|
| Temas de FC |
F. Benito González*, H. Expósito de Mena**
*Servicio de Otorrinolaringología y Patología Cráneo-Cervical. **Servicio de Pediatría. Unidad de vía aéreo-digestiva superior. Complejo Asistencial Universitario de Salamanca. Instituto de Investigación Biomédica de Salamanca
| Resumen
La vía aérea superior se extiende desde la abertura nasal hasta la subglotis y puede ser el asiento de múltiples tipos de malformaciones congénitas que conducen a su obstrucción anatómica o funcional. Esto puede causar problemas respiratorios graves, bien inmediatamente, en el momento del nacimiento, o bien en las primeras semanas de vida. En muchas ocasiones, se pueden asociar además problemas con la alimentación, como consecuencia de la obstrucción respiratoria o de una incoordinación de esta con la deglución, lo que puede conducir a una aspiración recurrente. Las anomalías congénitas de las vías respiratorias más frecuentes incluyen: laringomalacia, parálisis de las cuerdas vocales, estenosis subglótica, hendiduras laríngeas, hemangioma subglótico y atresia coanal. El conocimiento del desarrollo embriológico de la vía aérea superior y de las anomalías congénitas es de primordial importancia en la evaluación de la insuficiencia respiratoria del recién nacido. En la mayoría de los casos, el estudio endoscópico establece el diagnóstico. El manejo terapéutico se adaptará a cada condición y grado de severidad. |
| Abstract
The upper airway extends from the nasal aperture to the subglottis and can be the site of multiple types of congenital malformations leading to anatomical or functional obstruction. This can cause major respiratory problems either initially or during the first few weeks of life. Many such infants have associated feeding problems secondary to the airway obstruction and associating swallowing difficulties, which can lead to recurrent aspiration. More common upper airway congenital anomalies include laryngomalacia, vocal cord paralysis, subglottic stenosis, laryngeal clefts, subglottic hemangioma and choanal atresia. Knowledge of the embryological development of the upper airway and the congenital anomalies is of prime importance in assessing the newborn respiratory distress. In most cases the endoscopic study establishes the diagnosis. Management is tailored to each condition and its degree of severity. |
Palabras clave: Malformaciones congénitas vía aérea superior; Estridor; Respiración; Deglución; Aspiración
Key words: Congenital upper airway malformations; Stridor; Breathing; Swallowing; Aspiration
Pediatr Integral 2017; XXI (7): 465-473
Malformaciones congénitas frecuentes de la vía aérea superior
Introducción
Las malformaciones congénitas de la vía aérea superior pueden manifestarse tanto por problemas respiratorios, como por problemas para la alimentación, provocados por la obstrucción respiratoria o la incoordinación de esta con la deglución. Esto puede llevar a una aspiración recurrente que origine un daño pulmonar.
La vía aérea superior comprende la parte del tracto respiratorio situado entre la apertura nasal y la subglotis. Está constituida por las fosas nasales, la cavidad oral, la faringe y la laringe. Sus principales funciones son: la respiración, la deglución y la fonación. En el recién nacido, presenta una serie de características anatómicas que la hacen diferente de la del adulto: las fosas nasales son proporcionalmente más pequeñas; la lengua es más voluminosa y su tercio posterior se sitúa dentro de la cavidad oral, lo que hace, junto a un paladar óseo más cóncavo, que su movilidad esté más reducida; la faringe es más corta y la laringe se sitúa más alta y más anterior, lo que permite que el velo, la lengua y la epiglotis se aproximen, separándose funcionalmente el trayecto respiratorio del digestivo; y la laringe presenta una forma de embudo, de tal manera que la parte más estrecha de la vía aérea se encuentra a nivel del cartílago cricoides. Esta configuración condiciona la función, siendo la respiración del neonato fundamentalmente nasal, y facilitando el proceso de alimentación mediante la coordinación de los movimientos de succión, deglución y respiración.
La vía aérea superior puede ser el asiento de múltiples malformaciones congénitas que pueden originar problemas respiratorios agudos, bien inmediatamente al nacimiento o en los primeros días o semanas de vida. Muchos de los recién nacidos con estas alteraciones pueden sufrir, además, problemas con la alimentación, como consecuencia de la obstrucción respiratoria o de una incoordinación de esta con la deglución. Esto puede llevar a problemas en el crecimiento y, no infrecuentemente, a una aspiración recurrente que condicione un daño a corto o largo plazo en el tejido pulmonar.
Revisaremos las malformaciones congénitas más frecuentes en función del lugar anatómico de origen.
Anomalías laríngeas
Laringomalacia
La presencia de un estridor inspiratorio aislado no es criterio suficiente para el diagnóstico de una laringomalacia. Es necesaria la realización de un estudio endoscópico para poder concretar su diagnóstico.
La laringomalacia es la malformación congénita más frecuente de la laringe y la causa más frecuente de estridor en el neonato. Con la generalización del diagnóstico endoscópico, se postula que entre el 60 y el 70% de los casos de estridor neonatal secundarios a una malformación congénita de la laringe, son debidos a una laringomalacia(1).
Etiopatogenia
Se han descrito factores anatómicos, neurológicos e inflamatorios que contribuyen al desarrollo de una laringomalacia. Entre los factores anatómicos, se han identificado regularmente una serie de anomalías que incluyen: la epiglotis tubular, los repliegues ariepiglóticos cortos y la hipertrofia de la mucosa aritenoidea. Dentro de los neurológicos, se han enunciado: la inmadurez del sistema nervioso central o del control neuromuscular del soporte muscular de los cartílagos laríngeos. Los factores inflamatorios se relacionan con el reflujo gastroesofágico (observado en más del 60% de los casos), que induce un edema supraglótico que favorece la disminución del calibre laríngeo durante la inspiración, lo que, a su vez, incrementa la presión negativa intratorácica y el flujo retrogrado de contenido gástrico hacia el esófago y la faringe. La hipótesis de una inmadurez de los cartílagos laríngeos nunca ha sido verificada. Todas estas hipótesis son difíciles tanto de afirmar como de desmentir, debido principalmente al hecho de que bajo el término de laringomalacia se engloban entidades probablemente muy diferentes.
Clínica
Se caracteriza por un estridor inspiratorio de tonalidad aguda, vibrante y multifásico, que suele aparecer precozmente en las dos primeras semanas de vida. El estridor típicamente se incrementa con la agitación, el llanto, la alimentación o el decúbito supino y tiende a disminuir con la extensión de la columna cervical, la respiración calmada y el decúbito ventral. La sintomatología suele empeorar en los primeros meses de vida hasta estabilizarse en el sexto y después mejorar gradualmente, tendiendo a desaparecer hacia los 18-24 meses, aunque puede persistir durante años.
La mayoría de los casos de laringomalacia son leves (70-90%) y se manifiestan como un estridor aislado e intermitente, sin disnea y sin problemas de la deglución. Sin embargo, el 10-20% de los casos presentan signos de gravedad: disnea con tiraje intercostal o xifoideo importante y permanente, episodios de distrés respiratorio, apnea obstructiva durante el sueño, episodios de sofocación y cianosis durante la alimentación o dificultad para la misma y un retardo del crecimiento ponderal, que es el elemento probablemente más contributivo(2). Pueden asociarse lesiones laringotraqueales como: discinesias laríngeas, parálisis de las cuerdas vocales, estenosis subglóticas o traquebroncomalacias hasta en un 19% de los casos, si bien, son más frecuentes en las formas severas(2).
Diagnóstico
La presencia de un estridor inspiratorio aislado bien tolerado no es criterio suficiente para el diagnóstico de una laringomalacia. La existencia de un buen número de afecciones laríngeas que presentan esta clínica, algunas de las cuales precisan de un tratamiento específico, hace necesario la realización de un estudio endoscópico para poder concretar su diagnóstico.
La exploración laríngea de un lactante con un endoscopio flexible por vía nasal es un acto asequible en la consulta ORL y nos permite afirmar con certeza la presencia de una laringomalacia. Este gesto se realiza generalmente bajo una simple anestesia local de la fosa nasal y con el niño despierto, para poder valorar de forma dinámica, tanto la morfología como la movilidad. Los hallazgos endoscópicos que pueden ser observados de manera aislada o de forma combinada son: colapso más o menos completo del plano supraglótico durante la inspiración, que puede dificultar la visualización del plano glótico junto con unos repliegues aritenoepiglóticos cortos, colapso inspiratorio en báscula anterior y medial de la mucosa aritenoidea hipertrófica y desplazamiento inspiratorio posterior de la epiglotis hacia la pared faríngea posterior o inferiormente hacia las cuerdas vocales. La realización de un estudio endoscópico completo de la vía aérea, mediante una broncoscopia flexible y una endoscopia laríngea con óptica rígida bajo anestesia general, se reserva para los casos graves o con una disociación clínico-exploratoria(3).
Otras pruebas complementarias que pueden ser de utilidad en presencia de signos de gravedad son: una radiografía simple de tórax, un examen neurológico que descarte la presencia de patologías asociadas, monitorización con impedanciometría intraluminal multicanal – pH-metría (IIM-pH-metria) esofágica de 24 horas para descartar patología asociada a reflujo, una polisomnografía o un estudio de la deglución mediante observación de la ingesta bajo pulsioximetría, videofluoroscopia (VFSS) y/o fibroendoscopia (FEES).
Tratamiento
En los casos leves, se plantea una actitud expectante, con revisiones mensuales iniciales que pueden extenderse a cada 3-6 meses, según mejora la sintomatología. En los casos en los que se observan: tos, asfixia, regurgitación o dificultad para la alimentación, se establece la recomendación de iniciar una terapia de supresión ácida con inhibidores de la bomba de protones y/o antagonistas H2, si se demuestra reflujo ácido en IIM-phmetria, asociado a adaptación de la dieta con espesantes, tetinas y fórmulas especiales. En los casos en los que se observen signos de gravedad, además de dicha terapia, ha de valorarse el tratamiento quirúrgico mediante una supraglotoplastia, previo estudio endoscópico completo de la vía aérea(3). Algunos autores han propuesto la utilización de ventilación no invasiva (VNI), con diferentes interfases, reservándola para las laringomalacias aisladas o sindrómicas con signos de gravedad, cuando el tratamiento quirúrgico no puede ser propuesto o no ha sido suficientemente eficaz(2).
Diagnóstico diferencial
Es importante diferenciar las laringomalacias severas de las faringo-laringomalacias centrales. Esta entidad presenta un aspecto endoscópico característico con un colapso de las estructuras supraglóticas de apariencia anatómica normal, asociadas a un colapso faríngeo en inspiración y, frecuentemente, una glosoptosis. Clínicamente, presenta un distrés respiratorio que aumenta durante el sueño, asociado a problemas deglutorios con falsas vías y problemas de coordinación succión-deglución. El diagnóstico endoscópico precisa de la observación dinámica de la faringe y la laringe. El tratamiento quirúrgico endoscópico no ofrece mejoría, recomendándose el uso de VNI.
Estenosis subglótica
Los casos severos se presentan como un compromiso respiratorio en el nacimiento que requerirá intubación o traqueotomía. Los casos leves comienzan su clínica en relación con las infecciones de las vías aéreas y se asemejan a un crup laríngeo. La presencia de una sintomatología de crup prolongada o recurrente debe ponernos en alerta y plantearnos descartar su presencia.
La estenosis subglótica es una condición caracterizada por el estrechamiento de la vía aérea superior comprendida entre las cuerdas vocales y el borde inferior del cartílago cricoides. Pueden ser congénitas (20% de los casos), cuando se producen en ausencia de un trauma (intubación), o adquiridas (80% restante); si bien, un porcentaje de estas podrían considerarse mixtas, por la existencia de una estenosis previa a la intubación. Se considera que la estenosis subglótica congénita (ESGC) es la segunda malformación congénita de la laringe que más frecuentemente origina estridor(1).
Etiopatogenia
El cartílago cricoides deriva del sexto arco branquial y su luz se encuentra completamente canalizada hacia la 10ª semana de gestación. La existencia de un defecto embriogénico de recanalización de dicha luz puede dar lugar a un espectro de anomalías laríngeas, entre las que se incluyen: la atresia laríngea, la membrana laríngea y la estenosis subglótica.
Las ESGC pueden ser clasificadas histopatológicamente como membranosas y cartilaginosas. Las primeras presentan un incremento del tejido fibroso y/o una hiperplasia de las glándulas mucosas y tienen una típica forma circunferencial. Las cartilaginosas tienen una apariencia más variable, más comúnmente en forma elíptica, por prominencia de los laterales del cartílago, aunque, en ocasiones, podemos encontrar una apariencia normal, pero más estrecha.
Clínica
La clínica variará dependiendo de la severidad pero, en general, asocia estridor que, en este caso, es bifásico, tiraje, disfonía, tos perruna y problemas de alimentación. Los casos más severos suelen presentarse con compromiso respiratorio al nacimiento, que requerirá intubación o traqueotomía. Los casos leves suelen iniciar su sintomatología en relación con los procesos infecciosos de las vías respiratorias (que causan una inflamación que estrecha aún más la luz) y se asemejan a un crup laríngeo. La presencia de una sintomatología de crup prolongada o recurrente debe ponernos en alerta y plantearnos descartar la presencia de una ESGC(4).
Diagnóstico
El estudio endoscópico es clave ante su sospecha. La microlaringoscopia y broncoscopia con óptica rígida permite identificar el nivel de la obstrucción. Para valorar su severidad, utilizaremos la escala de graduación de Myer-Cotton(5). Esta escala se basa en la comparación entre el tubo endotraqueal más grande que puede ser colocado durante la intubación del paciente caso y que presenta una fuga de aire a su alrededor con una presión de ventilación de 20 cm de agua (objetivada mediante endoscopia) y el tubo endotraqueal apropiado para la edad de ese paciente. Así, se establece que una estenosis grado 1 corresponde a una luz <50%, grado 2 entre 51-70%, grado 3 entre 71-99% y grado 4 a una obstrucción completa.
Tratamiento
La actitud terapéutica variará dependiendo del grado de estenosis. Teniendo en cuenta que un gran número de estenosis congénitas se resuelven espontáneamente con el crecimiento, la actitud expectante es la norma habitual para las estenosis grado 1 y ciertas 2. Esto supone un seguimiento minucioso para descartar una evolución desfavorable, así como el uso de corticoides y/o terapia de supresión ácida para el tratamiento de las infecciones respiratorias y/o del reflujo gastroesofágico que puedan deteriorar la situación.
Los grados 3 y 4 y ciertos tipos 2 precisan una actitud intervencionista. En la actualidad, existen una amplia gama de procedimientos, tanto endoscópicos como abiertos, que puede ser considerados suplementarios. La dilatación laríngea con balones de alta presión es, hoy en día, una herramienta imprescindible en el manejo de las ESGC y ha reducido la necesidad del tratamiento abierto en más de un 80%(4). Los procedimientos quirúrgicos abiertos se pueden catalogar en tres grupos: cirugía de expansión (reconstrucción laringotraqueal con cartílagos costal o tiroideo y división cricoidea anterior), cirugía de resección (resección cricotraqueal) o traqueoplastia por deslizamiento.
Parálisis laríngea
Las formas bilaterales presentan una disnea inspiratoria con tiraje y estridor continuos, pudiendo llegar a comportarse como una urgencia vital. En las unilaterales, la disnea es rara, mientras que la disfonía es más significativa. Los problemas de alimentación están presentes en ambos tipos.
Se entiende por parálisis laríngea la alteración del control nervioso motor de la laringe, ya sea de origen central (tronco del encéfalo) o periférico (nervio vago o nervio recurrente), que afecta a una o ambas cuerdas vocales. Pueden ser de origen congénito o adquirido. Representan el 10% de las anomalías congénitas de la laringe y se consideran la tercera malformación congénita de la laringe que con más frecuencia origina estridor en el niño. La proporción de parálisis cordales uni y bilaterales es del 50%(1).
Etiología
Las formas bilaterales congénitas suelen ser de origen neurológico (relacionadas con: meningo-encefaloceles, malformaciones de Arnold-Chiari, agenesias de cuerpo calloso, sufrimiento cerebral neonatal, enfermedades de Werdnig-Hoffman, degeneraciones espino-musculares hereditarias o hidrocefalia congénita) o de origen idiopático. Las formas unilaterales congénitas suelen estar relacionadas con malformaciones cardiacas (dilataciones auriculares y/o ventriculares) o malformaciones vasculares (síndrome de Ortner), que inducen una parálisis por compresión nerviosa. Ciertos trastornos musculares, como puedan ser algunas miopatías (la enfermedad de Leigh o la miastenia congénita) pueden debutar por una parálisis laríngea unilateral.
Clínica
Las parálisis bilaterales se presentan como una disnea inspiratoria con tiraje y estridor continuos que se agravan con los esfuerzos, como la alimentación y el llanto. Pueden llegar a comportarse como una urgencia vital con cianosis, bradicardia y apnea. La fonación es prácticamente normal. En las parálisis unilaterales, los síntomas son menos llamativos. La disnea es rara y el estridor menos frecuente, mientras que la disfonía es más significativa. Los problemas de alimentación están presentes en ambos tipos pero, mientras que en los casos de las bilaterales se deben a una alteración de la coordinación deglución-respiración, en las unilaterales se relacionan con falsas vías por la asociación con la afectación del nervio laríngeo superior (sensitivo).
Diagnóstico
El examen nasofibroscópico de la laringe con el niño reactivo es el examen clave para el diagnóstico y nos mostrará la parálisis o la hipomovilidad cordal uni o bilateral. Una vez detectada, ha de completarse el estudio con una prueba de imagen cráneo-cérvico-torácico mediante RMN, para evaluar la posible asociación de anomalías cardiacas o neurológicas.
Tratamiento
Teniendo en cuenta que un porcentaje elevado de casos se resuelven bien espontáneamente o tras el tratamiento de la causa que las origina, se propone una actitud expectante durante, al menos, dos años. En los casos bilaterales con intensa disnea o asfixia, puede ser necesaria una traqueotomía de urgencia. Se han planteado diversas y variadas técnicas quirúrgicas para su reparación (lo que sugiere que ninguna es ideal) con el objetivo de mejorar el componente respiratorio sin deteriorar los problemas deglutorios preexistentes ni alterar significativamente la fonación.
Diastemas laríngeos
Ante la presencia de una tos en relación con la ingesta, la sospecha de una aspiración o una neumopatía recurrente no filiada, es necesario descartar una hendidura laríngea.
Los diastemas o hendiduras laríngeas posteriores son una malformación congénita rara (1 de cada 20.000 recién nacidos) que ocurre como consecuencia de un defecto de fusión de las dos láminas laterales del cartílago cricoides asociado o no a un defecto de cierre medio de la membrana traqueal. La mayoría de los casos son aislados, mientras que algunos asocian anomalías, como fístulas traqueoesofágicas, o síndromes, como el G/Opiz-Frias o el Pallister-Hall(6).
Clínica
En función de la extensión de la hendidura, podemos diferenciar cuatro tipos que presentarán diferencias clínicas. El tipo I supone un defecto interaritenoideo que alcanza el nivel de las cuerdas vocales, el tipo II se extiende parcialmente a través del cricoides, el tipo III lo hará completamente alcanzando la tráquea cervical y el tipo IV se extenderá a la tráquea torácica. En las tipo I, la sintomatología será moderada con presencia de falsas vías y tos principalmente con líquidos, con escasa o nula repercusión pulmonar. En las tipo II y III, la sintomatología será más marcada, con falsas vías masivas y neumonías recurrentes por aspiración. Por último, las tipo IV suponen un pronóstico sombrío debido a la dificultad respiratoria secundaria a la inundación bronquial por secreciones y a los problemas para asegurar la ventilación.
Diagnóstico
Su diagnóstico requiere de una alta capacidad de sospecha. Ante la presencia de una tos en relación con la ingesta, la sospecha de una aspiración o una neumopatía recurrente no filiada, se ha de descartar la presencia de una hendidura laríngea. La realización de una nasofibroscopia de la laringe puede no ser suficiente, ya que, en ocasiones, pasan desapercibidas, por lo que es obligado realizar una microlaringoscopia con óptica rígida con palpación sistemática de la región interaritenoidea, lo que permitirá afirmar el diagnóstico y determinar la profundidad de la hendidura. Otras pruebas de interés serían el estudio de la deglución mediante observación de la ingesta bajo pulsioximetría, videofluoroscopia (VFSS) y/o fibroendoscopia (FEES) y la TAC.
Tratamiento
Su objetivo conlleva, tanto la resolución de las limitaciones alimentarias como la minimización de las complicaciones respiratorias y comprende, tanto aspectos conservadores como procedimientos quirúrgicos. El tratamiento conservador engloba, tanto terapia nutricional (modificaciones de la consistencia y de la postura, y terapia orofacial por parte del logopeda especialista en rehabilitación miofuncional que enseñe las diferentes maniobras compensatorias de deglución), como optimización del estatus respiratorio o medicación antirreflujo si se precisa. La corrección quirúrgica puede realizarse mediante un abordaje endoscópico o por cirugía abierta. La sutura por vía endoscópica se ha convertido en la de elección para los tipos I, II y ciertos III, por sus excelentes resultados y su baja morbi-mortalidad, reservándose los abordajes mediante laringofisura anterior, faringotomía lateral o toracotomía para el resto de casos(6). A pesar de una alta tasa de éxito quirúrgico, una minoría sustancial de niños tienen disfunción persistente de la deglución después de la reparación de la hendidura laríngea, por lo que se recomienda realizar estudios de deglución después de su reparación(7).
Hemangiomas subglóticos
Sus síntomas pueden confundirse con facilidad con una laringitis subglótica recidivante del neonato de menos de seis meses.
Los hemangiomas subglóticos son una malformación vascular congénita benigna que deriva de los restos mesodérmicos. Son poco comunes y suponen el 1,5% de todas las anomalías congénitas de la laringe, con una preponderancia femenina. El 50% de los casos diagnosticados presentan lesiones cutáneas asociadas(1).
Clínica
Siguiendo la evolución natural de los hemangiomas infantiles, en el recién nacido suele ser asintomático, iniciándose la clínica en los primeros seis meses de vida, coincidiendo con la fase proliferativa. A partir de los 12-18 meses, comienza la fase de involución, lo que estabiliza la clínica, que acabará remitiendo a los 5-7 años de vida. Los síntomas incluyen estridor bifásico, accesos de tos tipo crup, disfonía variable y fallo de medro. La forma de presentación puede confundirse con facilidad con una laringitis subglótica recidivante del neonato de menos de seis meses.
Diagnóstico
La presencia de lesiones cutáneas o cérvico-faciales y la fibroendoscopia laríngea pueden hacer sospechar su diagnóstico. La microlaringoscopia con óptica rígida permitirá la confirmación, al observarse una masa subglótica submucosa, asimétrica, depresible, de aspecto angiomatoso o simplemente congestivo y de predominio lateral o posterolateral izquierdo, que no es necesario biopsiar. El estudio de imagen con RMN puede ayudar a concretar la extensión.
Tratamiento
Los excelentes datos publicados en los últimos años en el manejo de los hemangiomas subglóticos con propanolol y su bajo índice de complicación en comparación con los tratamientos quirúrgicos, han convertido a este en la primera línea de tratamiento(8). Una vez iniciado el tratamiento, debe monitorizarse la respuesta y mantenerse durante, al menos, 15 meses. Otros tratamientos, como los corticoides sistémicos a altas dosis, el interferón alfa-2a han quedado en desuso. La resección quirúrgica láser, con microdebridador o por cirugía abierta y la traqueotomía han quedado reservadas para los casos no respondedores.
Otras anomalías laríngeas
Las membranas laríngeas son malformaciones poco comunes causadas por un fallo en la recanalización de la laringe durante la embriogénesis. La mayoría afectan al plano glótico, aunque pueden tener una extensión subglótica. La sintomatología dependerá del tamaño y localización de la membrana, pudiendo variar desde una disfonía leve (en la mayoría de los casos que afectan a la región anterior) a una obstrucción de la vía aérea con estridor y disnea (en los casos que afecten a más del 75% de la luz o a la región posterior interaritenoidea). Su manejo variará en función de la clínica y la extensión, pudiendo plantearse una actitud expectante o una sección endoscópica con láser o bisturí frío, en las membranas glóticas anteriores finas, o una sección amplia con interposición de un stent en la quilla laríngea que evite recidivas, en las membranas más gruesas. Se ha planteado incluso la laringoplastia abierta si existe afectación subglótica.
La atresia laríngea es la forma extrema de estenosis y es incompatible con la vida. Se manifiesta al nacimiento como disnea obstructiva con signos de lucha. Asocia, habitualmente, anomalías traqueales y esofágicas. El diagnóstico ecográfico prenatal ha permitido la realización de procedimientos EXIT (ex-utero intrapartum therapy) descritos en la literatura(9).
Los quistes laríngeos son una patología mucho más rara que las estenosis y pueden ser divididos en dos grandes grupos: los quistes apendiculares, verdaderos quistes congénitos, que se desarrollan a partir del extremo anterior del ventrículo laríngeo, mostrándose como una masa a nivel de la banda ventricular; y los quistes canaliculares, debidos a la obstrucción del canal excretor de una glándula submucosa y que prevalecen a nivel de la epiglotis o el repliegue ari-epiglótico. La sintomatología variará en función del volumen del quiste y suele aparecer tardíamente, como un estridor progresivo asociado a una disnea que varía con la posición de la cabeza. El diagnóstico es endoscópico y el tratamiento consiste en la marsupialización o exéresis del mismo.
La reanimación de recién nacidos, cada vez más prematuros, conlleva la aparición cada vez más frecuente de quistes subglóticos de retención, que muestra una sintomatología similar a la de una estenosis postintubación(10). La microlaringoscopia con óptica rígida permite comprobar la presencia de uno o varios quistes en las paredes de la subglotis. Su tratamiento consiste en su marsupialización con láser.
Anomalías nasales
Atresia de coanas
Las formas bilaterales presentan un distrés respiratorio con disnea inspiratoria que empeora con la alimentación. Las unilaterales pueden pasar desapercibidas al nacimiento. Puede formar parte de síndromes polimalformativos, entre los que destaca la asociación CHARGE.
La atresia coanal (AC) es una malformación congénita, de tipo imperforación, de la parte posterior de la cavidad nasal. Su incidencia se estima entre 1 por 7.000 a 8.000 recién nacidos y es considerada la malformación nasal más frecuente. Es más prevalente en el sexo femenino. Alrededor de las dos terceras partes de los casos son unilaterales. El 50% de los casos de AC suele acompañarse de otro tipo de malformaciones y, en este caso, son más frecuentes las formas bilaterales(11).
Embriopatogenia
Se han propuesto diferentes teorías embriológicas, si bien, la más aceptada es la atribuida a un fallo en la reabsorción de la membrana buco-faríngea hacia la cuarta semana de desarrollo embriológico. Se estima que, entorno al 30% de ellas son puramente óseas y el 70% restantes mixtas, membranosas-óseas, negándose la existencia de formas puramente membranosas.
Clínica
En las bilaterales, el neonato presentará típicamente un distrés respiratorio con episodios de disnea inspiratoria que cederán con la apertura bucal. Esta situación se verá agravada por la alimentación y mejorará durante el llanto. Las unilaterales pueden pasar desapercibidas al nacimiento, sospechándose mucho más tarde ante la presencia de una rinorrea crónica no purulenta asociada a una obstrucción nasal completa unilateral. La AC puede formar parte de síndromes polimalformativos, entre los que destaca, por su frecuencia, la asociación CHARGE, o de otros síndromes como: Treacher-Collins, Apert, Di George, Crouzon, Kallmann, VATER o Velo-cardio-facial.
Diagnóstico
La práctica clínica actual desaconseja el paso sistemático de sondas nasogástricas para descartar atresias en el recién nacido sano, por lo que solo ha de realizarse esta ante una sospecha razonada. En este caso, se planteará su existencia ante la imposibilidad de avanzar una sonda de calibre 5 o 6 French más allá de 32 mm desde la narina. La nasofibroscopia flexible o la endoscopia nasal con óptica rígida de pequeño tamaño (2,7 mm) completan el diagnóstico. La TAC resulta obligada en el contexto preoperatorio y, además de confirmar la atresia, nos informará de su localización y del componente óseo y membranoso.
Tras el hallazgo de una AC, se debe realizar una valoración multidisciplinar en busca de otras malformaciones asociadas o síndromes polimalformativos. A la inversa, la sospecha de un síndrome polimalformativo evocador, como una asociación CHARGE, nos obliga a descartar la presencia de una AC.
Tratamiento
El tratamiento de la AC bilateral debe ser considerado una urgencia neonatal y puede abordarse mediante la inserción de una cánula de Guedel o un chupete de McGrovern para preservar la vía aérea y la utilización de sondas orogástricas para garantizar la alimentación. El tratamiento definitivo en ambos tipos es el quirúrgico. En las formas bilaterales, debe realizarse lo antes posible. Se han descrito dos vías principales de abordaje: la vía transnasal y la vía transpalatina. La vía transnasal con ayuda de instrumentos endoscópicos es la de elección en la actualidad, ya que permite una resección segura de la placa atrésica sin daño de las estructuras vecinas. Tradicionalmente, la intervención se sigue de la colocación de calibradores durante un periodo de varias semanas, aunque una alternativa es la realización de dilataciones neumáticas semanales durante 6 semanas. El abordaje transpalatino es más invasivo y se reserva para los fracasos del abordaje endonasal, dado el mayor riesgo de lesión vascular y de un desarrollo anómalo de la arcada dental superior. La tasa de re-estenosis, independientemente de la técnica utilizada, alcanza el 20%(12,13).
Otras causas de obstrucción nasal congénita
La hipoplasia congénita del orificio piriforme (apertura ósea anterior de la fosa nasal formada superiormente por los huesos nasales y lateral e inferiormente por el hueso maxilar) es una causa poco frecuente de obstrucción de la vía aérea, por lo que suele ser infradiagnosticada. Clínicamente, se presenta como un distrés respiratorio neonatal con respiración ruidosa que, al igual que la atresia de coanas, empeora con la alimentación y mejora con el llanto. Un 40% de los casos asocian otras malformaciones craneofaciales: megaincisivo central único, holoprosencefalia o anomalías de la pituitaria(14). La rinoscopia revela un estrechamiento anterior de la fosa nasal que imposibilita la realización de una nasofibroscopia. La TAC confirma el diagnóstico cuando la medida del orificio piriforme es inferior a 11 mm. Un manejo inicial con la aplicación de gotas de suero con adrenalina o, en su defecto, con el uso de una cánula de Guedel o de un chupete de McGrovern con colocación de una sonda orogástrica, puede ser útil en casos poco severos, en espera de su resolución con el crecimiento. El tratamiento quirúrgico consiste en la ampliación de la apertura piriforme mediante el fresado submucoso por vía sublabial del proceso nasal del maxilar(13).
Los quistes del conducto lacrimonasal se manifiestan por la asociación de dificultad respiratoria nasal con una dacriocistitis o una masa situada cerca del canto interno. El diagnóstico se realizará por la observación de una masa por debajo del cornete inferior en la nasofibroscopia y el tratamiento consistirá en la marsupialización endoscópica intranasal.
Síndromes congénitos que afectan a la cavidad oral
y a la faringe
La secuencia de Pierre-Robin se caracteriza por: micrognatia, glosoptosis y obstrucción de la vía aérea superior. Puede aparecer aisladamente o formando parte de síndromes craneofaciales, como el de Stickler, Nager y Treacher Collins.
Uno de los más frecuentes es la secuencia Pierre-Robin. Descrita en 1923 por Robin, se caracteriza por una tríada de síntomas que incluyen: micrognatia, glosoptosis y obstrucción de la vía aérea superior(15). Puede asociar hendidura palatina en un 50% de los casos. Se teoriza que esta colección de síntomas es el resultado de un evento de incitación y, por lo tanto, se refiere más como una secuencia que como un síndrome. No se ha comprobado su etiología, pero se cree que es debida a un defecto intrínseco o extrínseco del crecimiento mandibular. Esta alteración detiene la normal rotación y el avance de la lengua que, al no descender, impide el cierre palatino. Además, la lengua permanece presionada contra la faringe, lo que conduce a un compromiso respiratorio. Hasta la fecha, no ha habido ningún gen humano específico asociado con la secuencia de Pierre Robin. Sin embargo, la secuencia tiene una fuerte correlación con otros síndromes craneofaciales, en particular, con los de Stickler, Nager y Treacher Collins. La obstrucción de la vía aérea superior puede conducir a una hipoxia, una hipertensión pulmonar y una insuficiencia cardiaca derecha, además de a un fallo de medro, debido a los mayores requerimientos energéticos necesarios para sostener el trabajo respiratorio extra y a la dificultad de alimentación secundaria a los problemas de coordinación succión, deglución y respiración. El crecimiento facial, que se produce entre los 3 y 12 meses de edad, conduce a la resolución del problema respiratorio. Hasta entonces su manejo dependerá de la severidad. Se pueden plantear desde simples medidas posturales, como el decúbito prono, a opciones no quirúrgicas, como el uso de tubos nasofaríngeos, ventilación no invasiva y nutrición enteral por SNG (que, además, protegerá del riesgo de aspiración) o, en casos más severos, tratamientos quirúrgicos, como la distracción mandibular, la glosopexia, la hiomandibulopexia o, en último caso, la traqueotomía(16).
La macroglosia que caracteriza al síndrome de Beckwith-Wiedemann, junto con el onfalocele, la visceromegalia y la citomegalia del cortex adrenal, puede dar lugar a problemas respiratorios. En muchos pacientes, la macroglosia suele regresar. Si se precisa una actuación sobre la vía aérea superior es preferible la realización de una traqueotomía sobre cualquier cirugía de lengua durante los primeros años de vida(17).
Función del pediatra de Atención Primaria
Las anomalías de la vía aérea son poco frecuentes y su atención se realizará, en su gran mayoría, en centros hospitalarios pediátricos. Sin embargo, es importante su conocimiento por parte del pediatra de Atención Primaria para poder sospechar su diagnóstico. Efectivamente, la laringomalacia es la causa más frecuente de estridor laríngeo en el niño y puede no manifestarse hasta las primeras semanas de vida. No todo estridor inspiratorio aislado bien tolerado es criterio suficiente para el diagnóstico de una laringomalacia. Otros casos de malformaciones leves, como la estenosis subglótica, los diastemas laríngeos o los hemangiomas subglóticos pueden pasar desapercibidos en el momento del nacimiento y dar a medio-largo plazo una clínica de tos crónica, episodios repetidos de laringitis-crup, neumonías de repetición o asma de difícil control. La observación de una toma, a ser posible bajo pulsioximetría, en la consulta de Atención Primaria, puede ser útil al poner de manifiesto alteraciones sugerentes de aspiración como tos y/o desaturación. La presencia, tanto de una clínica respiratoria recurrente como de hallazgos en la observación de la ingesta, debe hacernos sospechar la presencia de una malformación de la vía aérea y, por tanto, remitir el caso a un centro hospitalario para realizar estudio etiológico que incluya un estudio endoscópico.
Bibliografía
Los asteriscos reflejan el interés del artículo a juicio del autor.
1.*** Gerber ME, Holinger LD. Congenital laryngeal anomalies. En: Bluestone CD, eds. Pediatric otolaryngology. Elsevier Science (USA); 2003. p. 1460-72.
2.*** Ayari S, Aubertin G, Girsching H, Van den Abbeele T, Mondain M. Laryngomalacie. En: Denoyelle F, Couloigner V, Froehlinch P, Nicollas R. Le larynx de l´enfant. Rapport de la Société Française d´Oto-rhino-laryngologie et de Chirurgie de la Face et du Cou; 2011. p. 303-32.
3.*** Carter J, Rahbar R, Brigger M, et al. International pediatric ORL Group (IPOG) laryngomalacia consensos recommendations. International Journal of Pediatric Otorhinolaryngology. 2016: (86): 256-61.
4.*** Jefferson ND, Cohen AP, Rutter MJ. Subglottic stenosis. Seminars in Pediatric Surgery. 2016; (25): 138-43.
5. Myer CM, O´Connor DM, Cotton RT. Proposed grading system for subglottic stenosis based on endotracheal tube size. Ann Otol Rhinol Laryngol. 1994; 103: 319-23.
6.*** Strychowsky JE, Rahbar R. Laryngotracheoesophageal clefts. Seminars in Pediatric Surgery. 2016; (25): 128-31.
7. Osborn AJ, de Alarcon A, Tabangin ME, Miller CK, Cotton RT, Rutter MJ. Swallowing function after laryngeal cleft repair: more than just fixing the cleft. Laryngoscope. 2014; 124: 1965-9.
8. Elluru RG, Fiess MR, Richter GT, et al. Multicenter evaluation of the effectiveness of systemic propanolol in the treatment of airway hemangiomas. Otolaryngol Head Neck Surg. 2015; 153: 452-60.
9. Shimabukuro F, Sakumoto K, Masamoto H, et al. A case of congenital high airway obstruction syndrome managed by ex utero intrapartum treatment: case report and review of the literature. Am J Perinatol. 2007; 24: 197-201.
10.*** Roger G. Malformations congénitales du Larynx. En: Garabédian EN, Bobin S, Montiel JP, Triglia JM, eds. ORL de l´enfant. Médicine-Sciences Flammarion; 2006. p. 221-8.
11.*** Triglia JM, Garabédian EN, Roman S. Atrésie choanal. En: Garabédian EN, Bobin S, Montiel JP, Triglia JM, eds. ORL de l´enfant. Medicine-Sciences Flammarion; 2006. p. 151-5.
12.*** Daniel SJ. The upper airway: Congenital malformations. Pediatric respiratory review. 2006; 7s: 260-3.
13.** Szeremeta W, Parikh TD, Widelitz JS. Congenital nasal malformations. Otolaryngol Clin North Am. 2007; 40: 97.
14.** Van Den Abbeele T, Triglia JM, François M, Narcy P. Congenital nasal pyriform apertura stenosis: diagnosis and management of 20 cases. Ann Otol Rhinol Laryngol. 2001; 1: 70-5.
15. Robin R. Backward lowering of the root of the tongue causing respiratory disturbances. Bull Acad Natl Med. 1923; 89: 37-41.
16.** Buchanan EP, Xue AS, Hollier LH Jr. Craneofacial syndromes. Plast Reconstr Surg. 2014; 134: 128-53.
17.** Muntz HR, Gray SD. Congenital malformations of the mouth and pharynx. En: Bluestone CD, eds. Pediatric otolaryngology. Elsevier Science (USA); 2003. p. 1149-61.
Bibliografía recomendada
– Bluestone CD, eds. Pediatric otolaryngology. Elsevier Science (USA). 2003.
Tratado de referencia ORL pediátrica en inglés, escrito por expertos mundiales, que desarrolla de forma amplia todas las enfermedades pediátricas con implicación ORL.
– Garabédian EN, Bobin S, Montiel JP, Triglia JM, eds. ORL de l´enfant. Medicine-Sciences Flammarion, 2006.
Libro de referencia en ORL pediátrica, escrito por la escuela francesa, en el que se desarrolla de forma escueta y sencilla las enfermedades ORL del paciente pediátrico.
– Denoyelle F, Couloigner V, Froehlinch P, Nicollas R. Le larynx de l´enfant. Rapport de la Société Française d´Oto-rhino-laryngologie et de Chirurgie de la Face et du Cou. 2011.
Ponencia de la Sociedad Francesa de ORL y Cirugía Cérvico-Facial en la que se desarrollan de forma extensa, las diferentes patologías de la laringe del niño.
– Carter J, Rahbar R, Brigger M, et al. International pediatric ORL Group (IPOG) laryngomalacia consensos recommendations. International Journal of Pediatric Otorhinolaryngology. 2016; (86): 256-61.
Reciente consenso en el que se enuncian algoritmos muy útiles para el manejo diagnóstico y terapéutico de la laringomalacia.
– Jefferson ND, Cohen AP, Rutter MJ. Subglottic stenosis. Seminars in Pediatric Surgery. 2016; (25): 138-43.
Completo artículo de revisión sobre la etiología, diagnóstico y tratamiento de las estenosis subglóticas.
– Strychowsky JE, Rahbar R. Laryngotracheoesophageal clefts. Seminars in Pediatric Surgery. 2016; (25): 128-31.
Interesante artículo sobre el diagnóstico y actitud terapéutica de las hendiduras laríngeas.
| Caso clínico |
|
Paciente de 3 años remitida por su pediatra a neumología infantil por episodios de bronconeumonías de repetición en los últimos 6 meses. Presenta episodios recurrentes consistentes en tos, mucosidad, fiebre alta, crepitantes e imágenes en la radiografía de tórax de aumento de densidad radiológica en lóbulo inferior derecho e izquierdo, que precisan para su resolución antibioterapia oral. Antecedentes personales Seguimiento en Unidad de Fibrosis Quística (FQ) hasta los 2 años por screening metabólico positivo (tripsina inmunorreactiva positiva, dos determinaciones), genotipo FQ y test del sudor por conductancia negativo (35 mmol/L). Desarrollo ponderoestatural y psicomotor adecuado. Contacto reciente con un caso de adenitis subaguda granulomatosa, compatible con infección con Micobacterium Avium Complex. Antecedentes familiares Padre fumador, en estudio actual por intradermorreacción tuberculina positiva (10 mm). En la exploración física, presenta constantes adecuadas (FC 90 lpm, FR 15 rpm y saturación: 97-98%) con buen estado general y de nutrición. Fenotipo normal, sin malformaciones torácicas evidentes. Eupneica. Auscultación pulmonar crepitante en base derecha. No hipertrofia adenoidea. Se solicita control analítico con serologías, quantiferon y mantoux, todo ello negativo. Test del sudor por cloridometria negativo (14 mmol/L). Detección de virus respiratorio sincitial en secreción respiratoria positivo. El estudio microbiológico del esputo muestra inicialmente una tinción de auramina para bacterias ácido alcohol resistentes (BAAR) positiva, por lo que se inicia tratamiento antituberculoso con pauta de 4 fármacos a la espera de cultivo definitivo. Continúa presentando exacerbaciones respiratorias recurrentes, con aislamiento de Heaemophilus Influenzae en esputo repetidamente, precisando numerosos ingresos hospitalarios. Se amplía estudio: inmunológico, cardiológico, secuenciación completa del gen de FQ, espirometría y pulsioximetría nocturna, todos dentro de la normalidad. Se mantiene tratamiento antituberculoso durante 3 meses hasta confirmación de cultivos de esputo negativos para micobacterias. Requiere ingreso en Unidad de Cuidados Intensivos pediátricos a los 4 años por empeoramiento para manejo respiratorio, con ventilación mecánica no invasiva. Se completa estudio con TAC torácico, donde se objetivan bronquiectasias basales bilaterales (Fig. 1) y fibrobroncoscopia con laringoscopia directa, donde se visualiza inflamación y secreción purulenta en árbol endobronquial y defecto interaritenoideo que alcanza el nivel de las cuerdas vocales.
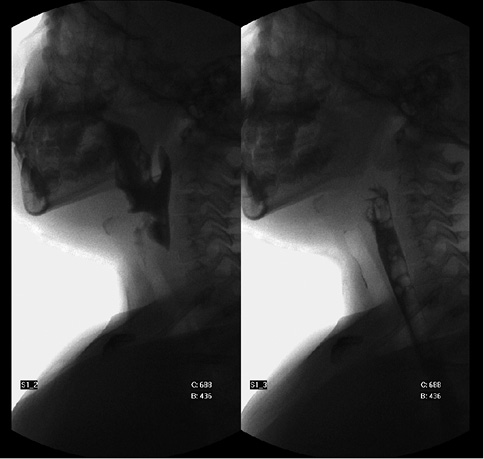
Figura 1. TAC torácico: bronquiectasias en la región posterobasal izquierda, con coexistencia de pequeños aumentos de densidad en posible relación con sobreinfección de las mismas y pequeñas bronquiectasias en base derecha. Ante estos hallazgos, se realiza videofluoroscopia, constatando paso de contraste líquido a la vía aérea con tos y desaturación asociada (Fig. 2).
Figura 2. Videofluroscopia: paso de contraste líquido a vía aérea. Se realiza corrección quirúrgica mediante microsutura endoscópica del defecto por parte del servicio de ORL. La evolución posterior fue favorable, disminuyendo el número de exacerbaciones respiratorias y con normalización de la videofluoroscopia.
|