pulmonar con irrigación sistémica y no conectado al árbol traqueobronquial; si es de localización intralobar, tiene alto riesgo de complicaciones, especialmente infecciones recurrentes.
Es una anomalía poco frecuente; supone el 1-6 %. Se caracteriza por un tejido pulmonar displásico no funcionante sin comunicación con el árbol bronquial; además, presenta la característica de estar irrigado por una arteria sistémica anómala procedente de la aorta descendente.
Se clasifica en secuestro intralobar (SPI) y secuestro extralobar (SPE). El SPI se localiza dentro de un lóbulo pulmonar normal y carece de pleura visceral propia. Representa, aproximadamente, el 75 % de todos los secuestros pulmonares(23). El SPE se localiza fuera del pulmón normal y posee su propia pleura visceral. La gran mayoría se localizan en hemitórax izquierdo, siendo la localización más frecuente entre el lóbulo inferior izquierdo y el hemidiafragma. En ocasiones, se localiza debajo del diafragma o en el retroperitoneo, en particular en la glándula suprarrenal, donde puede simular un neuroblastoma suprarrenal(24). Representa, aproximadamente, el 25 % y es más probable que se asocie con otras anomalías congénitas, como hernia diafragmática congénita, anomalías vertebrales, cardiopatías congénitas, hipoplasia pulmonar, duplicación colónica y MCVAP.
La presentación clínica es variable y depende del tipo, tamaño y localización de la lesión. Muchas de estas lesiones se diagnostican de forma prenatal. La mayoría de los recién nacidos son asintomáticos, pero, en ocasiones, presentan dificultad respiratoria en periodo neonatal. En el caso de SPI y las formas híbridas, se manifiestan en etapa de lactante y escolar, como neumonías de repetición, tos crónica, hemoptisis, etc., pero también como hallazgo casual en una Rx tórax. Las complicaciones de los secuestros pulmonares son raras e incluyen insuficiencia cardiaca, debido al excesivo flujo a través de la arteria aberrante, o sangrado masivo(2).
Ante un diagnóstico de sospecha, la primera prueba a realizar es una Rx tórax, que muestra los secuestros pulmonares como una masa densa dentro de la cavidad torácica o parénquima pulmonar. Las técnicas de imagen avanzadas, como la Angio-TC (Fig. 4) o Angio-RM, demuestran la arteria sistémica que lo irriga, y tienen como objetivo la confirmación del diagnóstico, aunque el diagnóstico definitivo se realiza con la anatomía patológica tras la resección quirúrgica.

Figura 4. Área radiolucente en lóbulo inferior izquierdo con irrigación dependiente de aorta descendente, compatible con secuestro pulmonar intraparenquimatoso.
Manejo
La indicación de cirugía, lobectomía o resección segmentaria, se establece en aquellos secuestros pulmonares sintomáticos y en los SPI asintomáticos, por el riesgo de infección, o en las lesiones híbridas. Para aquellas lesiones con arteria sistémica bien definida e insuficiencia cardiaca de alto gasto importante, el tratamiento con embolización arterial podría ser una opción terapéutica inicial.
Algunos autores recomiendan la observación en lugar de la cirugía para aquellos pacientes asintomáticos, en particular si la lesión es pequeña, no quística y es compatible con SPE(16,25).
Atresia bronquial
Interrupción total del desarrollo bronquial, con área distal hiperinsuflada con bajo riesgo de complicaciones, por lo que se suele optar por manejo conservador.
Se caracteriza por la interrupción intraútero de un bronquio lobar, segmentario o subsegmentario; como consecuencia, se produce una hiperinsuflación del segmento afecto e impactación mucosa, originando el mucocele o broncocele. Suele presentarse de manera incidental, al realizar una Rx tórax, como una zona hiperinsuflada o hiperlúcida que puede comprimir el tejido adyacente y provocar un desplazamiento del mediastino. El lóbulo más afectado es el superior izquierdo. La mayoría de los pacientes se encuentran asintomáticos. La infección es poco frecuente dada la ausencia de comunicación con el árbol traqueobronquial. Por ello, la actitud expectante es la opción más razonable.
Quiste broncogénico
Lesión quística con tendencia a la sobreinfección o crecimiento, con compresión de estructuras vecinas, por lo que suele ser recomendable la exéresis quirúrgica.
Es una lesión quística que se origina de un defecto en el desarrollo embrionario del árbol bronquial. Pueden aparecer en cualquier localización, siendo la más frecuente en mediastino (85 %), paratraqueal, hiliar o subcarinal.
Muchos pacientes con esta patología se encuentran asintomáticos y, con frecuencia, se descubren de manera incidental al realizar estudios por otros motivos. En caso de ser sintomáticos, suelen presentarse, como tos recurrente, sibilancias, neumonías de repetición, dificultad respiratoria o disfagia. Dichos síntomas están en relación con la infección del quiste o con crecimiento de este, ocasionando compresión de vía respiratoria y/o digestiva.
En la Rx tórax se presentan como masas redondeadas bien definidas de densidad acuosa o cavidades hidroaéreas, en caso de sobreinfección (Fig. 5A).
La TACAR suele mostrar masas mediastínicas quísticas de tejido blando bien delimitadas o atenuación hídrica (Fig. 5B).
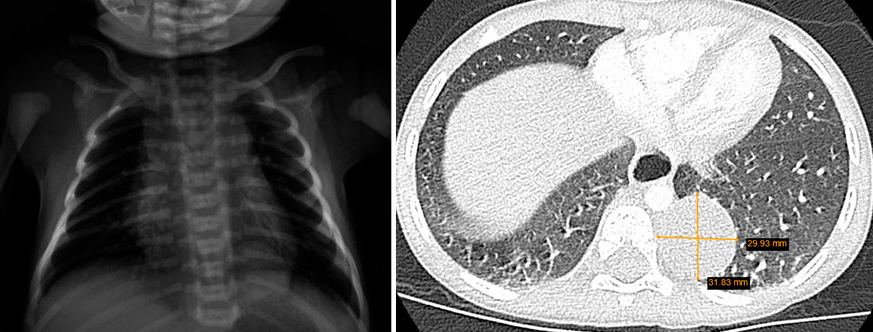
Figura 5. A. Rx tórax con imagen redondeada retrocardiaca. B. Tomografía axial computarizada de alta resolución (TACAR) con masa redondeada bien delimitada de 3 x 3 cm, densidad agua, compatible con quiste broncogénico.
El manejo de un quiste broncogénico consiste en la escisión quirúrgica por el riesgo de infección o compresión de la vía aérea o digestiva.
Función del pediatra de Atención Primaria
La función del pediatra de Atención Primaria (AP) es clave en el manejo de las malformaciones congénitas pulmonares y de la vía aérea (v. Algoritmo al final del artículo). Por una parte, jugarán un papel fundamental en el diagnóstico de aquellas patologías con clínica larvada que pueden escapar al screening prenatal y no hacerse evidentes en los primeros días de vida. Tener un alto grado de sospecha clínica es muy importante para detectar cuanto antes estas patologías y derivar de manera precoz a atención especializada.
Además, una vez realizado el diagnóstico, el manejo, en muchos casos, dependerá de la repercusión que las diferentes malformaciones tengan en el paciente, siendo de especial importancia el pediatra de AP en el seguimiento de estos pacientes. El pediatra de AP tomará decisiones en cuanto al manejo, basándose en una evaluación integral del paciente, prestando especial atención a aspectos, como el desarrollo ponderoestatural, el número de infecciones respiratorias y su repercusión en la vida del paciente o síntomas respiratorios que pueden no ser evidentes sin una adecuada anamnesis.
Por último, conocer las diferentes entidades y la patología asociada puede requerir, en muchos casos, estudios complementarios de manera programada o derivación a otros especialistas que puede realizarse desde AP.
Conflicto de intereses
No hay conflicto de interés en la elaboración del presente manuscrito ni fuente de financiación.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.*** Hegde BN, Tsao K, Hirose S. Management of Congenital Lung Malformations. Clin Perinatol. 2022; 49: 907-26. Disponible en: https://doi.org/10.1016/j.clp.2022.08.003.
2.** Pederiva F, Rothenberg SS, Hall N, Ijsselstijn H, Wong KKY, von der Thüsen J, et al. Achiron R, Pio d´Adamo A, Schnater JM. Congenital lung malformations. Nat Rev Dis Primers. 2023; 9: 60. Disponible en: https://doi.org/10.1038/s41572-023-00470-1.
3. Burge D, Wheeler R. Increasing incidence of detection of congenital lung lesions. Pediatric Pulmonol. 2010; 45; 103. Disponible en: https://doi.org/10.1002/ppul.21150.
4.* Lee EY, Dorkin H, Vargas SO. Congenital pulmonary malformations in pediatrics: review and update on etiology, classification, and imaging findings. Radiol Clin North Am. 2011; 49: 921-48. Disponible en: https://doi.org/10.1016/j.rcl.2011.06.009.
5. Rierdlinger WF, Vargas SO, Jennings RW, Extroff JA, Barnewolt CE, Lillehei CW, et al. Bronchial atresia is common to extralobar sequestration, intralobar sequestration, congenital cystic adenomatoid malformation and lobar emphysema. Pediatr Dev Pathol. 2006; 9: 361-73. Disponible en: https://doi.org/10.2350/06-01-0023.1.
6.** Fichera G, Cavaliere A, Causin F, Zuliani M, Bisogno G, Rea F, et al. Pediatric congenital pulmonary malformations: key findings at imaging. Clin Transl Imaging. 2024; 12: 457-66. Disponible en: https://doi.org/10.1007/s40336-024-00632-5.
7. Priest JR, Williams GM, Hill DA, Dehner LP, Jaffé A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009; 44: 14-30. Disponible en: https://doi.org/10.1002/ppul.20917.
8. Oermann CM. Congenital lobar emphysema. En: UpToDate. 2025.
9. Thakral CL, Maji DC, Sajwani MJ. Congenital lobar emphysema: experience with 21 cases. Pediatr Surg Int. 2001; 17: 88-91. Disponible en: https://doi.org/10.1007/s003830000506.
10. Gatt D, Lapidus-Krol E, Chiu PPL. High comorbidity rates in congenital lobar emphysema and the effect on clinical presentation. Eur J Pediatr. 2024; 183: 4573-7. Disponible en: https://doi.org/10.1007/s00431-024-05684-3.
11. Zobel M, Gologorsky R, Lee H, Vu L. Congenital lung lesions. Semin Pediatr Surg. 2019; 28: 150821. Disponible en: https://doi.org/10.1053/j.sempedsurg.2019.07.004.
12. Gornall AS, Budd JL, Draper ES, Konje JC, Kurinczuk JJ. Congenital cystic adenomatoid malformation: accuracy of prenatal diagnosis, prevalence and outcome in a general population. Prenat Diagn. 2003; 23: 997-1002. Disponible en: https://doi.org/10.1002/pd.739.
13. Kantor N, Wayne C, Nasr A. Symptom development in originally asymptomatic CPAM diagnosed prenatally: a systematic review. Pediatr Surg Int. 2018; 34: 613-20. Disponible en: https://doi.org/10.1007/s00383-018-4264-y.
14. Egloff A, Bulas DI. Congenital pulmonary airway malformation: Prenatal diagnosis and management. En: UpToDate, WilKins-Haug L. (Ed). 2025.
15. Cass DI, Olutoye OO, Cassady CL, et al. Prenatal diagnosis and outcome of fetal lung lesions. J Pediatric Surg. 2012; 47: 147-53.
16.** Lamberti R, Canali G, Romero A, Ghezzi M, Zuccotti GV, D’Auria E. Congenital pulmonary airways malformation: state of the art review. Pediatric Respiratory Journal. 2023; 1: 120-263. Disponible en: https://doi.org/10.56164/PediatrRespirJ.2023.26.
17. Stanton M, et al. Lobectomy for congenital lung malformations: a comparison of open thoracoscopic approaches. J Pediatr Surg. 2003; 38: 1026-30.
18. Cabezalí Barbancho D, Cano Novillo I, García Vázquez A, López Díaz M, Tejedor Sánchez R, Benavent Gordo M, et al. Thoracoscopic Lobectomy in patients with congenital cystic adenomatoid malformation. Cir Pediatr. 2008; 21: 107-10.
19. Stanton M, Njere I, Ade-Ajayi N, Davenport M. Survey of management of congenital lung lesions in Europe. Eur J Pediatric Surg. 2017; 27: 404-9.
20.* Mondéjar López P, Alfonso Diego J. Malformaciones pulmonares. Tratado de Neumología Pediátrica 2019.
21. Oermann CM. Congenital pulmonary airway malformation. En: UpToDate. 2025.
22. Pattillo JC, Sáez J, Vuletin SF, Montero JI. Actualización y controversias en el tratamiento de las malformaciones congénitas de la vía aérea pulmonar. Neumol Pediatr. 2021; 16: 41-7.
23.* Oermann CM. Bronchopulmonary sequestration. En: UpToDate Redding G(Ed). 2025.
24. Laje P, Martínez-Ferro M, Grisoni E, Dudgeon D. Intraabdominal pulmonary sequestration. A case series and review of the literature. J Pediatr Surg. 2006; 41: 1309. Disponible en: https://doi.org/10.1016/j.jpedsurg.2006.03.049.
25.* Stanton M. The argument for a non-operative approach to asymptomatic lung lesions. Semin Pediatr Surg. 2015; 24: 183-6. Disponible en: https://doi.org/10.1053/j.sempedsurg.2015.01.014.
26. Benito González F, Expósito de Mena H. Malformaciones congénitas frecuentes de la vía aérea superior. Pediatr Integral. 2017; 7: 465-73. Disponible en: https://www.pediatriaintegral.es/publicacion-2017-10/malformaciones-congenitas-frecuentes-de-la-via-aerea-superior/.
Bibliografía recomendada
– Hegde BN, Tsao K, Hirose S. Management of Congenital Lung Malformations. Clin Perinatol. 2022; 49: 907-26. Disponible en: https://doi.org/10.1016/j.clp.2022.08.003.
Revisa el enfoque actual para el diagnóstico y manejo de las malformaciones congénitas pulmonares en recién nacidos y niños. Se abordan las principales malformaciones, como la MCVAP, el secuestro pulmonar, el enfisema lobar congénito y los quistes broncogénicos. Se analizan las opciones de tratamiento prenatal y posnatal, incluyendo cuándo se justifica la cirugía, y las técnicas quirúrgicas disponibles (especialmente la toracoscopia).
– Pederiva F, Rothenberg SS, Hall N, Ijsselstijn H, Wong KKY, von der Thüsen J, et al. Achiron R, Pio d´Adamo A, Schnater JM. Congenital lung malformations. Nat Rev Dis Primers. 2023; 9: 60. Disponible en: https://doi.org/10.1038/s41572-023-00470-1.
Este artículo ofrece una revisión de las malformaciones congénitas, aborda su origen, diagnóstico, manejo clínico y aspectos moleculares. Describe la controversia sobre tratar o vigilar las lesiones asintomáticas, así como la importancia del seguimiento a largo plazo por el posible riesgo de complicaciones.
– Lee EY, Dorkin H, Vargas SO. Congenital pulmonary malformations in pediatrics: review and update on etiology, classification, and imaging findings. Radiol Clin North Am. 2011; 49: 921-48. Disponible en: https://doi.org/10.1016/j.rcl.2011.06.009.
Revisa todas las malformaciones, enfocándose en su origen, clasificación y hallazgos por imagen, con énfasis en el papel diagnóstico de las técnicas avanzadas, tomografía y resonancia en la práctica clínica.
– Oermann CM. Bronchopulmonary sequestration. En: UpToDate Redding G(Ed). 2025.
Revisión donde se aborda la clasificación, diagnóstico por imagen y las opciones terapéuticas del secuestro pulmonar.
– Stanton M. The argument for a non-operative approach to asymptomatic lung lesions. Semin Pediatr Surg. 2015; 24: 183-6. Disponible en: https://doi.org/10.1053/j.sempedsurg.2015.01.014.
El artículo defiende la estrategia de la observación frente a la cirugía en lactantes con malformaciones congénitas asintomáticas, evaluando los riesgos del tratamiento quirúrgico.
– Mondéjar López P, Alfonso Diego J. Malformaciones pulmonares. Tratado de Neumología Pediátrica 2019.
Describe las malformaciones pulmonares congénitas, su epidemiología, las diferentes entidades, retos de definición y clasificación. Expone las controversias en el manejo de los casos asintomáticos.
– Lamberti R, Canali G, Romero A, Ghezzi M, Zuccotti GV, D’Auria E. Congenital pulmonary airways malformation: state of the art review. Pediatric Respiratory Journal. 2023; 1: 120-263. Disponible en: https://doi.org/10.56164/PediatrRespirJ.2023.26.
El artículo revisa el diagnóstico, tratamiento y seguimiento de la malformación congénita de la vía aérea pulmonar (MCVAP), así como sus formas clínicas y la controversia del manejo de los casos asintomáticos.
| Caso clínico |
|
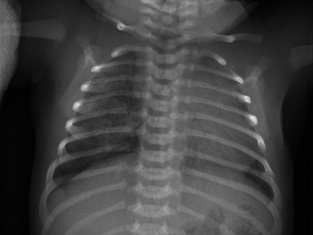
Gestante de 44 años a la que, en el control ecográfico del segundo trimestre, detectan una masa quística pulmonar de 21 x 19 x 17 mm, sin otras malformaciones asociadas. Cribado de cromosomopatías negativo y resto del embarazo sin incidencias, salvo hipotiroidismo subclínico. En controles subsiguientes persiste la masa con leve disminución de su tamaño sin asociar otros hallazgos patológicos. Nace un varón a término con llanto espontáneo, adecuado tono y coloración, con exploración física a las 8 horas de vida sin alteraciones. Ante hallazgos en ecografía prenatal, se solicita una radiografía de tórax que evidencia un aumento de densidad de morfología nodular, de unos 2 cm de diámetro, localizado en campos medios-inferiores derechos, que presenta imágenes radiolucentes milimétricas en su interior (Fig. 6). Es dado de alta sin incidencias a las 48 horas de vida.
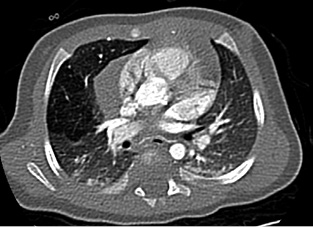
Figura 6. Radiografía del paciente en el primer día de vida. Se valora de forma presencial en consulta de neumología al mes de vida. El paciente no ha presentado infecciones respiratorias ni episodios de cianosis. Realiza tomas de manera correcta. Adecuado desarrollo ponderoestatural. Patrón respiratorio con rachas de polipnea sin tiraje. Para filiar mejor la lesión, se solicita TACAR, que se realiza a los 2 meses de vida, en el que se observa hipoatenuación e hipovascularización en lóbulo medio sin lesiones quísticas definidas. No efecto masa sobre estructuras mediastínicas ni parénquima pulmonar adyacente. Hallazgos compatibles con enfisema lobar (Fig. 7).
Figura 7. TACAR del paciente a los 2 meses de vida. Controles trimestrales durante el primer año de vida. El paciente permanece asintomático. Se deriva a cardiología para descartar cardiopatía estructural asociada. A los 9 meses, radiografía de control que muestra disminución del área hiperdensa respecto al estudio previo.
|


 Acute bronchiolitis and bronchitis in pediatrics
Acute bronchiolitis and bronchitis in pediatrics