 |
| El Rincón del Residente |
Coordinadores:
M. García Boyano*, S. Criado Camargo*, J.A. Soler Simón**, L. García Espinosa*
**Hospital Universitario Infantil La Paz. Madrid. **Hospital Universitario Infantil Niño Jesús. Madrid.
Autores:
V. Martí Enrique*, J.M. Olmos García**
*Médico Interno Residente Pediatría y sus áreas específicas. Hospital Mare de Déu dels Lliris. Alcoy. Alicante.
**Facultativo Especialista de Pediatría y sus áreas específicas. Hospital Mare de Déu dels Lliris. Alcoy. Alicante
|
El Rincón del Residente es una apuesta arriesgada de Pediatría Integral. No hemos querido hacer una sección por residentes para residentes. Yendo más allá, hemos querido hacer una sección por residentes para todo aquel que pueda estar interesado. Tiene la intención de ser un espacio para publicaciones hechas por residentes sobre casos clínicos e imágenes entre otras. |
Pediatr Integral 2024; XXVIII (2): 126.e1 – 126.e2
Imagen en Pediatría Clínica.
Haz tu diagnóstico
Adolescente con piel sucia persistente
Niño de 13 años que acude a consulta por manchas cutáneas marronáceas que presenta desde la infancia. Refiere que en los últimos meses se han vuelto más oscuras y extensas, asociando prurito de forma ocasional.
Como antecedentes obstétricos, nació mediante parto vaginal con expulsivo dificultoso y prolongado, aunque sin signos de sufrimiento fetal; test de Apgar: 9/10. Al nacimiento no se objetivaron lesiones cutáneas ni criptorquidia ni otras malformaciones asociadas. El paciente no tiene otros antecedentes personales de interés para el caso. Entre los antecedentes familiares no existen antecedentes de lesiones cutáneas, tiene una hermana sana y los padres no son consanguíneos.
A la exploración física, presenta placas marronáceas ictiosiformes poligonales en tronco y extremidades, más abundantes en región extensora de extremidades. Aspecto seco e hiperqueratosis asociada. Se objetiva en pliegues cutáneos aspecto de suciedad debido a la confluencia de dichas placas.
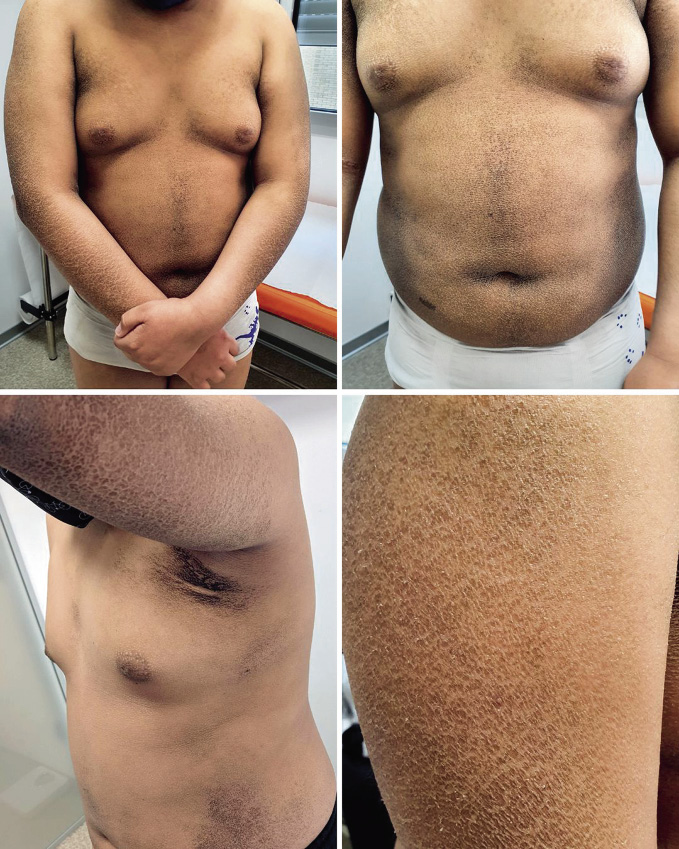
Figura 1. Lesiones oscuras con escamas e hiperqueratosis en zonas extensoras, axilas y tronco.
¿Cuál es el diagnóstico?
a. Ictiosis vulgar.
b. Síndrome de Gaucher tipo 2.
c. Ictiosis ligada al cromosoma X.
d. Acantosis nigricans.
e. Síndrome de la piel exfoliada.
Respuesta correcta: c. Ictiosis ligada al cromosoma X.
Comentario
El caso descrito se corresponde con una ictiosis, ya que el paciente tiene hiperqueratosis e hiperpigmentación con sequedad y descamación, que simulan las “escamas” características del diagnóstico.
El principal diagnóstico diferencial es la acantosis nigricans, que se caracteriza por engrosamiento cutáneo e hiperpigmentación; sin embargo, las lesiones son papilomatosas y aterciopeladas al tacto, con ausencia de descamación. Aparecen en el cuello, la región inguinal, antecubital o poplítea. Se relaciona con la resistencia a la insulina principalmente, aunque también con síndromes genéticos o tumores malignos, siendo estos casos excepcionales(1).
Las ictiosis comprenden un grupo heterogéneo de enfermedades cutáneas. Las no sindrómicas son más frecuentes y no asocian manifestaciones extracutáneas, se incluyen: la ictiosis vulgar, la ligada al cromosoma X, las ictiosis congénitas autosómicas recesivas (AR) y las ictiosis epidermolíticas. Las ictiosis sindrómicas tienen mayor repercusión, afectan a varios órganos y sistemas, como el síndrome Gaucher tipo 2(2).
La ictiosis vulgar es la forma más común, presenta un patrón de herencia autosómico dominante. Se caracteriza por xerosis y descamación generalizada de coloración blanquecina que respeta pliegues y áreas de flexión(3). A diferencia de nuestro paciente, este tipo de ictiosis suele respetar las zonas de la cara, el cuello y las flexuras. También es característica la afectación palmo-plantar que no se objetiva en nuestro caso(4).
El síndrome de Gaucher tipo 2 se puede manifestar en el periodo neonatal con bebé colodión: piel fina, brillante, con aspecto de “envoltura por papel de celofán” y que suele evolucionar a una descamación y xerosis cutáneas. Así mismo, asocia hidrops fetal, hepatoesplenomegalia y deterioro neurológico grave. El patrón de herencia es AR y su esperanza de vida suele ser inferior a dos años(3). Nuestro paciente no había presentado durante su desarrollo manifestaciones extracutáneas, por lo que se orientó el diagnóstico diferencial hacia entidades no sindrómicas.
El síndrome de la piel exfoliada con herencia AR, a diferencia de nuestro paciente, suele presentar descamación exfoliativa blanquecina superficial generalizada de curso estacional que simula la descamación posterior a una quemadura solar, tiene afectación palmo-plantar y onicomadesis(2).
En cuanto a la ictiosis ligada al cromosoma X, es una enfermedad poco frecuente que afecta a varones. Los pacientes sufren hiperqueratosis con escamas grandes y oscuras por inhibición de la descamación cutánea en zonas extensoras, cuello, axilas y tronco (como podemos observar en las imágenes), por un déficit enzimático en la epidermis que produce un aumento del sulfato de colesterol, que inhibe la descamación. Estos hallazgos pueden aparecer desde las primeras semanas de vida(5). Estos pacientes presentan una disminución de estrógenos en la placenta, lo que produce una dilatación cervical insuficiente, alargando el trabajo de parto, como el caso descrito(3).
Ante la sospecha de dicha entidad, se solicitó estudio genético para despistaje de ictiosis no sindrómicas que reveló una delección en la región cromosómica Xp22.31 confirmando el diagnóstico.
Aunque se trata de una enfermedad crónica, si se realizan medidas de higiene y se aplican productos que restablecen las propiedades de elasticidad y tersura de la piel (emolientes) tiene buena evolución. Además, si aparece hiperqueratosis como en nuestro caso, resulta conveniente añadir agentes queratolíticos(6). El paciente ha presentado mejoría clínica significativa con dicho tratamiento.
Palabras clave
Enfermedades de la piel; Ictiosis; Ictiosis ligada al cromosoma X.
Skin disease; Ichthyosis; X-Linked Ichthyosis.
Bibliografía
1. González M, Triana M, Molina MA. Imagen en Pediatría Clínica. Haz tu diagnóstico. Pediatr Integral. 2013; XVII: 574-80. Disponible en: https://www.pediatriaintegral.es/numeros-anteriores/publicacion-2013-10/imagen-en-pediatria-clinica-haz-tu-diagnostico-2013-10/.
2. Metze D, Traupe H, Süßmuth K. Ichthyoses-A clinical and pathological spectrum from heterogeneous cornification disorders to inflammation. Dermatopathology (Basel). 2021 ;8: 107-23.
3. Vega Almendra N, Aranibar Duran L. Ictiosis hereditaria: desafío diagnóstico y terapéutico. Rev Chil Pediatr. 2016; 87: 213-23.
4. Jaffar H, Shakir Z, Kumar G, Ali IF. Ichthyosis vulgaris: An updated review. Skin Health Dis. 2022; 3: e187.
5. Takeichi T, Akiyama M. Inherited ichthyosis: Non‐syndromic forms. J Dermatol. 2016; 43: 242-51.
6. Süßmuth K, Traupe H, Metze D, Oji V. Ichthyoses in everyday practice: management of a rare group of diseases. J Dtsch Dermatol Ges. 2020; 18: 225-43.

 Neurological emergencies in Pediatrics
Neurological emergencies in Pediatrics