|
| Historia de la Medicina y la Pediatría |
M. Zafra Anta*, V.M. García Nieto**
*Servicio de Pediatría del Hospital Universitario de Fuenlabrada, Madrid. Miembro del Grupo de Historia de la Pediatría de la AEP. **Coordinador del Grupo de Historia de la Pediatría de la AEP. Director de Canarias Pediátrica
Pediatr Integral 2022; XXVI (8): 515.e1 – 515.e8
Enfermedades pediátricas que han pasado a la historia (14). Fiebre reumática aguda en países y ámbitos desarrollados con un adecuado sistema de salud pública
“No te asustes; espero que no será grave (–la fiebre escarlatina–). Miré en el libro de mamá y noté que comienza con dolor de cabeza y de garganta, y sensaciones extrañas como las mías; tomé belladona y me siento mejor
–dijo Beth, poniendo sus manos frías sobre su frente caliente, y tratando de aparentar que estaba bien”.
En la novela Mujercitas (1868), de Louisa May Alcott
Introducción
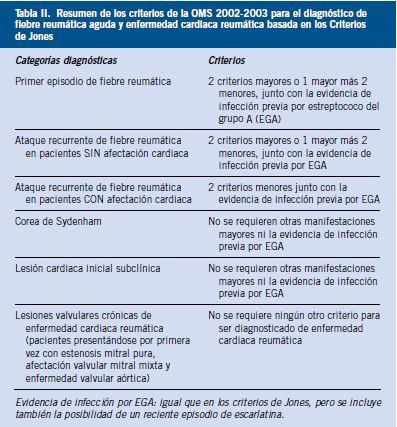
La fiebre reumática aguda (FRA), CIE-10: 100, es una enfermedad inflamatoria, no supurativa, producida por una respuesta autoinmune, por el sistema inmunitario de algunas personas predispuestas frente a los antígenos de la bacteria Streptococcus pyogenes o Estreptococo betahemolítico del Grupo A (EBHGA, también se cita como EGA o, con sus siglas en inglés, GAS). La FRA puede afectar a las articulaciones, el corazón, los vasos sanguíneos, el cerebro y la piel. La enfermedad cardiaca reumática (ECR) o cardiopatía reumática, CIE-10: 109.9, es la que produce lesiones que pueden hacerse permanentes y graves. La cardiopatía reumática crónica puede ser el resultado de un episodio único de FRA o de múltiples recurrencias, y es la determinante del pronóstico(1-5).
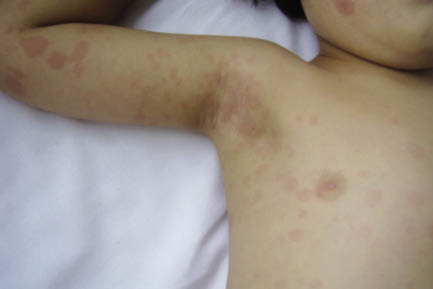
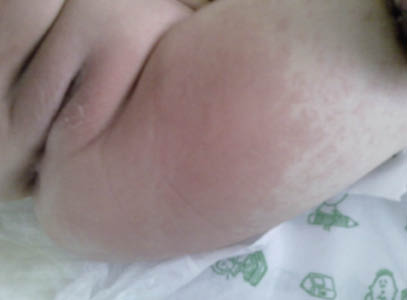
El periodo latente de la FRA es de 2 a 3 semanas tras una infección estreptocócica, en general una faringoamigdalitis, hasta que aparecen los primeros signos y síntomas, que incluyen: fiebre, poliartritis, carditis, erupción cutánea, nódulos subcutáneos y corea, no todos se presentan en cada paciente. Todos los síntomas, con o sin tratamiento, desaparecen en semanas (la corea a veces se prolonga), excepto el daño cardiaco que puede persistir, sobre todo en forma de daño valvular.
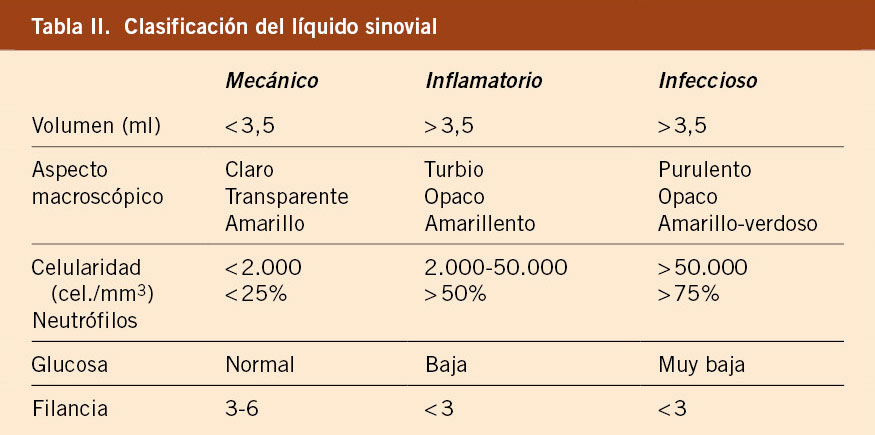
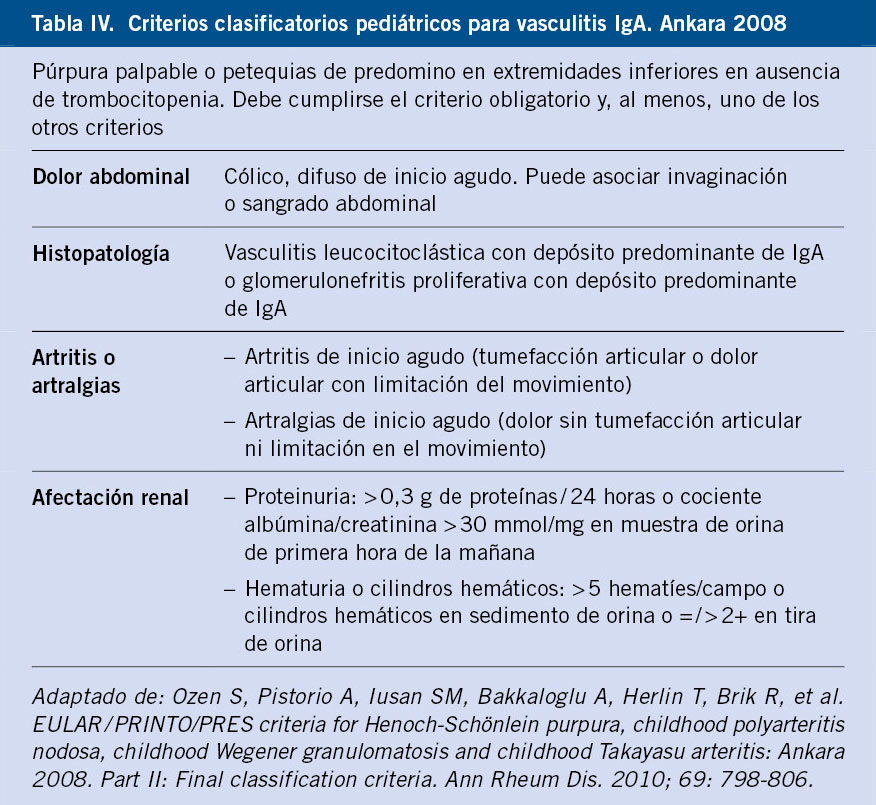
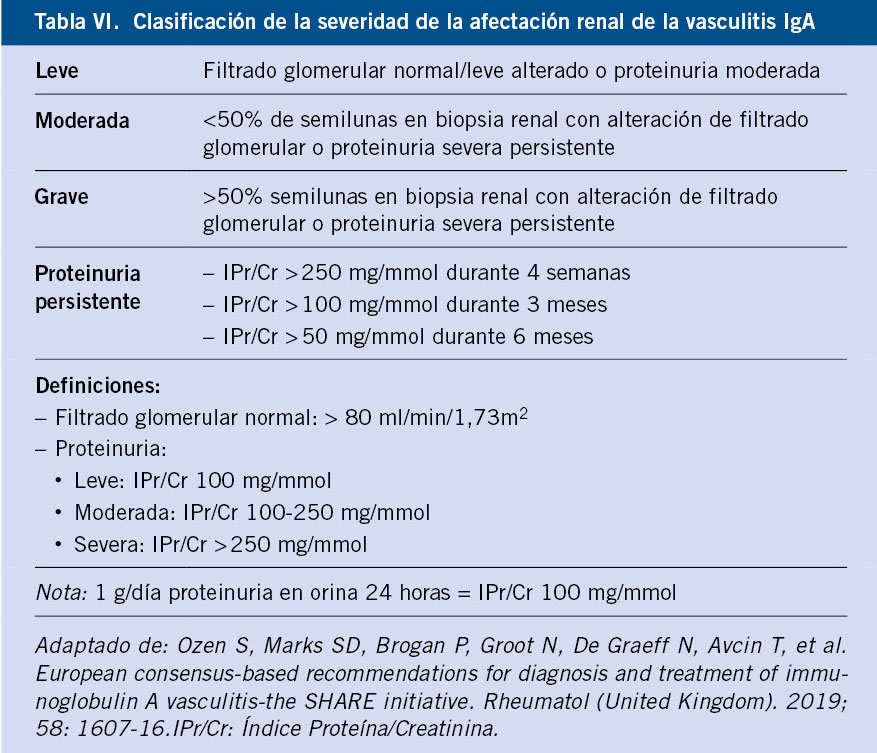
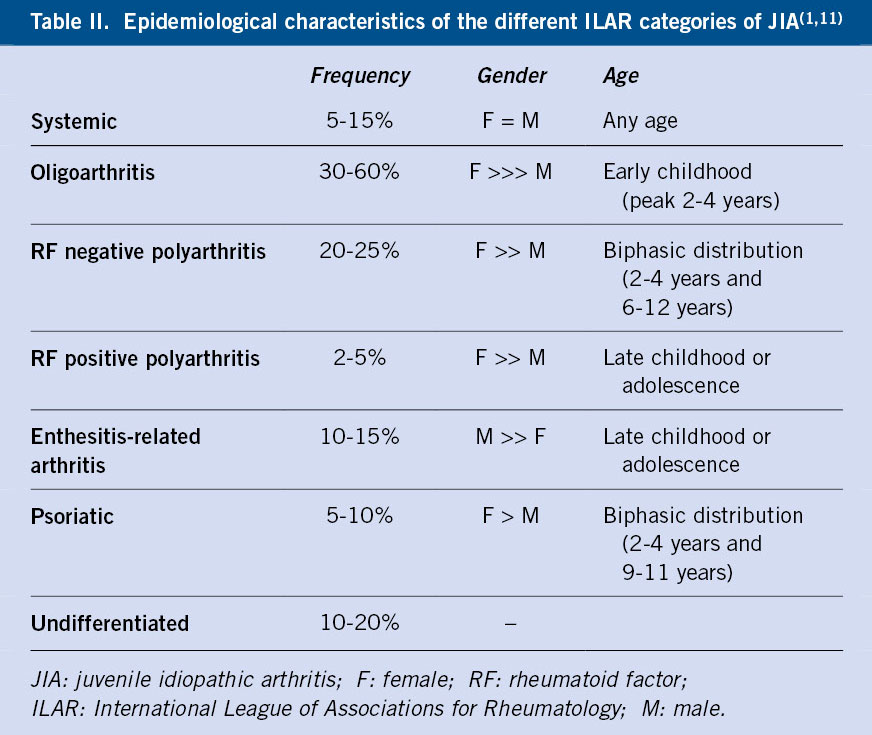
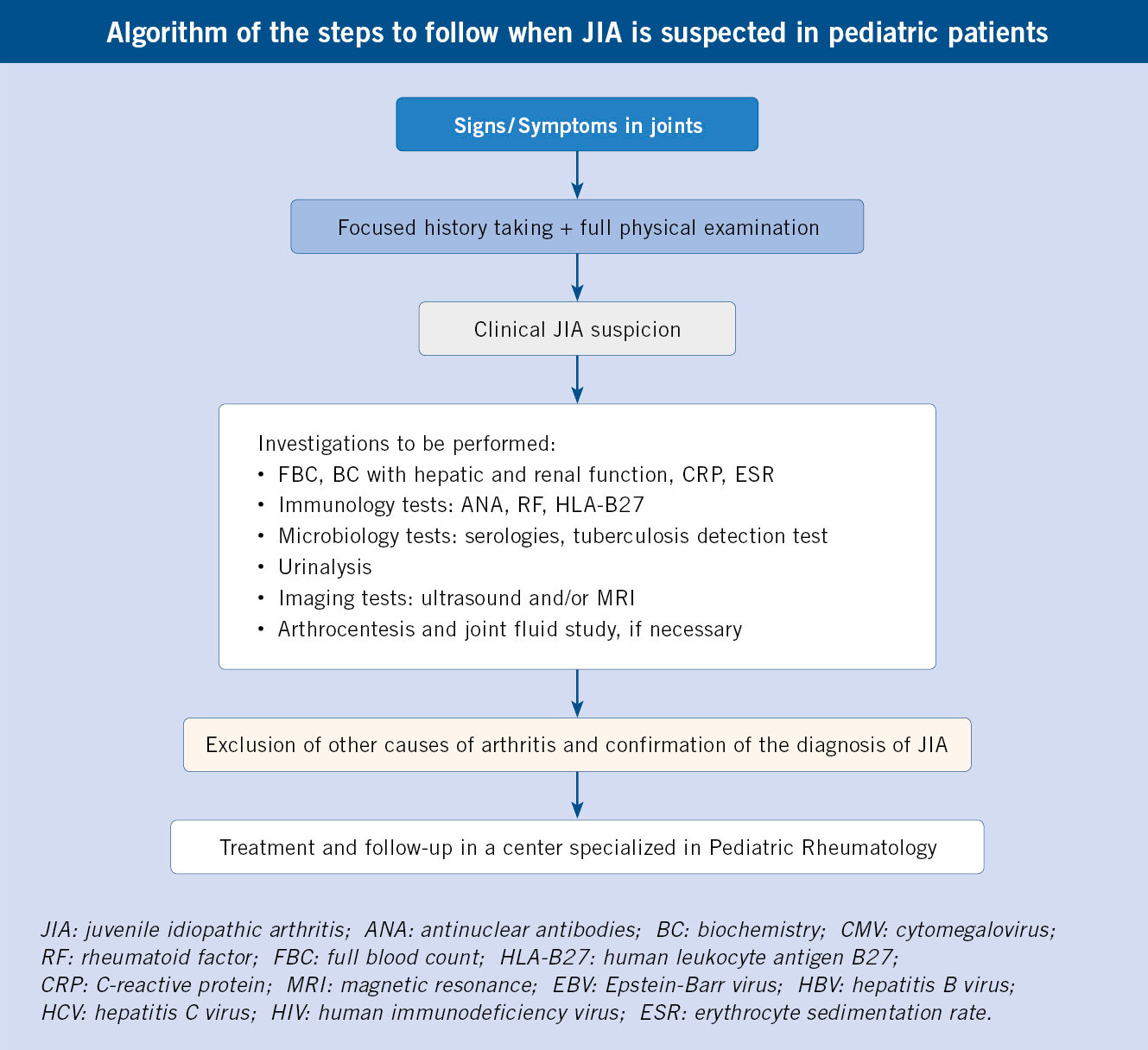
No se conoce completamente la patogenia de la FRA. Fue una enfermedad muy común en todo el mundo durante el siglo XIX y primera mitad del XX. Durante el siglo XX, en los países llamados desarrollados, la incidencia y la carga de la enfermedad descendió significativamente hasta virtualmente desaparecer(1,3,6-8) (Figs. 1 y 2). Aun así, la FRA continúa siendo un problema de salud pública en los países de bajos ingresos y también en zonas marginales de países de medianos y altos, con el fenómeno de la migración puede haber personas afectas de ECR en países desarrollados. Existe una clara asociación entre la inequidad social y la prevalencia de ECR(9,10).
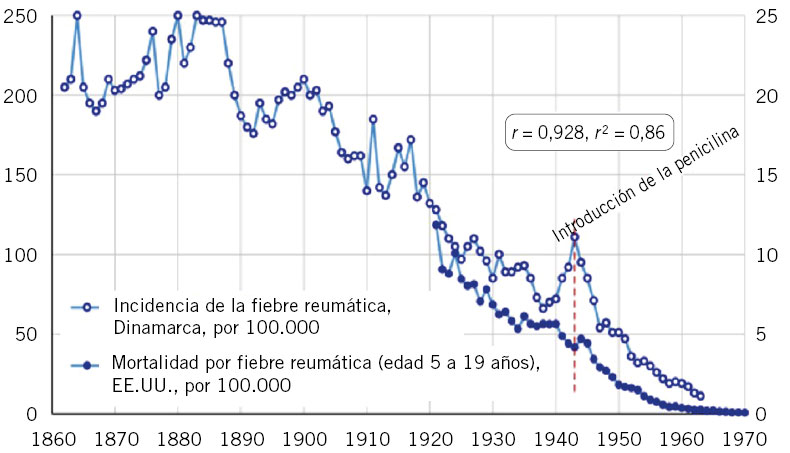
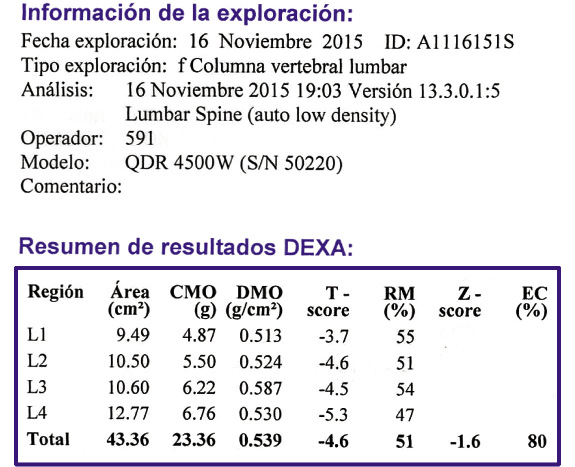
Figura 1. El declive internacional histórico de la fiebre reumática, con datos de incidencia de Dinamarca (1862-1963) y datos de mortalidad de EE.UU. (1921-1970). Correlación: r = 0,928, r2 = 0,81. La penicilina se introdujo en 1943, en EE.UU. y en Dinamarca. Imagen tomada de Alm(7), traducida.

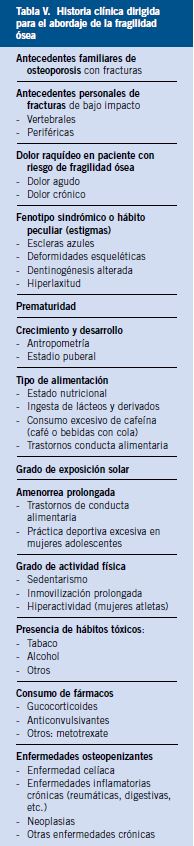
Figura 2. Caída en el tiempo de la mortalidad por fiebre reumática. Tasas estandarizadas totales. España-EE.UU. Edad: 5-19 años (España, 1951-1985; EE.UU. 1951-1978). Tomado de P. Cortina(8).
En este trabajo revisaremos la historia de la enfermedad y sus enseñanzas. Se resume la actualidad y se plantea la necesaria actitud vigilante que se necesita tener con las infecciones por EBHGA y sus complicaciones supurativas y no supurativas (fiebre reumática, carditis reumática, artritis postestreptocócica y glomerulonefritis postestreptocócica)(1-3,6). Por otro lado, en la historia de la infección por EBHGA se ha establecido una asociación con trastornos neuropsiquiátricos autoinmunes (PANDAS), los tics, el trastorno obsesivo compulsivo (TOC), aunque ello está en debate aún(11). Todo ello en cierta relación con la corea de Sydenham, manifestación neurológica de la FRA. El diagnóstico de PANDAS no está aún aceptado como una entidad independiente y tampoco se contempla en la CIE-10, ni en el DSM-IV. La DSM 5 lo cita en TOC relacionados a otra condición médica, pero dice que es “controvertido, aunque hay un cuerpo de evidencia”.
Es importante señalar aquí que la fiebre reumática aguda fue una enfermedad de declaración obligatoria (EDO) desde 1951(8) hasta 1996, en que dejó de ser EDO en España; véase BOE nº 21, del 24 de enero de 1996, p. 2153. Si bien, algunas Comunidades Autónomas la mantuvieron como EDO unos años más, como por ejemplo Castilla y León hasta 2006 (v. Anexo I de la Orden SAN/2128/2006, del 27 de diciembre, por la que se regula el Sistema de Enfermedades de Declaración Obligatoria de Castilla y León).
Resumen de la actualidad de la fiebre reumática aguda
Epidemiología
Si bien hay casos y brotes descritos en todo tipo de población, la FRA y la ECR en la actualidad son enfermedades de la pobreza, de los económicamente desfavorecidos. Es una de las principales causas de muerte cardiovascular durante las primeras cinco décadas de la vida en países subdesarrollados(1,9,10), entorno en el que además se infradiagnostica y se infradeclara. Anualmente, en el mundo, hay hasta 470.000 nuevos casos de FRA, y más de 275.000 muertes atribuibles a cardiopatía reumática. No es, pues, una enfermedad desaparecida globalmente.
En países desarrollados la incidencia media es < 2 casos por 100.000 escolares, es mayor en países con bajos o medios recursos, y donde hay población indígena como en Australia (en ellos hasta 380 casos por 100.000). A mediados del siglo XX se producían casos de FRA en un 3 % de las faringitis agudas estreptocócicas no tratadas, actualmente la incidencia es mucho menor (< 1 %). Esta cifra no es uniforme en todos los pacientes con infecciones no tratadas, es una enfermedad con una afectación también determinada genéticamente. La mayoría de los casos de FRA ocurren en niños de 5 a 15 años de edad.
Patogénesis
El mecanismo patogénico todavía no es conocido completamente. Se necesita una infección faríngea previa y susceptibilidad genética. Hay brotes de FRA tras epidemias de faringitis estreptocócica o escarlatina asociada a faringitis.
El declinar en la FRA tiene relación con el descenso de las cepas de EBHGA denominadas reumatógenas (las emm: 3, 5, 6, 14, 18, 19, 24 y 29), que pertenecen al patrón A y C. Aunque puede haber cepas diferentes también implicadas(1,4,6). Incluso, en zonas tropicales parece que infecciones dermatológicas por estreptococo podrían desencadenar esta patología.
La similitud o “mimetismo” molecular parece que es la vía patogénica que está envuelta en la alteración tisular, tanto en el desarrollo de la carditis reumática (miosina, laminina) como en el corea (lisogangliósido de mamíferos)(1,4,7). Es conocido que la infección por Streptococcus pyogenes origina superantígenos que determinarían una activación general del sistema inmune. Se activan células B y T autorreactivas, con antígenos del EBHGA. En el corea de Sydenhan hay tendencia a producir anticuerpos frente a CaMK II, que aumenta la transmisión de dopamina o anticuerpos frente al receptor de dopamina D2.
También hay datos que apuntan a que coinfecciones virales con el EBHGA incrementan citokinas pro-inflamatorias y pueden potenciar las respuestas autoinmunes.
Hay susceptibilidad genética, parece que poligénica. Se encuentra concordancia de la FRA en gemelos monocigóticos más que entre dicigóticos.
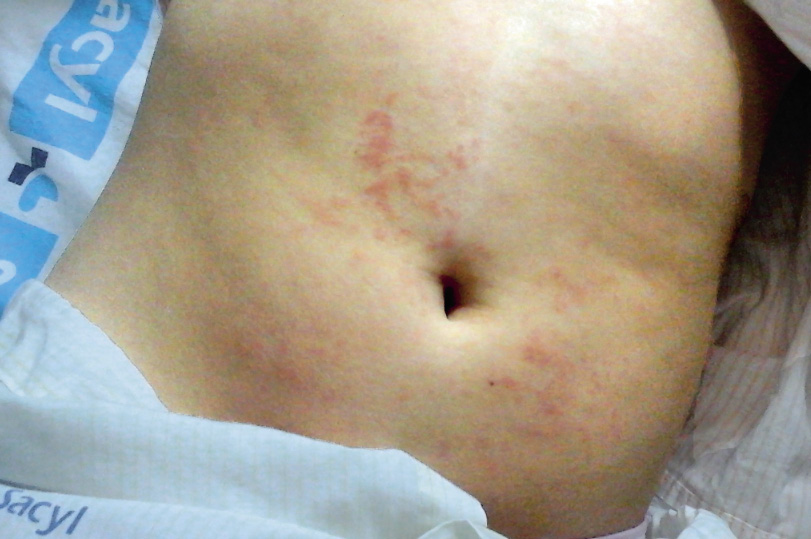
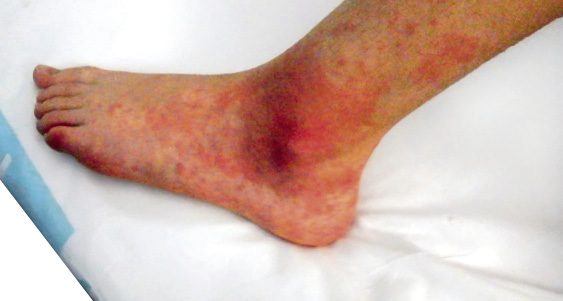
La FRA en la fase aguda produce generalmente fiebre. En un 50-80 %, hay una pancarditis, que puede ser asintomática; en la fase crónica se produce la enfermedad cardíaca reumática. Afecta también: a la piel (eritema marginado, <6 % de casos); a las articulaciones (60-80 %, poliartritis migratoria, que responde rápidamente a antiinflamatorios); al sistema nervioso central (corea de Sydenham o corea menor, 10-30 %); y al tejido celular subcutáneo (nódulos subcutáneos, 0-10 %).
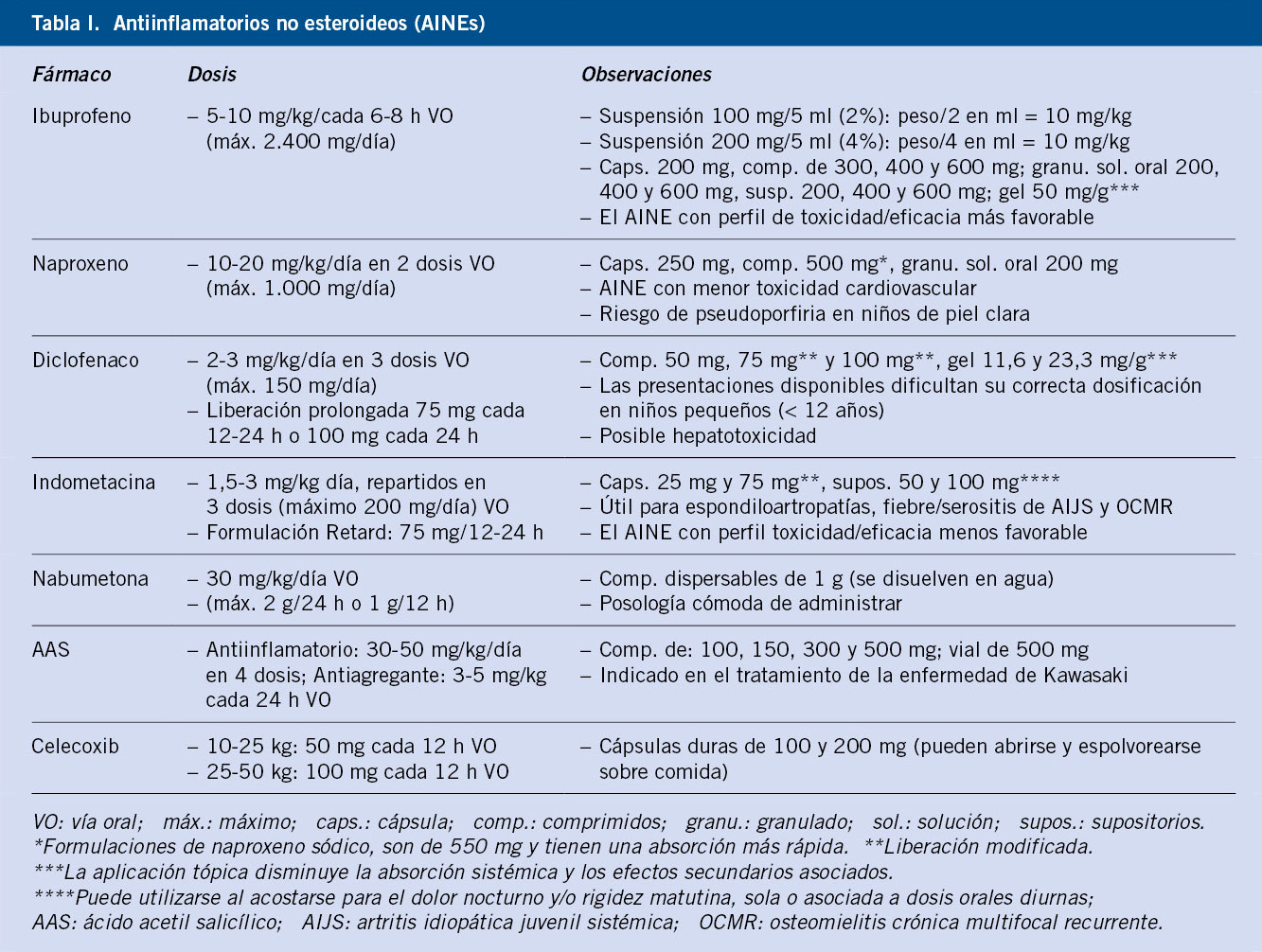
Las manifestaciones clínicas(1,3,5) se clasifican en manifestaciones mayores y menores, según los criterios de Jones, publicados por este autor(12) en 1944.
• Criterios mayores: artritis, carditis, afectación del SNC, como corea, nódulos subcutáneos y eritema marginado.
• Criterios menores: artralgias, fiebre, elevación de reactantes de fase agua (VSG) e intervalo PR prolongado en el ECG.
Hasta que se aceptaron unánimemente los criterios de T.D. Jones, cada autor sistematizaba el diagnóstico de la enfermedad siguiendo concepciones personales o de escuela, haciendo mayor hincapié en la clínica y/o en las pruebas complementarias. Jones afirmó que en las circunstancias en que coincidieran artralgia, fiebre y alguna anomalía de laboratorio, sería de valor para establecer el diagnóstico de fiebre reumática la existencia de historia de infección respiratoria previa o de exposición al estreptococo hemolítico epidémico y/o el desarrollo de anticuerpos antiestreptococo hemolítico. Es interesante señalar que los criterios originales no incluían pruebas de apoyo de una infección estreptocócica previa. De hecho, solo hay dos menciones a las infecciones estreptocócicas en el artículo de Jones, y más bien como observaciones de pasada(12,13).
A lo largo de los años, los comités de la Asociación Americana del Corazón han realizado cambios relativamente menores pero significativos en los criterios originales. La última actualización es de 2015, que incluye niveles de evidencia científica, la clasificación en poblaciones de riesgo y el apoyo de la ecografía cardiaca; la anterior actualización era de 1992(2,3,13-15).
Criterios de Jones revisados
• Para todos los pacientes con evidencia de EBHGA precedente, diagnóstico de FRA inicial o recurrente.
• Criterios mayores. En poblaciones de bajo riesgo:
– Carditis. Clínica y/o subclínica (incluye valvulitis ecográfica).
– Artritis. Poliartritis solo.
– Corea.
– Eritema marginado.
• Criterios menores. En poblaciones de bajo riesgo.
– Poliartralgia.
– Fiebre (≥ 38,5ºC).
– VSG ≥ 60 mm y/o PCR ≥ 3,0 mg/dL.
– Intervalo PR prolongado teniendo en cuenta la variabilidad por edad.
El diagnóstico de carditis puede ser clínico o subclínico y se puede hacer por ecocardiografía(2,3,15). La ecografía es útil, además, para valorar la insuficiencia mitral o aórtica subclínicas.
Población de bajo riesgo: aquella con incidencia ≤ 2 por 100.000 escolares o ≤ 1 por 1.000 por población general de todas las edades con enfermedad cardiaca reumática.
La probabilidad diagnóstica de FRA es alta si en el contexto de infección por EBHGA se suceden dos criterios mayores o uno mayor y dos menores.
Estos criterios se modifican ligeramente si el entorno es de población de moderado-alto riesgo (monoartritis con poliartralgia, fiebre menos elevada, una VSG o PCR ligeramente menos elevada).
Hay dos situaciones donde el diagnóstico de FRA se plantea, y hay que hacer controles cardiológicos:
• Corea como única manifestación.
• Carditis indolente o subclínica como única manifestación.
La secuela tardía más común es la ECR, que ocurre a los 10 a 20 años tras la enfermedad inicial. Es la causa más común de valvulopatía cardíaca adquirida en el mundo.
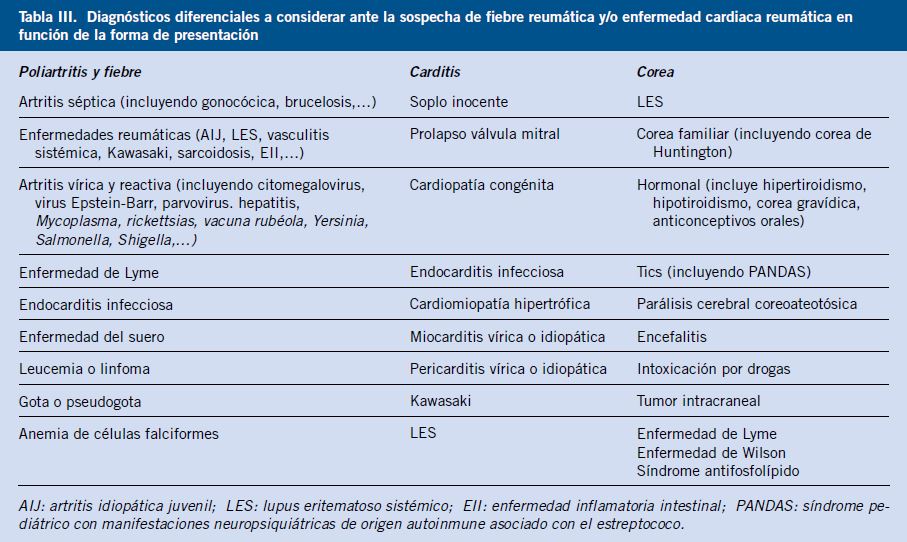
Diagnóstico diferencial con: artritis poliarticular en niños y adultos, incluyendo infecciosa, inmune postinfecciosa, enfermedad autoimmune o autoinflamatorio, neoplasia o enfermedades sistémicas.
Otras complicaciones no supurativas del EBHGA son: la escarlatina, la artritis postestreptocócica y la glomerulonefritis postestreptocócica.
El tratamiento de la FRA requiere antibióticos, terapia antiinflamatoria, manejo de la insuficiencia cardiaca y seguimiento con prevención secundaria y educación sanitaria. Se recomienda ingreso hospitalario en el episodio agudo(2,3-5).
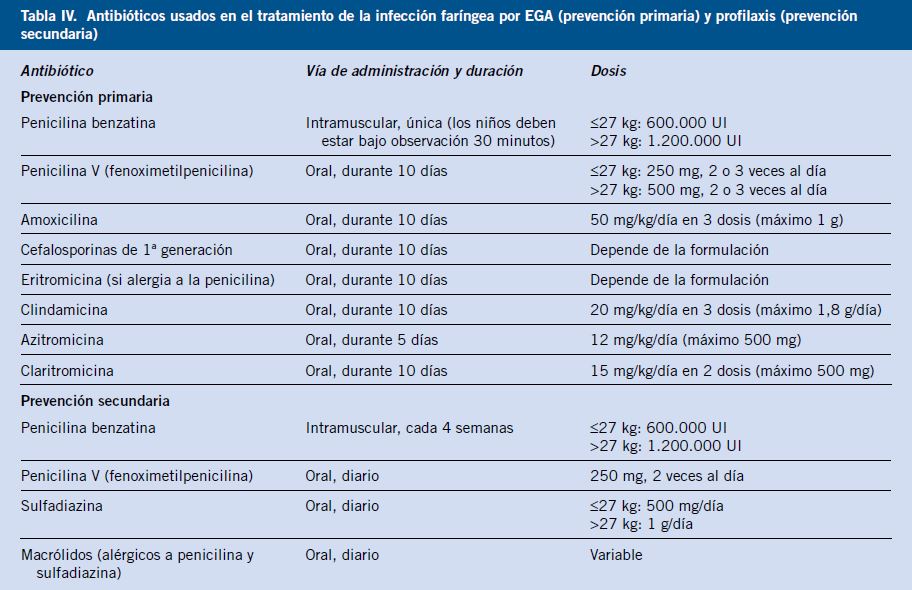
La penicilina demostró, en estudios controlados en los años 50 y 60, reducciones en la proporción de FRA y de recurrencias. Esto está apoyado por décadas de experiencia clínica. La duración del tratamiento recomendado es de 10 días si se busca erradicar la bacteria de la orofaringe (hay porcentajes menores en el tratamiento de 5 días: 80,4 %, frente al de 10 días: 90,7 %)(16).
Tras un episodio de FRA se recomienda la profilaxis a largo plazo de la infección por EBHGA. Con penicilina benzatina IM mensual, sobre todo en zonas endémicas; la penicilina oral puede ser útil en zonas no endémicas. La duración de la profilaxis depende de si ha habido carditis en el episodio inicial de FRA y si hay secuelas de esta. Al menos, hasta cumplir los 21 años de edad o que transcurran diez años sin recurrencia, si no hubo secuelas. Además, se busca la erradicación de los portadores de EBHGA. En los alérgicos el tratamiento se detalla en las referencias(2,3,5).
Antiinflamatorios: naproxeno de elección actualmente. Ibuprofeno es una alternativa en niños pequeños. Aspirina es una alternativa, y glucocorticoides a bajas dosis.
Direcciones útiles:
• AHA: Scientific statement on the revision of the Jones criteria for the diagnosis of acute rheumatic fever in the era of Doppler echocardiography (2015).
• AHA: Scientific statement on the prevention of rheumatic fever and diagnosis and treatment of acute streptococcal pharyngitis (2009).
• European Society of Cardiology (ESC)/European Association for Cardio-Thoracic Surgery (EACTS): Guidelines for the management of valvular heart disease (2021).
El diagnóstico, tratamiento y manejo de la FRA y de la ECR está consensuado a nivel mundial. Se ha organizado un esfuerzo desde Naciones Unidas para desarrollar estrategias de prevención, detección y seguimiento de la fiebre reumática y sus secuelas. También hay importantes esfuerzos actuales para la vacunación contra el S. pyogenes(6).
Por otro lado, no hay un modelo experimental animal adecuado para el estudio de la fiebre reumática. Solo recientemente, Rafeek y cols.(17) han publicado un modelo con ratas de Lewis, en las que las proteínas M de diferentes especies de estreptococos (EBHGA, y también Streptococcus dysgalactiae subespecie equisimilis) podrían iniciar y promover el daño tisular cardíaco mediado por la autoinmunidad, así como las anormalidades neuroconductuales, comparables a los que determinan algunos síntomas cardinales observados en los pacientes con FRA/ECR(17).
Hitos en la historia de la fiebre reumática y cardiopatía reumática
La Reumatología es el estudio y tratamiento de las afecciones clínicas que se manifiestan generalmente por inflamación de las articulaciones de las extremidades. Es una especialidad médica, no quirúrgica, del aparato locomotor y del tejido conectivo.
Fiebre reumática en la Antigüedad
Los componentes léxicos de “Reumatología” proceden del griego y son: rheuma, rheumatos (flujo, manantial, catarro) y logos (estudio). Se refiere al humor mórbido que fluye a través del cuerpo, por gravedad, de la cabeza al cuerpo, y causa una enfermedad. Aunque algunos de los aforismos de Hipócrates ya se referían a enfermedades articulares y la palabra “reuma” fue introducida en el siglo I d.C., el concepto de “reumatismo” como síndrome musculoesquelético corresponde a la Era Moderna(5,18,19). Dioscórides, Celsius, Galeno, Aretaeus, Avicena, Abucasim utilizaban indiscriminadamente las palabras gota, artritis y reuma. Durante el siglo XVII se empieza realmente a diferenciar la gota, del reumatismo agudo (en la mayoría de los casos sería fiebre reumática) y del reumatismo crónico (artritis reumatoide); la palabra artritis realmente se conocía poco y se aplicaba a las afecciones articulares. La palabra Gota (del latín gutta o humor viciado que fluía gota a gota) en las articulaciones de las manos o pies (cheiragro y podagro) se utilizaba para inflamación articular.
Fiebre reumática en la Edad Moderna-Renacimiento(13,18-20)
Guillaume de Baillou, en latín Ballonius (1538-1616). Médico francés. Es considerado uno de los padres de la reumatología y el primer epidemiólogo tras Hipócrates. Hizo también descripciones de la tosferina y la difteria.
Sigismundo Albicus (hacia 1347-1427). Para algunos historiadores de la medicina fue el primer médico en hablar de reuma. Fue arzobispo de Praga y médico de la corte. Albicus, en su libro Régimen contra reumata (siglo XV), al parecer, es el que mejor describe la palabra reuma y fue el primero en utilizarla, y no Baillou.
Thomas Sydenham (1624-1689), famoso médico inglés, describió en 1685 una artritis febril aguda, y en 1686 describió el chorea sancti viti “baile de San Vito”, posteriormente llamado corea de Sydenham o corea menor; que no asoció a la artritis febril. El término “Corea” procede del latín y este del griego (“danza”). La corea menor fue tipificada en 1625 por el médico y anatomista alemán Gregor Horstius (1578-1636).
Raymond Vieussens (hacia 1635 o 1641-1715), iatroquímico y anatomista francés, publicó a principios del XVIII la descripción necrópsica de la estenosis mitral y la insuficiencia aórtica.
Herman Boerhaave (1668-1778), médico holandés, separó la gota del reumatismo.
Fiebre reumática en la Edad Contemporánea
Siglo XIX
En 1853 Jean-Martin Charcot (1825-1893) realizó una excelente diferenciación entre la artritis reumatoide, la gota, la fiebre reumática y la osteoartritis.
David Dundas (1749-1826), inglés, sargento cirujano del rey y baronet. Describe en 1808 casos de “peculiar enfermedad del corazón, con necropsia. En no menos de 9 casos que he visto la enfermedad sucede a uno o más ataques de fiebre reumática… las valvas mitrales tenían un borde esponjoso”. Matthe Baillie (1761-1823), médico inglés y anatomopatólogo, en su texto de Patología de 1797 escribió sobre una osificación o engrosamiento de las válvulas cardiacas en pacientes que habían padecido reumatismo agudo.
William Charles Wells (1757-1817), médico escocés, en 1812 planteó la asociación entre fiebre reumática y carditis y describió los nódulos subcutáneos. Jean-Baptiste Bouillaud (1796-1881), médico francés, en 1836, así como Walter Butler Cheadle (1836-1910) pediatra inglés, en 1889, publicaron sendos estudios extensos acerca de la asociación de la fiebre reumática y las secuelas cardiacas. Los diccionarios médicos franceses siguen denominando la endocarditis reumatoide aguda como “enfermedad de Bouillaud”; también se conoció así a la fiebre reumática a principios del XX.
James Kingston Fowler (1852-1934), inglés, en 1880 señaló la asociación entre inflamación de garganta y fiebre reumática en Lancet, 1880.
Arthur Newsholme hizo el primer estudio epidemiológico sobre FRA. Señaló, entre otros datos, la presentación de la FRA en brotes epidémicos. Publicado en Lancet, 1885.
Siglo XX
Karl Ludwig Aschoff (1866-1942) anatomopatólogo alemán, en 1904 describió la lesión miocárdica especifica de la fiebre reumática, que se conoce como “nódulo de Aschoff”.
Carey Franklin Coombs (1879-1932), británico, cardiólogo especialista en fiebre reumática. Publicó el libro, “Rheumatic Heart Diseases” (1924). El soplo de Carey Coombs es un soplo corto mesodiastólico, que mejora o se resuelve con la recuperación de la enfermedad aguda.
Poynton y Paine en 1900 (Lancet) aislaron un estreptococo de la faringe de un paciente que padecía fiebre reumática aguda con amigdalitis, y con este organismo produjeron artritis experimental.
Homer Fordyce Swift (1881-1953), médico estadounidense y cols. anticiparon en 1928 la teoría de la patogénesis de la fiebre reumática como hipersensibilidad a estreptococo (estreptococo o productos diseminados, los tejidos reaccionan resultando el cuadro característico de la enfermedad).
Los trabajos epidemiológicos de Alvin F. Coburn (1899-1976), médico estadounidense y W.R.F Collis (Inglaterra), publicados en 1931, dirigieron la investigación final hacia el EBHGA como iniciador del proceso.
E.W. Todd introdujo en 1932 el test de antiestreptolisina. Estaba elevada en el 90 % de los enfermos. La antiestreptolisina O disminuye en la fase de inactividad.
Rebecca Craighill Lancefield (1895-1981), en 1933 introdujo la clasificación de los grupos de estreptococos, basándose en la naturaleza antigénica de los hidratos de carbono de su pared celular. Se afirma que “puso en orden” a los estreptococos.
Thomas Duckett Jones (1899-1954). Dedicó su vida al tratamiento y la investigación en fiebre reumática. Publicó en JAMA, en 1944(12), criterios para ayudar en el diagnóstico de la fiebre reumática aguda. Desde entonces se denominan: Criterios de Jones, con revisiones. Fue, entre otros cargos, presidente de la Asociación Americana de Reumatismo (Fig. 3).

Figura 3. Thomas Duckett Jones. Tomado de Denny(14).
Las primeras publicaciones sobre el uso de antibióticos para tratar la fiebre reumática, ya en 1945, de B.F. Massell y cols., en New England Journal of Medicine, y en JAMA, de F.W. Denny y cols. También en 1948 se comunica la capacidad de erradicar al EBHGA con penicilina por B.F. Massell, J.W. Dow y T.D. Jones en JAMA.
Sobre la prevención de la fiebre reumática, Rammelkamp en 1953 demostró que la fiebre reumática se podía prevenir tratando las faringitis estreptocócicas con antibióticos.
Fiebre reumática y algunos personajes históricos
En el estudio de las enfermedades en la Historia se tiene la dificultad de que se trata de un diagnóstico a posteriori; además, dicho diagnóstico no es solo clínico, sino que precisaría otras exploraciones complementarias, incluido un estudio anatomopatológico. Se ha citado la posibilidad de que tuvieran afectación por FRA diversos personajes de la cultura, en los que además habría influido en el desarrollo de su arte: Wolfgang Amadeus Mozart (1756-1791), Lord Byron (1788-1824), Andy Warhol (1928-1987), Gustav Mahler (1860-1911), Bertolf Brecht (1898-1956)(21), Carson McCullers (1917-1967), etc.
Gustav Mahler padecía un trastorno del movimiento, pero su naturaleza sigue siendo indeterminada. Hay publicada una carta de Freud a Theodore Reik refiriéndose a Gustav Mahler sobre su neurosis obsesiva. A Mahler se le diagnosticó una valvulopatía en 1907 y murió de endocarditis bacteriana subaguda en 1911. Estas características sugieren, pues, la hipótesis de que el compositor sufrió de FRA en la infancia con carditis y corea de Sydenham, que le dejó como secuelas valvulopatía, comportamiento obsesivo-compulsivo y corea persistente(21-22).
Evolución de la fiebre reumática en el mundo
Durante el siglo XX en los países llamados desarrollados, la incidencia de la enfermedad por FRA descendió significativamente hasta virtualmente desaparecer. El descenso de mortalidad por FRA y por ECR comenzó en muchos países ya en los años 20-30 del siglo XX y se acentuó a partir de los años 40, coincidiendo con la introducción de la antibioterapia. Parece que los principales factores asociados a esta dinámica fueron: la higiene, la mejora en las condiciones de vida de la infancia, evitar el hacinamiento (con hermanos en la casa, en barracones militares), la mejora en el acceso a los servicios de salud, la optimización de las herramientas de diagnóstico y la disponibilidad de los antibióticos en los países desarrollados(1,6,7,13,19,23,24).
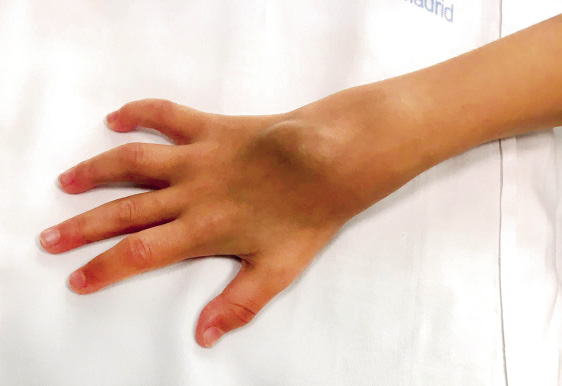
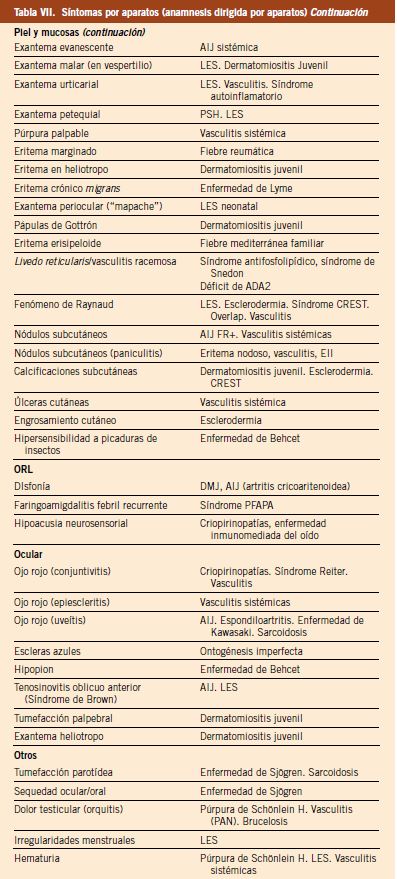
Con respecto a los últimos 25 años, se estima que la carga sanitaria de la cardiopatía reumática está disminuyendo en todo el mundo. D.A. Watkins y cols. publicaron en New England Journal of Medicine un estudio sobre prevalencia global de la enfermedad y la mortalidad por cardiopatía reumática en el periodo 1990-2015(25), estas fechas son concretamente antes de la pandemia del SARS-CoV 2. Los datos de enfermedad y mortalidad se obtuvieron de los registros nacionales y de cohortes según clasificación CIE-10. Hay que tener en cuenta que la caída de prevalencia por ECR es anterior en el tiempo a la de la caída de mortalidad por ECR. El descenso de la carga sanitaria de la ECR fue alrededor de un 8,1 % (de 2,7 a 13,5 con un intervalo de incertidumbre de 95 %). Pero las tasas de enfermedad siguen siendo elevadas en algunas de las regiones más pobres, especialmente en África, Asia Central, Caribe y Oceanía(23,25). Ofrecemos aquí dos figuras de esta publicación de Watkins (Figs. 4 y 5).

Figura 4. Prevalencia de cardiopatía reumática en el mundo en los años 1990 y 2015. En la prevalencia estandarizada por edad, los territorios con mayor carga de enfermedad son: Oceanía, Asia Central, Caribe y África(25).

Figura 5. Clasificación de los países según tengan un patrón endémico o no endémico de cardiopatía reumática en el año 2015. Se clasificó a un país como con un patrón endémico de la enfermedad si su mortalidad infantil estimada por cardiopatía reumática era superior a 0,15 muertes por 100.000 habitantes entre los niños de 5 a 9 años de edad(25).
Evolución histórica de mortalidad y morbilidad de la fiebre reumática en España
En cuanto a la situación en España, hay que advertir que no existen datos, ni oficiales ni de otras fuentes, en cuanto al número de casos de FRA anteriores a 1930. Existen datos de morbilidad por FRA y ECR desde 1951(8). La mortalidad por FRA es mínima actualmente en España(3,8), como en otros territorios con un adecuado sistema de salud pública.
P. Cortina y cols. analizaron en 1991 la evolución epidemiológica en España en la segunda mitad del XX, esto es, mortalidad y morbilidad por FRA y ECR(8). Este estudio recogió los datos de mortalidad de España por FRA y ECR del “Movimiento Natural de la Población Española”, publicados por el Instituto Nacional de Estadística (INE), de los años 1951 a 1988, basados en la Clasificación Internacional de Enfermedades (CIE). El material utilizado para el estudio de morbilidad por FRA en España, de 1951 a 1988, se obtuvo a partir de los Boletines Epidemiológicos Semanales. Se objetivó que en nuestro país el descenso de mortalidad por FRA y ECR se inició con posterioridad a otros países, como EE.UU. y la mortalidad fue claramente descendente en ese período, si bien con un cierto repunte en los años 60. En cambio, aunque la morbilidad por fiebre reumática registró una tendencia decreciente, todavía mantenía valores considerables y presentó un notable aumento a finales de los años 80, lo que señalaba que la FRA volvía a ser un problema de Salud Pública en esa década en España (Fig. 6).
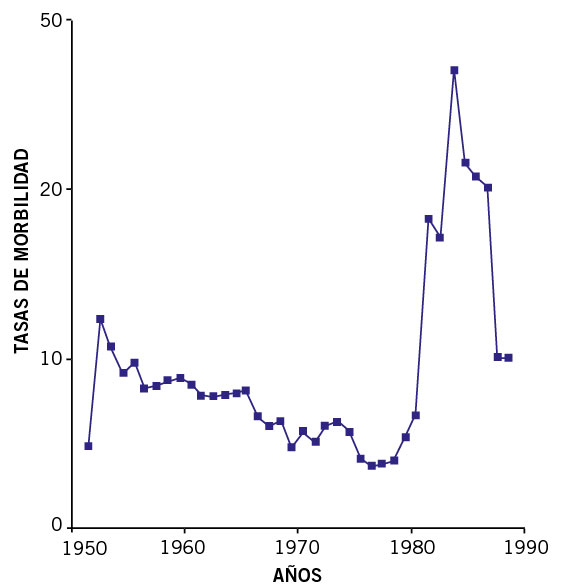
Figura 6. Morbilidad por fiebre reumática. Tasas por 100.000 habitantes. Totales. España, 1951-1988. Tomado de P. Cortina(8).
Encontramos el tema de la FRA y ECR en primeras publicaciones en revistas pediátricas españolas, tras la Guerra Civil:
• Profilaxis y tratamiento de la infección reumática en la infancia. Boix Barrios J. En la Rev Esp Pediatr. 1946; 2: 492-512.
• Etiopatogenia del reumatismo cardioarticular. Sandalio M. Rev Esp Pediatr. 1946; 2: 485-91.
• Prevención de la fiebre reumática en los niños. Gallego Barcina F. Acta Pediatr Esp. 1951; 9:143-51.
Hay que destacar en España en los años 30, el papel de la Medicina Interna: Carlos Jiménez Díaz y Gregorio Marañón (1934): “Vocabulario de la terminología reumática”, en: Once lecciones sobre el reumatismo. 269-74.
¿La fiebre reumática aguda y la infección por Streptococcus pyogenes tienen relación con la tartamudez?
Los estudios históricos sobre la fiebre reumática nos pueden aportar hipótesis en patología médica que en un primer momento pueden parecer incluso sorprendentes a los que se incorporan con profundidad al tema, como su relación con la tartamudez o disfemia (trastorno del habla). Este tema lo aporta y revisa en profundidad el estudio publicado por Alm(7).
En la década de 1930 se llevó a cabo una exhaustiva investigación sobre tartamudez por la Dra. Mildred Freburg Berry (1902-1993), del Rockford College, IL, EE.UU. (Berry, 1938). Se utilizó como fuente el historial médico de los niños que tartamudeaban. Ella declaró que la motivación para este trabajo le llegó de un comentario que era familiar para los terapeutas del habla en ese momento: ‘’mi hijo comenzó a tartamudear inmediatamente después de una grave enfermedad”. Mildred F. Berry, doctora en logopedia, fue una mujer pionera en la investigación, en el trabajo académico y profesional de los trastornos del lenguaje en los niños. El estudio de Berry de 1938, citado en Alm(7), señaló con solidez que hubo un tipo de evento médico en particular, que precedió a la aparición de la tartamudez infantil con una frecuencia inesperada: las enfermedades relacionadas con las infecciones de garganta por EBHGA.
Se ha propuesto(7) que la infección por EBHGA fue una de las principales causas subyacentes de la tartamudez hasta mediados del siglo XX, interactuando con factores genéticos. En particular, esto incluía la amigdalitis y la escarlatina, pero especialmente la fiebre reumática. Los datos y trabajos históricos disponibles sobre los cambios de la prevalencia de la tartamudez en la infancia indican un paralelismo sorprendente entre la tartamudez y la incidencia de la fiebre reumática, con descenso desde principios de 1900; y más marcado descenso desde la introducción de la penicilina a mediados de los años 40, alcanzando un nivel más estable en los años 60. En el estudio de Alm se encuentra una correlación muy alta, de aproximadamente 0,945 (Fig. 7).
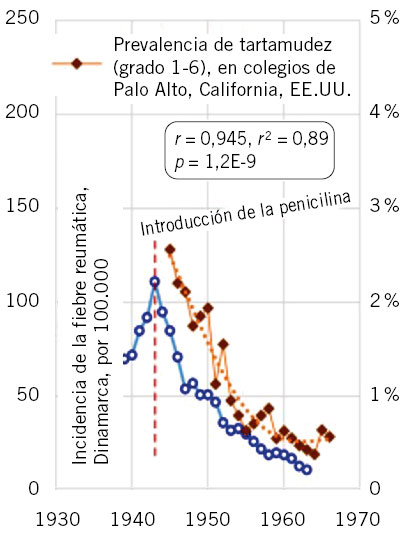
Figura 7. La disminución paralela de la tartamudez y la fiebre reumática en diferentes continentes, tras la introducción de la penicilina en 1943, en EE.UU. y en Dinamarca. La correlación es r = 0,945, r2 = 0,89, p = 1,2E-9. Imagen tomada de Alm(7), traducida y modificada únicamente para mostrar el periodo en años con los datos de tartamudez.
La relación temporal histórica FRA-tartamudez no demuestra la asociación causal. Pero hay más datos que apoyan esto, como casos clínicos más recientes. El mecanismo patogénico podría ser una reacción autoinmune de la amigdalitis, dirigida a moléculas específicas, en los ganglios basales. Distinto mecanismo que el de la corea menor. Recientemente, el aumento de la susceptibilidad a tener tartamudez se ha relacionado con la presencia del alelo C del polimorfismo rs6277 del gen dopaminérgico DRD2, y con mutaciones de genes involucrados en el metabolismo lisosomal(4,7,11).
Epílogo
Todavía no tenemos un conocimiento completo de la etiología de la fiebre reumática, no podemos explicar definitivamente su declive, ni los cambios en ciertos periodos históricos, ni su persistencia en subgrupos poblacionales en ciertas regiones del mundo, así como en zonas desarrolladas(1,7,13,25). Esto no se explica solo por la atención médica y los antibióticos. Aunque el EBHGA es el agente etiológico fundamental de la FRA, la enfermedad puede tener un origen multifactorial. Este hecho debería impedir cualquier relajación o complacencia por nuestra parte en lo que respecta a la fiebre reumática.
Hay una tendencia actual a realizar el tratamiento de la faringoamigdalitis durante 5-7 días(16,26). Esto evita generalmente las complicaciones supurativas, pero no erradica la bacteria. Hay que tener esto en cuenta al menos ante población considerada de riesgo, o en las que se precise la erradicación del EBHGA como prioridad, así como estar vigilantes por si se presentan brotes de enfermedad. Recordemos aquí que la FRA actualmente no es una EDO en España.
Desde el punto de vista clínico, el bajo riesgo de FRA en la actualidad, sugiere que tal vez deberíamos reconsiderar las políticas actuales relativas al tratamiento de la infección por EBHGA y nuestra determinación de erradicarlo. Pero sin conocer exactamente las razones del declive, no se pueden anticipar completamente los posibles resultados de cualquier relajación en la política antibiótica. En cualquier caso, dicha relajación sería completamente inapropiada para los países en desarrollo, en los que los riesgos de fiebre reumática y cardiopatía reumática siguen siendo tan elevados.
Bibliografía
1. Steer A, Gibofsky A. Acute rheumatic fever. Epidemiology and pathogenesis. UpToDate. 2022. Disponible en: www.uptodate.com.
2. Steer A, Gibofsky A. Acute rheumatic fever. Clinical manifestations and diagnosis. UpToDate. 2022. Disponible en: www.uptodate.com.
3. Mosquera Angarita JM, Antón López J. Fiebre reumática y artritis posestreptocócica. Protoc Diagn Ter Pediatr. 2020; 2: 295-309. Disponible en: https://www.aeped.es/sites/default/files/documentos/26_fiebre_reumatica.pdf.
4. Sika-Paotonu D, Beaton A, Raghu A, Steer A, Carapetis J, Ferretti JJ, et al. Acute Rheumatic Fever and Rheumatic Heart Disease. 2017. En: Ferretti JJ, Stevens DL, Fischetti VA, eds. Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet]. Oklahoma City (OK): University of Oklahoma Health Sciences Center; 2016. Bookshelf. Disponible en: www.ncbi.nlm.nih.gov/books/NBK425394/pdf/Bookshelf_NBK425394.pdf
5. Carceller-Blanchard A. Fiebre reumática aguda. An Pediatr (Barc). 2007; 67: 1-4.
6. Cannon JW, Abouzeid M, de Klerk N, Dibben C, Carapetis JR, Katzenellenbogen JM. Environmental and social determinants of acute rheumatic fever: a longitudinal cohort study. Epidemiol Infect. 2019; 147: e79. Disponible en: https://doi.org/10.1017/S0950268818003527.
7. Alm PA. Streptococcal Infection as a Major Historical Cause of Stuttering: Data, Mechanisms, and Current Importance. Front. Hum. Neurosci. 2020; 14: 569519. DOI: 10.3389/fnhum.2020.569519.
8. Cortina Creus P, Alfonso Sánchez JL, Cortés Vizcaíno D, Smeyers Durá P, González Arráez JI. Evolución epidemiológica de la fiebre reumática y cardiopatía reumática en España (1951-1986). Rev San Hig Pub. 1991; 65: 17-24.
9. Yacoub M, Mayosi B, ElGuindy A, Carpentier A, Yusuf S. Eliminating acute rheumatic fever and rheumatic heart disease. Lancet. 2017; 390: 212-13.
10. Karthikeyan G, Guilherme L. Acute rheumatic fever. Lancet. 2018; 392: 161-74.
11. Orefici G, Cardona F, Cox CJ, Cox CJ, Cunningham MW. Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections (PANDAS). En: Ferretti JJ, Stevens DL, Fischetti VA, eds. Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet]. Oklahoma City (OK): University of Oklahoma Health Sciences Center; 2016. Bookshelf. Disponible en: https://www.ncbi.nlm.nih.gov/books/.
12. Jones TD. The diagnosis of rheumatic fever. JAMA. 1944; 126: 481-4.
13. Gordis L. The virtual disappearance of rheumatic fever in the United States: lessons in the rise and fall of disease T. Duckett Jones Memorial Lecture. Circulation. 1985; 72: 1155-62.
14. Denny FW. T. Duckett Jones and rheumatic fever in 1986. T. Duckett Jones Memorial Lecture. Circulation. 1987; 76: 963-70.
15. Gewitz MH, Baltimore RS, Tani LY, Sable CA, Shulman ST, Carapetis J, et al, American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of Doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015; 19; 131: 1806-18.
16. Ståhlgren GK, Tyrstrup M, Edlund C, Giske CG, Mölstad S, Norman Ch, et al. Penicillin V four times daily for five days versus three times daily for 10 days in patients with pharyngotonsillitis caused by group A streptococci: randomised controlled, open label, non-inferiority study. BMJ. 2019; 367: l5337. DOI: 10.1136/bmj.I5337.
17. Rafeek RAM, Hamlin AS, Andronicos NM, Lawlor CS, McMillan DJ, Sriprakash KS, et al. Characterization of an experimental model to determine streptococcal M protein–induced autoimmune cardiac and neurobehavioral abnormalities. Immunol Cell Biol. 2022; 100: 653-66.
18. Iglesias-Gamarra A, Quintana G, Restrepo Suárez JF. Prehistoria, historia y arte de la Reumatología, Gota y espondilitis anquilosante. Rev Colombiana Reumat. 2006; 13: 120-41.
19. Manson G. A History of Rheumatic Fever. Henry Ford Hospital Medical Bulletin. 1959; 7: 145-55. Disponible en: https://scholarlycommons.henryford.com/hfhmedjournal/vol7/iss3/2.
20. Shulmann ST. T. Duckett Jones and his criteria for the diagnosis of acute rheumatic fever. Pediatr Annals. 1999; Disponible en: https://doi.org/10.3928/0090-4481-19990101-04.
21. Parker S. Diagnosing Bertolt Brecht. Lancet. 2011; 377: 1146-47.
22. Cardoso F, Lees AJ. Historical Review Did Gustav Mahler Have Sydenham’s Chorea? Movement Disorders. 2006; 21: 289.92.
23. Oliver J, Osowicki J, Cordell B, Hardy M, Engelman D, Steer AC. Incidence of acute rheumatic fever and rheumatic heart disease in Melbourne, Australia from 1937 to 2013. J Paediatr Child Health. 2020; 56: 1408-13.
24. Quinn RW. Comprehensive review of morbidity and mortality trends for rheumatic fever, streptococcal disease, and scarlet fever: the decline of rheumatic fever. Rev. Infect. Dis. 1989; 11: 928-53.
25. Watkins DA, Karthikeyan G, Beaton A, Bukhman G, Forouzanfar M, Longenecker CT, et al. Global, Regional, and National Burden of Rheumatic Heart Disease, 1990-2015. N Engl J Med. 2017; 377: 713-22.
26. Salinas Salvador B, Moreno Sánchez A, Carmen Marcén G, Molina Herranza D, Arana Navarro T, García Vera C. Estudio retrospectivo sobre la efectividad y seguridad de la pauta antibiótica reducida a 5-7 días en la faringoamigdalitis aguda estreptocócica comparada con la pauta clásica de 10 días. An Pediatr. 2022. Disponible en: https://doi.org/10.1016/j.anpedi.2022.07.001.