|
| Temas de FC |
E. Urbaneja Rodríguez, P. Solís Sánchez
Unidad de Inmunología y Reumatología Pediátricas. Servicio de Pediatría. Hospital Clínico Universitario de Valladolid
| Resumen
La Artritis Idiopática Juvenil (AIJ) es la enfermedad reumática más frecuente en Pediatria (quitar en la infancia) y una importante causa de morbilidad infantil. El término AIJ se define como una artritis que debe aparecer antes de los 16 años y persistir, al menos, 6 semanas, excluyendo otras causas de artritis típicas de la infancia, y engloba a un grupo heterogéneo de artritis dividido en 7 categorías principales (oligoarticular, poliarticular con factor reumatoide negativo, poliarticular con factor reumatoide positivo, sistémica, artritis relacionada con entesitis, artritis psoriásica y artritis indiferenciada), según la última clasificación vigente. La forma sistémica asocia sintomatología general (fiebre, exantema, hepatoesplenomegalia, linfadenopatías, serositis) y tiene una clara base autoinflamatoria; mientras que en el resto de categorías predominan los síntomas articulares y tienen un origen autoinmune. El diagnóstico de la enfermedad es clínico, aunque las pruebas de laboratorio y de imagen pueden ayudar a confirmarlo. La principal complicación asociada es la aparición de uveítis crónica anterior, por lo que siempre será necesario un seguimiento conjunto y periódico, con Oftalmología, de estos pacientes. El tratamiento de la enfermedad depende de múltiples factores y ofrece numerosas posibilidades, siendo, en general, el metotrexato el primer fármaco de elección utilizado para conseguir la remisión de la enfermedad, aunque también se disponen en la actualidad de otras muchas estrategias terapéuticas (fármacos biológicos), que han supuesto toda una revolución en el pronóstico de esta patología. |
| Abstract
Juvenile Idiopathic Arthritis (JIA) is the most frequent rheumatic disease in children and an important cause of childhood morbidity. The term JIA is defined as an arthritis that should appear before 16 years old and persist for at least 6 weeks, excluding other causes of arthritis typical in childhood, and encompasses a heterogeneous group of arthritis divided into 7 main categories (oligoarticular, poliarticular with negative rheumatoid factor, poliarticular with positive rheumatoid factor, systemic, arthritis related to enthesitis, psoriatic arthritis and undifferentiated arthritis) according to the last classification. The systemic form associates general symptomatology (fever, exanthema, hepatosplenomegaly, lymphadenopathy, serositis) and has a clear autoinflammatory base, while in the other categories joint symptoms predominate and have an autoimmune origin. The diagnosis of the disease is clinical, although laboratory |
Palabras clave: Artritis idiopática juvenil; Uveítis; Metotrexato; Terapia biológica
Key words: Juvenile Idiopathic Arthritis; Uveitis; Metotrexato; Biological therapy
Pediatr Integral 2017; XXI (3): 170-182
Artritis idiopática juvenil
Introducción
La artritis idiopática juvenil es la enfermedad reumática más frecuente en la infancia, una de las enfermedades crónicas más prevalentes en niños y una importante causa de morbilidad infantil.
La Artritis Idiopática Juvenil (AIJ) es la enfermedad reumática crónica más frecuente en niños y una de las enfermedades crónicas más frecuentes en la infancia(1). El término AIJ engloba a un grupo heterogéneo de artritis crónicas de causa desconocida que, como premisas fundamentales, deben aparecer antes de los 16 años de edad y persistir durante, al menos, 6 semanas para su diagnóstico, habiendo excluido previamente otras causas conocidas de artritis.
La primera descripción de la enfermedad se realizó por George Frederic Still en 1897, quién detalló en 22 niños un patrón de artritis crónica, asociando algunos de ellos un comienzo sistémico con fiebre, adenopatías y hepatoesplenomegalia(2). Posteriormente, numerosos autores hicieron referencia a la aparición de diferentes formas de la enfermedad, planteándose si se trataba de una única entidad o de varias patologías agrupadas bajo un mismo término.
La denominación actual de AIJ está aceptada internacionalmente y fue acuñada a partir de 1995 por la International League of Associations for Rheumatology (ILAR), suplantando a otros nombres que esta enfermedad recibió con anterioridad: “Artritis Reumatoide Juvenil”, término propuesto en Estados Unidos por el American College of Rheumatology (ACR) y “Artritis Crónica Juvenil”, término utilizado en Europa por la European League Against Rheumatism (EULAR)(3). El uso actual de estos dos últimos términos como sinónimos de AIJ conduce a error y no son equivalentes, ya que en ellos fueron utilizados otros criterios de clasificación, por lo que no deben usarse.
La AIJ se divide, a su vez, en diferentes subtipos según unos criterios unificados de clasificación de la ILAR, revisados por última vez en el 2001 en Edmonton(4). Cada forma clínica presenta unos criterios de clasificación bien definidos (forma clínica, criterios de inclusión y criterios de exclusión), ya que cada categoría presenta diferentes manifestaciones clínicas, evolución, respuesta al tratamiento y pronóstico. Está clasificación está en proceso de validación y continua revisión, con el fin de obtener una categorización lo más perfecta posible que permita agrupar a los pacientes de forma homogénea y, a su vez, facilite la investigación. Los criterios de clasificación actualmente vigentes, que presentan algunas limitaciones, se verán modificados próximamente, cuando exista suficiente evidencia científica que así lo aconseje.
Epidemiología
La prevalencia e incidencia reales de la AIJ son desconocidas, aunque se sabe que la AIJ oligoarticular es el subtipo más frecuente en todas las poblaciones.
La AIJ presenta una distribución mundial, aunque su incidencia y prevalencia reales son desconocidas, probablemente debido a un infradiagnóstico de la enfermedad. En los últimos estudios publicados por Thierry et al., se estima en Europa una incidencia de 1,6-23/100.000 niños menores de 16 años y una prevalencia de 3,8-400/100.000 niños menores de 16 años(5).
En nuestro medio, la AIJ oligoarticular es el subtipo más frecuente (50%), seguido de la AIJ poliarticular (25%), AIJ sistémica (5-15%), artritis relacionada con entesitis (10-15%) y artritis psoriásica (2%). Al aplicar la clasificación de la ILAR, el porcentaje de artritis indiferenciadas puede llegar hasta un 10%. La edad de aparición es variable y característica de cada subtipo de AIJ(3). Existe predominio del sexo femenino en las formas oligoarticular, poliarticular y psoriásica, no hay diferencias en cuanto al sexo en la forma sistémica, y predomina el sexo masculino en la artritis relacionada con entesitis (Tabla I).

Clasificación
La AIJ engloba a un conjunto heterogéneo de artritis, dividido en 7 categorías principales según la última clasificación vigente (Edmonton, 2001), con características mutuamente excluyentes y fenotípicamente bien diferenciadas.
Según la última clasificación vigente de la ILAR, que ya se comentó anteriormente (Edmonton, 2001), se distinguen 7 categorías dentro de la AIJ(4). Dicha categorización en subtipos de la enfermedad se realizó con el fin de clasificar a los pacientes en grupos homogéneos con características similares. La clasificación se realiza a los 6 meses del diagnóstico de la enfermedad, sobre la base de unos criterios bien definidos. Cada categoría de AIJ es mutuamente excluyente, existiendo unos criterios de exclusión aplicados a cada categoría que se definen a continuación de forma general y, posteriormente, según categorías.
Criterios de exclusión
a. Psoriasis en paciente o en familiar de primer grado.
b. Artritis en varón mayor de 6 años HLA-B27 positivo.
c. Antecedentes de: espondilitis anquilosante, artritis relacionada con entesitis, sacroileítis asociada a enfermedad inflamatoria intestinal, síndrome de Reiter o uveítis anterior aguda en paciente o en familiar de primer grado.
d. Presencia de factor reumatoide (FR) en, al menos, dos determinaciones separadas entre sí 3 meses.
e. Presencia de AIJ sistémica en paciente.
Características principales de cada categoría de AIJ
• AIJ oligoarticular: es la categoría más frecuente y la que tiene mejor pronóstico. Se define como una artritis de 4 o menos articulaciones durante los 6 primeros meses de enfermedad. Puede ser persistente (si a lo largo de la evolución de la enfermedad no se afectan más de 4 articulaciones) o extendida (si a lo largo de la evolución de la enfermedad se afectan más de 4 articulaciones). Criterios de exclusión: a, b, c, d y e.
• AIJ poliarticular factor reumatoide negativo: artritis de 5 o más articulaciones durante los 6 primeros meses de enfermedad. La determinación del factor reumatoide es negativa. Criterios de exclusión: a, b, c, d y e.
• AIJ poliarticular factor reumatoide positivo: está categoría se asemeja a la artritis reumatoide del adulto. Se define como una artritis de 5 o más articulaciones durante los 6 primeros meses de enfermedad. La determinación del factor reumatoide debe ser positiva en, al menos, dos determinaciones separadas entre sí 3 meses. Criterios de exclusión: a, b, c y e.
• AIJ sistémica: artritis en una o más articulaciones y fiebre diaria de, al menos, dos semanas de duración (objetivada, al menos, 3 días) con uno o más de los siguientes criterios:
1. Exantema eritematoso evanescente.
2. Adenopatías.
3. Hepatomegalia y/o esplenomegalia.
4. Serositis.
Criterios de exclusión: a, b, c y d.
• AIJ artritis relacionada con entesitis: artritis y entesitis, o artritis o entesitis y dos o más de los siguientes criterios:
1. Dolor sacroilíaco y/o dolor inflamatorio lumbosacro.
2. Presencia de HLA-B27 positivo.
3. Aparición de síntomas en varón mayor de 6 años.
4. Uveítis anterior aguda.
5. Antecedentes de: espondilitis anquilosante, artritis relacionada con entesitis, sacroileítis asociada a enfermedad inflamatoria intestinal, síndrome de Reiter o uveítis anterior aguda en familiar de primer grado.
Criterios de exclusión: a, d y e.
• AIJ artritis psoriásica: artritis y psoriasis, o artritis y dos o más de los siguientes criterios:
1. Dactilitis.
2. Afectación ungueal: lesiones puntiformes en uñas u onicolisis.
3. Familiar de primer grado afecto de psoriasis.
Criterios de exclusión: b, c, d y e.
• AIJ artritis indiferenciada: artritis que no cumple criterios de ninguna categoría o cumple criterios de varias categorías.
A pesar de que los criterios de inclusión y exclusión de cada subtipo de AIJ están bien establecidos, no todos los pacientes se encuentran correctamente clasificados con los criterios actuales, por lo que siguen existiendo aspectos mejorables, siendo cada vez más necesaria una revisión de la clasificación actual que se llevará a cabo en un futuro cercano(6).
Etiopatogenia
La AIJ sistémica es una enfermedad autoinflamatoria, mientras que el resto de formas de AIJ son enfermedades autoinmunes. Estas diferencias etiopatogénicas son la clave de la utilización de diferentes estrategias terapéuticas según el subtipo de la enfermedad.
La AIJ tiene una etiopatogenia multifactorial. Se describen mecanismos inmunológicos y ambientales que actúan en un individuo genéticamente predispuesto(7). Salvo el subtipo de AIJ sistémica, que tiene una base autoinflamatoria con alteración de la inmunidad innata, el resto de formas de AIJ poseen un claro componente autoinmune y se asocian a alteraciones en la inmunidad adaptativa, tanto humoral como celular (Tabla II).

Actualmente, se conoce bien que la AIJ sistémica se produce como consecuencia de una disregulación del sistema inmune innato, caracterizada por una activación anómala de fagocitos que conduce a un aumento de citoquinas proinflamatorias (IL-1, IL-6, IL-18, proteínas S100) que son las responsables de sus manifestaciones clínicas específicas, englobándose, por tanto, dentro de los trastornos autoinflamatorios. Se han descrito polimorfismos en genes que codifican IL-6 y el factor inhibidor de macrófagos en esta forma de AIJ.
En las formas oligo y poliarticulares, existe una respuesta autoinmune mediada por linfocitos T CD4+, que son activados por autoantígenos, produciendo un desequilibrio entre células Th1 y Th17 (aumentadas) y células T reguladoras (disminuidas) que causa un fallo en la tolerancia frente a dichos autoantígenos y un incremento de citoquinas proinflamatorias (IFN-γ, IL-17) con inhibición de citoquinas antiinflamatorias (IL-10), provocando, todo ello, la inflamación sinovial. Existe una cierta asociación de estas formas de AIJ con algunos antígenos leucocitarios humanos, como: HLA-A2 (forma oligoarticular), HLA-DRB1 (forma poliarticular con FR negativo), HLA-DR4 (forma poliarticular con FR positivo), aunque con una asociación menos fuerte que en otros tipos de artritis.
En el caso de las formas de artritis relacionada con entesitis y artritis psoriásica (englobadas ambas dentro de las espondiloartropatías juveniles), existe una respuesta predominante de linfocitos T CD8+, con mayor expresión celular de TNF-α. En la artritis relacionada con entesitis, existe además una fuerte asociación con la presencia de HLA-B27, molécula perteneciente al complejo mayor de histocompatibilidad tipo I, determinando una mayor frecuencia de casos dentro de una misma familia. En ambos subtipos, se han descrito polimorfismos de IL23R-, que codifica el receptor de IL-23, favoreciendo la aparición de estos dos subtipos de AIJ.
Numerosos factores ambientales podrían actuar como desencadenantes de las diferentes formas de la enfermedad, existiendo en los últimos años numerosas publicaciones que intentan relacionar la aparición de AIJ con: antecedentes infecciosos y vacunales, estrés psicológico, lactancia materna, tabaquismo materno durante embarazo, traumatismos, etc.
Un mejor conocimiento en los mecanismos inmunológicos de la AIJ y en los biomarcadores implicados en cada categoría de la enfermedad, podrá facilitar en los próximos años nuevos avances en su inmunopatología e investigar nuevas dianas terapéuticas(8).
Clínica
Es necesario que exista artritis para poder diagnosticar a un niño de AIJ, aunque pueden existir otras múltiples manifestaciones clínicas.
En general, en la mayoría de casos, la AIJ tiene un comienzo lento e insidioso. El niño suele presentar cojera ocasional con rigidez matutina (está peor después del reposo prolongado y mejora a lo largo del día), marcando la descripción de un ritmo inflamatorio, disminuye su actividad física y la tumefacción articular no es muy evidente. Cuando el debut de la enfermedad se produce de forma precoz en época de lactante, estos suelen estar irritables y se niegan a caminar. Cuando la enfermedad avanza, las articulaciones afectas aparecen: inflamadas, calientes al tacto, limitadas en movimiento y, en ocasiones, puede existir dolor a la palpación o con la movilización, aunque el dolor no es un síntoma predominante en la AIJ.
Cada subtipo de AIJ presenta una forma de afectación articular característica(9), que se describe de forma gráfica en la tabla I.
Si se trata de una AIJ oligoarticular, se afectan al inicio del cuadro ≤ 4 articulaciones, siendo lo más frecuente la artritis de grandes articulaciones de extremidades inferiores, sobre todo, rodilla y tobillo (Fig. 1).

Figura 1. Artritis de rodilla derecha en paciente con AIJ oligoarticular.
La afectación aislada de articulaciones de miembros superiores es menos frecuente. En función de si aparecen anticuerpos antinucleares (ANA) positivos o negativos, existirá un mayor o menor riesgo de uveítis en este grupo de pacientes, siendo la categoría de AIJ que, en general, tiene un mayor riesgo de uveítis.
La AIJ poliarticular se caracteriza por afectar a ≥ 5 articulaciones, generalmente de forma simétrica, pudiendo aparecer en miembros superiores e inferiores, afectando incluso a pequeñas articulaciones de dedos de manos y pies (Fig. 2).
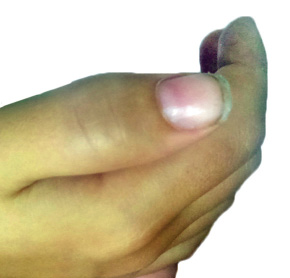
Figura 2. Artritis de articulación interfalángica de 1er dedo mano derecha en paciente con AIJ poliarticular.
Si asocia factor reumatoide positivo su evolución se asemejará a una artritis reumatoide del adulto. La aparición de nódulos reumatoides no es frecuente en la infancia y se asocia a formas graves con factor reumatoide positivo. Es típico de esta categoría la aparición de micrognatia secundaria a la afectación crónica de la articulación temporomandibular. También, es posible la afectación de las vértebras cervicales, ocasionando una disminución de la extensión del cuello y la afectación de caderas.
En la artritis relacionada con entesitis, existe artritis periférica y asimétrica de grandes articulaciones de miembros inferiores, sobre todo: rodilla, tobillo y cadera, pudiendo aparecer entesitis (inflamación en la zona de inserción de un tendón, ligamento, fascia o unión de cápsula articular al hueso, que generalmente afecta a las inserciones de la fascia plantar, tendón de Aquiles y tendón rotuliano) o tarsitis (Fig. 3).
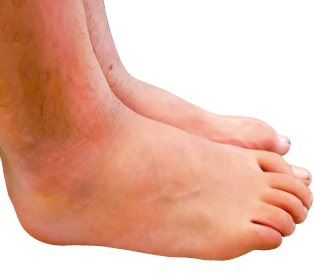
Figura 3. Tarsitis bilateral en paciente varón de 14 años con artritis relacionada con entesitis.
La afectación del esqueleto axial (articulaciones sacroilíacas) es menos frecuente en la infancia y suele aparecer durante la edad adulta.
En la artritis psoriásica, aparece artritis asimétrica que puede afectar a grandes y pequeñas articulaciones, siendo muy característico de este subtipo la aparición de dactilitis o dedo en salchicha (Fig. 4).
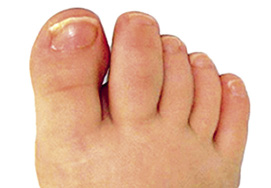
Figura 4. Dactilitis de 2º dedo pie derecho, sugestiva de forma de AIJ asociada a psoriasis
Puede haber psoriasis en hasta el 50% de casos, siendo poco frecuente su presentación simultánea (Fig. 5).
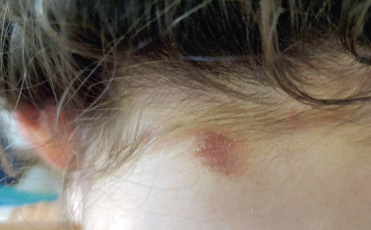
Figura 5. Psoriasis vulgar en niña con AIJ. Recordar su típica aparición en forma de placas eritematosas descamativas que afectan a zonas de extensión y cuero cabelludo.
También, es frecuente la afectación ungueal en forma de pitting u onicolisis.
En algunas ocasiones, el debut de la AIJ no es puramente articular y comienza de forma brusca y grave con participación visceral, ocasionando un cuadro de afectación del estado general, con aparición de fiebre diaria en picos ≥ 39ºC, exantema maculopapuloso de la AIJ sistemica (no pruriginoso, transitorio, predominante en tronco, raíz de miembros y flexuras), que puede acompañarse de fenómeno de Koebner, linfadenopatías generalizadas, hepatoesplenomegalia y pleuritis, pericarditis o peritonitis, además de la asociación de artritis, que definen, en este caso, una AIJ sistémica (Figs. 6 y 7).
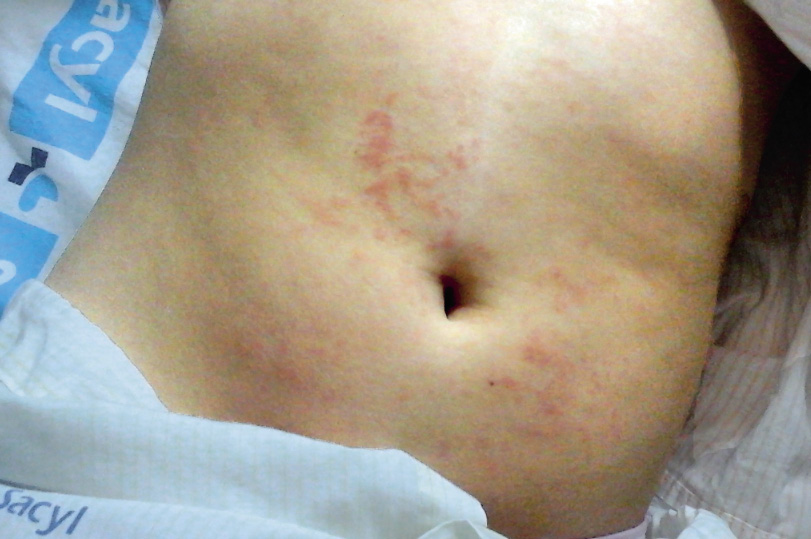
Figura 6. Exantema asalmonado evanescente en paciente con AIJ sistémica.
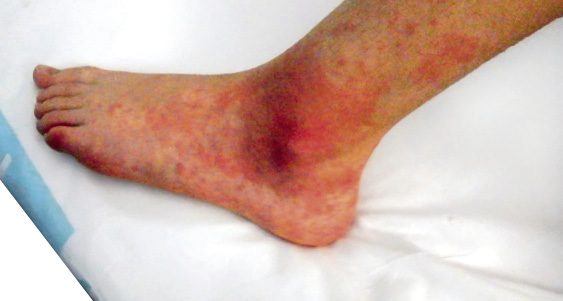
Figura 7. Exantema asalmonado evanescente en paciente con AIJ sistémica.
Inicialmente, la artritis puede afectar a una o varias articulaciones e incluso aparecer con posterioridad al resto de síntomas, pero clásicamente evoluciona a un patrón poliarticular en hasta un 30% de casos. La AIJ sistémica cursa de forma monocíclica con resolución completa del cuadro tras un primer episodio en la mitad de casos, pero en el resto, aparecen brotes repetidos de la enfermedad.
Complicaciones
La uveítis crónica anterior es la manifestación extraarticular más frecuente de la AIJ y su principal complicación.
En pacientes con AIJ, puede existir afectación ocular en forma de uveítis crónica anterior en un 10-30% de casos, sobre todo, durante los primeros cuatro años después del diagnóstico de AIJ y en aquellos pacientes con formas oligoarticulares y ANA positivos, aunque puede presentarse en cualquier momento de evolución de la enfermedad e incluso preceder a la aparición de la artritis. En la mayoría de las ocasiones, la uveítis es asintomática o silente, bilateral y recurrente, por lo que todo paciente con AIJ deberá tener un seguimiento ocular periódico en función de las recomendaciones actualmente vigentes(10,11).
La primera visita al oftalmólogo debería realizarse de forma precoz tras el diagnóstico de AIJ (preferiblemente en el primer mes) y las visitas de seguimiento se realizarán en función del riesgo de uveítis: cada 3 meses en pacientes con alto riesgo, cada 6 meses si riesgo moderado o cada 12 meses si bajo riesgo (Tabla III).
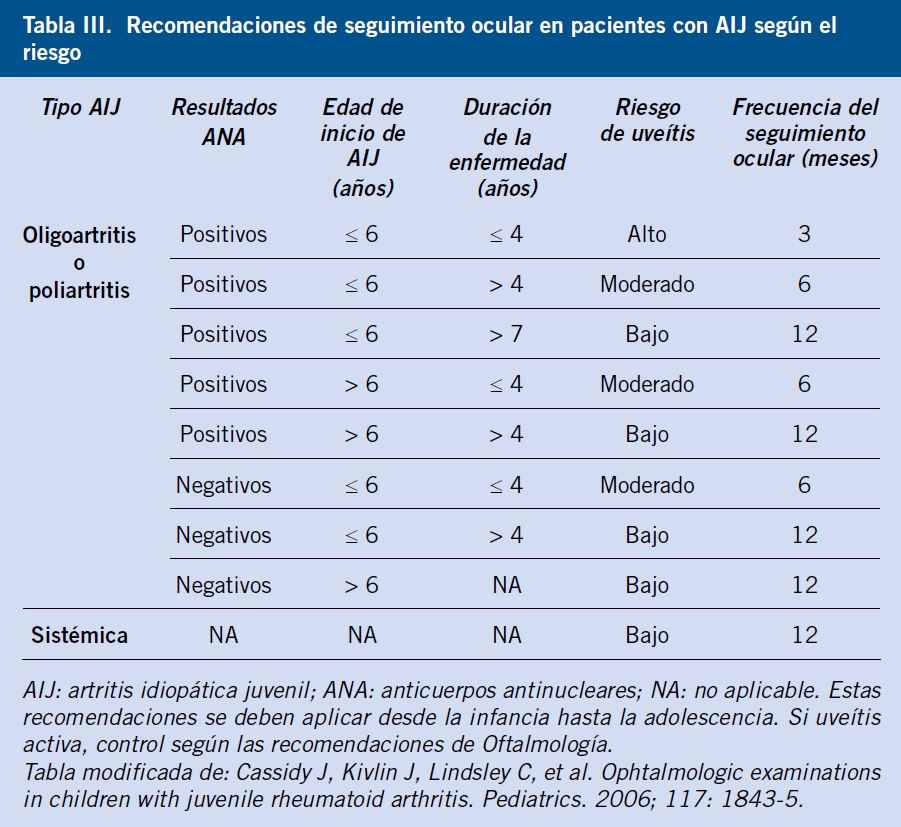
La exploración oftalmológica deberá realizarse siempre con lámpara de hendidura para detectar los signos típicos de uveítis (presencia de células en cámara anterior, debido a la rotura de la barrera hematoacuosa) y establecer el grado de actividad según el grupo Standardization of Uveitis Nomenclature (SUN)(12).
El objetivo fundamental de dicho seguimiento es evitar sus posibles complicaciones: sinequias, cataratas, glaucoma, queratopatía en banda, edema macular quístico y disminución de la agudeza visual. Si aparece uveítis, se deberá iniciar tratamiento específico de forma precoz y conjunta entre oftalmólogos y reumatólogos pediátricos(13).
Otra complicación mucho menos frecuente, pero grave y potencialmente mortal es el síndrome de activación del macrófago (SAM), que aparece, sobre todo, en los casos de AIJ sistémica. El SAM es un término utilizado para describir al grupo de linfohistiocitosis hemofagocíticas secundarias a enfermedades reumáticas. Puede aparecer en un 10% de pacientes con AIJ sistémica y, de forma subclínica, en hasta un 30-40% de casos e incluso puede ser su primera manifestación de una AIJ sistémica y aparecer en forma de recidivas. También, se han descrito casos de SAM secundarios a lupus eritematoso sistémico, enfermedad de Kawasaki y síndromes de fiebre periódica(14). Se caracteriza por una activación y proliferación descontrolada de linfocitos T y macrófagos que sintetizan de forma masiva citoquinas proinflamatorias, cuyo resultado es la llamada “tormenta citoquímica” responsable de sus alteraciones clínicas y analíticas.
Habrá que sospecharlo, cuando un paciente con AIJ sistémica presente de forma aguda un empeoramiento del estado general, con: fiebre alta persistente, hepatoesplenomegalia, linfadenopatías, alteraciones neurológicas (cefalea, irritabilidad, desorientación, obnubilación, convulsiones, coma), tendencia al sangrado (aparición de petequias/púrpura, sangrado en puntos de venopunción, epistaxis) y alteraciones analíticas características (citopenias, elevación de transaminasas, ferritina, lactato deshidrogenasa, triglicéridos y dímero D, con disminución del fibrinógeno y de la velocidad de sedimentación globular). El SAM puede llegar a tener una mortalidad del 8% según los últimos estudios publicados, siendo fundamental un reconocimiento precoz de esta entidad para iniciar un tratamiento adecuado en el menor tiempo posible.
Los niños con AIJ pueden presentar también retraso en el crecimiento y disminución de la masa ósea (osteopenia), secundarios a un estado de inflamación crónico y al tratamiento con corticoides.
Diagnóstico
No existe ninguna prueba de laboratorio o de imagen que confirme el diagnóstico de AIJ. Se trata, por tanto, de un diagnóstico clínico tras exclusión de otras causas de artritis en la infancia.
El diagnóstico diferencial de la AIJ es amplio (causas infecciosas, tumorales, traumáticas y otras enfermedades del tejido conectivo) y está basado fundamentalmente en una buena anamnesis y exploración física.
Datos clave para sospechar una AIJ en la anamnesis
Siempre habrá que diferenciar si los síntomas del niño tienen características mecánicas o inflamatorias (dato cardinal de AIJ). Para ello, debemos preguntar de forma específica si el niño está peor por las mañanas al levantarse después del reposo, presenta rigidez matutina y si va mejorando a lo largo del día. La detección de un ritmo inflamatorio obligará al despistaje de AIJ, entre otras enfermedades reumáticas. Además, será necesario investigar antecedentes familiares de patología reumática, enfermedad inflamatoria intestinal, uveítis y/o psoriasis en familiares de primer grado.
Datos clave para sospechar una AIJ en la exploración física
Siempre buscar lesiones cutáneas y ungueales compatibles con psoriasis (que pueden determinar el diagnóstico de AIJ psoriásica) o exantema maculopapuloso evanescente asalmonado (típico de AIJ sistémica), así como la presencia de hepatoesplenomegalia y/o adenopatías (que pueden acompañar a AIJ sistémica). Respecto al aparato locomotor, deberemos examinar, una a una, todas las articulaciones del niño, buscando datos de tumefacción, movilidad limitada y/o dolorosa. Además, evaluaremos la fuerza muscular, marcha y presencia de dismetrías (la articulación inflamada crece más que la sana).
Pruebas complementarias
Pruebas de laboratorio: ante la sospecha de una AIJ, siempre se deberán solicitar: hemograma, bioquímica (que incluya perfil hepático y renal) y determinación de reactantes de fase aguda (PCR y VSG). La determinación de ASLO, ácido úrico, enzimas musculares, serologías víricas y complemento puede ayudarnos a diferenciar otras causas de artritis. No existe ningún marcador de laboratorio que sea diagnóstico de AIJ; no obstante, la determinación de los anticuerpos antinucleares (ANA), HLA-B27 y factor reumatoide (FR) puede ayudar a la clasificación en subtipos de la enfermedad. Es importante destacar que dichos marcadores pueden estar presentes en niños sanos, no siendo específicos ni diagnósticos de enfermedad reumática. Los ANA positivos determinan, como ya se comentó previamente, un mayor riesgo de uveítis y pueden aparecer en hasta el 70% de formas oligoarticulares, 30-50% de formas psoriásicas y 25% de formas poliarticulares con FR negativo. La realización de una artrocentesis con obtención de líquido de características inflamatorias (líquido amarillento, turbio y poco viscoso, con aumento de celularidad, glucosa algo disminuida y proteínas aumentadas) y cultivo negativo, apoya el diagnóstico y debe realizarse siempre que sea posible.
Pruebas de imagen: la mejor técnica de imagen por su inocuidad, fácil realización y bajo coste es la ecografía articular. En ella, se puede distinguir sinovitis o presencia de derrame articular, características típicas de una articulación afecta de AIJ. Una radiografía ósea será necesaria para descartar procesos tumorales o traumatológicos, con los que es necesario realizar un diagnóstico diferencial al inicio del cuadro, pero no es demasiado útil en el seguimiento del paciente, ya que las lesiones óseas típicas de la AIJ aparecen en la radiografía de forma tardía, provocando una disminución del espacio articular, crecimiento óseo y erosiones. La resonancia magnética es una técnica con alta sensibilidad y especificidad para la detección de artritis, pero, en ocasiones, no está disponible en todos los centros. Existen una serie de recomendaciones sobre las pruebas de imagen más útiles en el diagnóstico y seguimiento de la AIJ, publicadas recientemente que pueden orientar el criterio del médico(15).
Diagnóstico de SAM
El diagnóstico de SAM no es fácil, debido a que sus manifestaciones clínicas y analíticas pueden, en ocasiones, solaparse inicialmente a las de brotes de AIJ sistémica o cuadros infecciosos/sepsis. En el 2016, se han publicado unos criterios conjuntos por EULAR, ACR y PRINTO para facilitar el diagnóstico de SAM en pacientes con AIJ sistémica(16), definiendo una serie de indicadores que se resumen en la Tabla IV.

Es útil remarcar, que para sospechar un SAM es importante que el pediatra sepa reconocer un empeoramiento clínico rápido y un cambio relativo en los valores de laboratorio en un paciente con sospecha de AIJ sistémica o con una AIJ sistémica ya confirmada, para poder hacer un diagnóstico temprano del mismo. En ocasiones, la biopsia de médula ósea no muestra signos de hemofagocitosis de los macrófagos inicialmente, por lo que se recomienda iniciar tratamiento específico del mismo de forma precoz, una vez que la sospecha clínica esté bien establecida.
Tratamiento
El tratamiento de la AIJ debe iniciarse de forma precoz, ya que se dispone de una “ventana terapéutica” en la que se obtiene una mejor respuesta, y realizarse de forma individualizada, dependiendo del subtipo de la enfermedad, la edad al debut, el tipo de afectación y su gravedad.
El tratamiento de la AIJ es complejo y siempre debe realizarse en centros especializados en el manejo de niños con enfermedades reumáticas. Las posibilidades terapéuticas en la AIJ son amplias y, de forma general, podrían entenderse como los peldaños de una escalera en la que se asciende progresivamente en función de las características del paciente y la evolución de su enfermedad (Fig. 8).
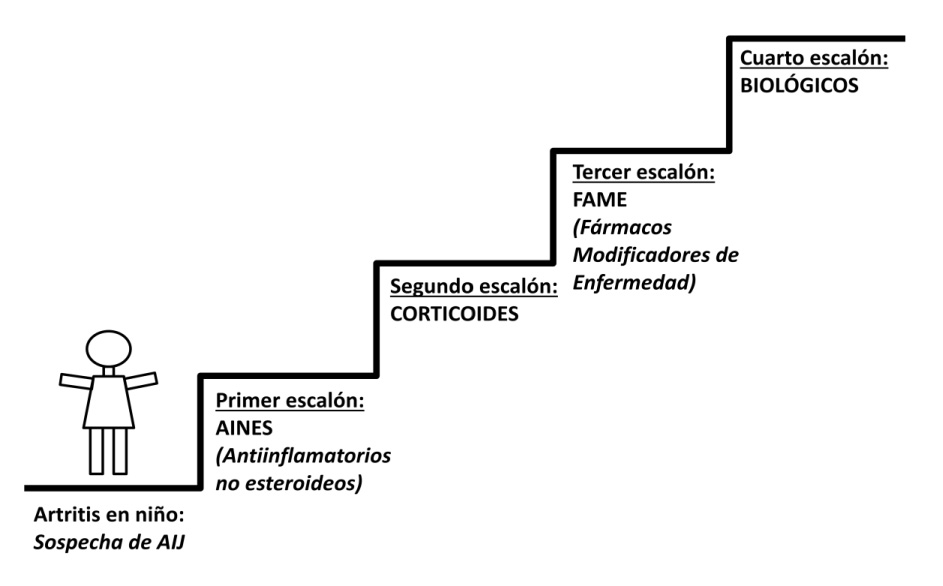
Figura 8. Posibilidades de tratamiento en la AIJ: escalera terapéutica.
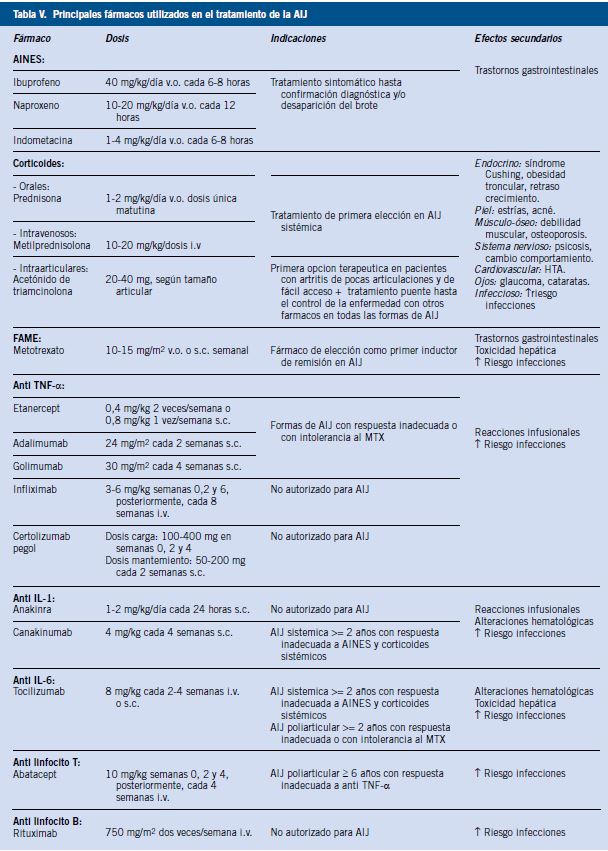
La actitud conservadora en su tratamiento ha cambiado desde hace ya más de una década, imponiéndose actualmente el empleo precoz de fármacos que controlen la actividad de la enfermedad y eviten el daño a largo plazo (Tabla V).
Primer escalón terapéutico: antiinflamatorios no esteroideos (AINES)
Se utilizan únicamente como tratamiento sintomático, ya que tienen efectos analgésico, antiinflamatorio y antipirético, al inhibir la ciclooxigenasa (COX) e interferir en la síntesis de prostaglandinas. No modifican la evolución de la enfermedad. Generalmente, se administran de forma precoz en cualquier subtipo de AIJ, ya sea en el debut de la enfermedad hasta su confirmación diagnóstica, como ante brotes de la misma hasta su adecuado control. Son la mejor opción terapéutica para administrar desde Atención Primaria a un niño en el que se sospeche una AIJ, hasta que pueda ser valorado de forma precoz por pediatras especialistas en este campo.
Segundo escalón terapéutico: corticoides
En la AIJ pueden utilizarse de forma oral, intravenosa o intraarticular. Presentan acción antiinflamatoria e inmunosupresora. Cuando existe artritis de una o varias articulaciones y estas se pueden infiltrar fácilmente, el uso de acetónido de triamcinolona disminuye la actividad de la enfermedad, aunque su efecto no dura más de varios meses. En general, los corticoides suelen utilizarse como terapia puente hasta que se obtiene la efectividad de otros fármacos y son una de las terapias de elección para formas graves y con afectación sistémica. Por sus efectos secundarios, se prefiere su uso durante períodos cortos de tiempo y es importante recalcar que los pacientes que realicen un tratamiento con corticoides sistémicos deben recibir suplementos de calcio y vitamina D.
Tercer escalón terapéutico: fármacos modificadores de enfermedad (FAME)
El más usado y con el que se tiene más experiencia en niños es el metotrexato (MTX), un antagonista del ácido fólico que inhibe competitivamente a la enzima dihidrofolato reductasa, con actividad antiinflamatoria e inmunosupresora (a dosis bajas) y antiproliferativa (a dosis altas). Es el fármaco de elección en la AIJ como primer inductor de remisión de la enfermedad y constituye el eje terapéutico fundamental en estos pacientes. En el 2016, se ha publicado en nuestro país un documento de consenso realizado por expertos en el campo de la Reumatología Pediátrica sobre el uso de metotrexato en pacientes con AIJ y sus principales recomendaciones(17). Puede usarse por vía subcutánea u oral, aunque por esta segunda vía su biodisponibilidad es un 10% menor. Tarda en hacer efecto unas 6-8 semanas, alcanzando su máxima efectividad a partir de los 3 meses de tratamiento. Para reducir sus efectos secundarios, se debe administrar ácido fólico unas 24-48 horas después del MTX.
Otros FAME, como la sulfasalazina o leflunomida, pueden usarse en ciertas formas de AIJ, aunque no suelen emplearse mucho en Pediatría.
Cuarto escalón terapéutico: fármacos biológicos
Han supuesto toda una revolución terapéutica en el campo de la Reumatología Infantil. Actúan de forma específica contra las moléculas que producen la respuesta inflamatoria (moléculas de adhesión, citoquinas, linfocitos B o T, etc.) y su uso ha cambiado radicalmente el pronóstico de esta enfermedad. Son fármacos de prescripción hospitalaria y se aplican de forma subcutánea o endovenosa. Se describen, a continuación, los mejor conocidos en Pediatría:
• Antagonistas del Factor de Necrosis Tumoral Alfa (TNF-α): representan el grupo más numeroso y con el que se tiene más experiencia. Existen dos categorías:
– Anticuerpos antirreceptor soluble del TNF: Etanercept.
– Anticuerpos anti TNF: Infliximab, Adalimumab, Golimumab y Certolizumab pegol.
• Antagonistas de interleucina 1 (IL-1): Anakinra y Canakinumab.
• Antagonista de interleucina 6 (IL-6): Tocilizumab.
• Antagonista de linfocitos T: Abatacept.
• Antagonista de linfocitos B: Rituximab.
Debemos saber que antes del inicio de cualquier terapia con MTX o con fármacos biológicos, habrá que solicitar un Mantoux y realizar serologías frente a virus de la hepatitis B, C y VIH (descartar infecciones latentes, ya que pueden reactivarlas). Además, debemos realizar controles analíticos periódicos, con hemograma y bioquímica (detección de posibles citopenias, alteraciones de perfil hepático, autoanticuerpos séricos). Debido a un mayor riesgo de infecciones, será recomendable que estos pacientes mantengan actualizado su calendario vacunal con administración de vacuna antigripal anual (al niño y a sus convivientes) e incluyendo vacunas frente a organismos encapsulados. No deben administrarse vacunas vivas durante dicho tratamiento.
Existen numerosas guías (Americana del 2011, Canadiense del 2016), en las que se explica de forma detallada diferentes posibilidades terapéuticas con sus peculiaridades, según cada categoría de AIJ. Por su extensión, no se describen en este artículo, aunque pueden resultar de interés si se quiere obtener una orientación sobre el tratamiento específico de una situación clínica determinada(18,19,20).
Tratamiento de la uveítis asociada a AIJ
Inicialmente, está basado en la administración de corticoides y ciclopléjicos tópicos, aunque a menudo se requiere una segunda línea de tratamiento con MTX y, si este no es eficaz, se añade terapia biológica con anti TNF-α (generalmente, Adalimumab), pudiendo utilizar otros fármacos como tratamiento de rescate en casos refractarios (Abatacept, Tocilizumab, Golimumab, Rituximab, etc.), de forma individualizada y en función de la experiencia de cada centro. En el año 2015, se ha publicado un documento de consenso elaborado por expertos en uveítis asociada a AIJ, donde se propone un algoritmo terapéutico y se resume de forma detallada cada opción terapéutica(10).
Tratamiento del SAM
Por la gravedad del cuadro, se recomienda iniciar de forma precoz bolos intravenosos de metilprednisolona a 10-30 mg/kg/día durante 3-5 días consecutivos, seguidos de prednisona oral a 1,5-2 mg/kg/día, con descenso progresivo posterior. Además, son necesarias medidas de soporte como en cualquier situación de riesgo vital. Si no se obtiene respuesta clínica en 24-48 horas desde el inicio del cuadro, se deberá añadir al tratamiento: ciclosporina A oral o endovenosa a dosis de 4-8 mg/kg/día o etopósido a 150 mg/m2/día, siendo, en este caso, todo un reto terapéutico. Cada vez hay más casos publicados con buena respuesta al tratamiento con fármacos biológicos (anti TNF-α y anti IL-1).
Tratamientos complementarios en la AIJ
Se recomienda realizar ejercicio físico de forma periódica y actividades propias de la edad del niño, para mejorar la movilidad articular y potenciar la musculatura. Además, la fisioterapia ocupa un lugar fundamental en la recuperación funcional de estos pacientes. Es importante vigilar también aspectos psicológicos relacionados con cualquier enfermedad crónica, insistiendo en la importancia del apoyo social. Los padres de niños con AIJ deben recibir información detallada de la enfermedad para poder comprender su naturaleza y evolución.
Dentro de la página web de la Sociedad Española de Reumatología Pediátrica (SERPE), se ofrece información rigurosa sobre la AIJ dirigida a profesionales sanitarios, pacientes y familiares, que puede ser muy útil para comprender mejor la enfermedad y ayudar a estos niños.
Evolución y pronóstico
El pronóstico de la AIJ ha mejorado en los últimos años, debido a los múltiples avances en dicho campo, no obstante, varía mucho en función del subtipo de la enfermedad, el grado de afectación y la respuesta al tratamiento.
El pronóstico de la enfermedad ha mejorado mucho durante las últimas décadas, debido fundamentalmente a:
• Creación de unidades especializadas en el manejo de estos pacientes y derivación precoz a las mismas.
• Utilización racional de corticoides.
• Inicio precoz de tratamiento sistémico de la enfermedad.
• Generalización del uso de fármacos biológicos.
Existen diferentes instrumentos de medida (ACR 30/50/70/90, criterios de remisión de Wallace, JADAS 10; 27; 71) de la actividad de la AIJ que se pueden aplicar para evaluar y monitorizar a los pacientes a lo largo de la evolución de su enfermedad. Como ya se ha comentado, la AIJ oligoarticular es la forma que tiene mejor pronóstico, con un 50% de remisiones a los 5-10 años del inicio del cuadro. Las AIJ poliarticulares suelen tener un curso más agresivo y no remiten espontáneamente, sobre todo, las que tienen un factor reumatoide positivo. La artritis relacionada con entesitis tiene un curso variable y puede evolucionar a espondilitis anquilosante en hasta un 25% de pacientes. La artritis psoriásica remite en el 35% de casos. La AIJ sistémica es la forma más grave y suelen ser pacientes con actividad inflamatoria prolongada y de difícil control, en muchas ocasiones.
Función del Pediatra de Atención Primaria
• El reconocimiento precoz de esta enfermedad es fundamental por parte del Pediatra de Atención Primaria para evitar secuelas y complicaciones a largo plazo.
• Cualquier caso sospechoso de AIJ deberá ser enviado de forma precoz para estudio y seguimiento en centro especializado en Reumatología Pediátrica. Hasta que el niño sea valorado en dicho centro, el Pediatra de Atención Primaria podrá iniciar el estudio de artritis en el niño, realizando un exhaustivo diagnóstico diferencial y pautando tratamiento con AINES. No es adecuado el uso de corticoides, ya que pueden enmascarar otros diagnósticos.
• Es necesario realizar un seguimiento estrecho del calendario vacunal de estos enfermos, teniendo en cuenta que en todo paciente con AIJ que recibe tratamiento inmunosupresor, no se deben administrar vacunas vivas y están especialmente recomendadas la vacunal antigripal anual y vacunas contra organismos encapsulados.
• Es fundamental la vigilancia de las infecciones en pacientes con AIJ, ya que estas pueden ser más frecuentes y graves (si los pacientes están sometidos a tratamiento inmunosupresor) e incluso desencadenar brotes de la enfermedad.
• Los pacientes con AIJ sometidos a tratamiento inmunosupresor deberán realizarse analítica sanguínea cada 3 meses, para monitorización de posibles citopenias y elevación de transaminasas.
• Asegurar de que el paciente con AIJ realiza un seguimiento ocular periódico por oftalmólogo experto en uveítis.
• Acompañar al paciente y sus familiares en el largo camino de esta enfermedad crónica, explicando dicha patología, su evolución y posibles tratamientos, siempre de forma coordinada y conjunta con el especialista en Reumatología Pediátrica.
Bibliografía
Los asteriscos reflejan el interés del artículo según los autores.
1.** Kliegman, Stanton, St. Geme, Schor. Nelson Tratado de Pediatría. 20ª Ed. Elsevier. 2016.
2. Still GF. On a form of chronic joint disease in children. Med Chir Trans 80 (1897) 47. Reprinted in Am J Dis Child. 1978; 132: 195-200.
3.*** Petty, Laxer, Lindsley, Wedderburn. Textbook of Pediatric Rheumatology. 7ª Ed. Elsevier. 2016.
4.*** Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J Rheumatol. 2004; 31: 390-2.
5. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014; 81: 112-7.
6. Martini A. It is time to rethink juvenile idiopatic arthritis classification and nomenclature. Ann Rheum Dis. 2012; 71: 1437-39.
7. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011; 377: 2138-49.
8. Swart, Prakken. Understanding inflammation in juvenile idiopathic arthritis: How immune biomarkers guide clinical strategies in the systemic onset subtype. Eur J Immunol. 2016; 46: 2068-77.
9.*** Gowdie PJ, Tse SM. Juvenile Idiopathic Arhtritis. Pediatr Clin North Am. 2012; 59: 301-27.
10. Bou R, Adán A, Borrás F, et al. Clinical management algorithm of uveitis associated with juvenile idiopathic arthritis: interdisciplinary panel consensus. Rheumatol Int. 2015; 35: 777-85.
11.** Cassidy J, Kivlin J, Lindsley C, et al. Ophtalmologic examinations in children with juvenile rheumatoid arthritis. Pediatrics. 2006; 117: 1843-5.
12. Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of uveitis nomenclature for reporting clinical data. Results of the first international workshop. Am J Ophthalmol. 2005; 140: 509-16.
13. Clarke SL, Sen ES, Ramanan AV. Juvenile idiopathic arthritis associated uveitis. Pediatr Rheumatol Online J. 2016; 14: 27.
14. Remesal Camba A, Merino Muñoz R. Síndrome de activación del macrófago. Protoc diagn ter pediatr. 2014; 1: 49-56.
15. Colebatch-Bourn AN, Edwards CJ, Collado P, et al. EULAR-PRES points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015; 74: 1946-57.
16. Ravelli A, Minoia F, Davi S, et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopatic Arthritis: A European League Against Rheumatism/American Collegue of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. 2016; 75: 481-9.
17. Calvo I, Antón J, López Robledillo JC, et al. Recommendations for the use of methotrexate in patients with juvenile idiopatic arthritis. An Pediatr (Barc). 2016; 84: 177.e1-8.
18. Beukelman T, Patkar NM, Saag KG, et al. 2011 American Collegue of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: iniciation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res. 2011; 63: 465-82.
19. Cellucci T, Guzman J, Petty RE, Batthish M, Benseler SM, Ellsworth JE, et al. Management of Juvenile Idiopathic Arthritis 2015: A position statement from the Pediatric Committee of the Canadian Rheumatology Association. J Rheumatol. 2016; 43: 1773-6.
20. Shenoi S, Wallace CA. Diagnosis and treatment of Systemic Juvenile Idiopathic Arthritis. J Pediatr. 2016; 177: 19-26.
21. Solís P. Artritis idiopática juvenil (AIJ). Pediatr Integral 2013; XVII(1): 24-33.
Bibliografía recomendada
- Kliegman, Stanton, St. Geme, Schor. Nelson Tratado de Pediatría. 20ª Ed. Elsevier. 2016.
Libro de referencia internacional en Pediatría. Su última edición disponible en español, aborda en los capítulos 155 y 156 las principales características y formas clínicas de la AIJ.
- Petty, Laxer, Lindsley, Wedderburn. Textbook of Pediatric Rheumatology. 7ª Ed. Elsevier. 2016.
Libro de referencia internacional en el campo de la Reumatología Pediátrica. Su última edición en inglés, aborda de forma detallada todas las enfermedades reumáticas infantiles con las últimas actualizaciones en su etiopatogenia y tratamiento.
- Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J Rheumatol. 2004; 31: 390-2.
Artículo que hace referencia a la clasificación de la AIJ utilizada hasta el momento actual.
- Gowdie PJ, Tse SM. Juvenile Idiopathic Arhtritis. Pediatr Clin North Am. 2012; 59: 301-27.
Excelente revisión en inglés de los aspectos más relevantes de esta enfermedad.
- Cassidy J, Kivlin J, Lindsley C, et al. Ophtalmologic examinations in children with juvenile rheumatoid arthritis. Pediatrics. 2006; 117: 1843-5.
Documento en el que se basa a nivel internacional, el seguimiento ocular de los niños afectos de AIJ.
| Caso clínico |
|
Niña de 12 años derivada a la consulta desde Atención Primaria en Enero del 2016, por artritis de rodilla derecha y artralgias de dedos de ambas manos de larga evolución. Anamnesis Antecedentes familiares: padres sanos. No hermanos. Abuela materna con artritis reumatoide. Prima con enfermedad celíaca. No psoriasis. No enfermedad inflamatoria intestinal. No otros antecedentes reumáticos ni autoinmunes. Antecedentes personales: embarazo, parto y período neonatal normales. Lactancia materna 6 meses. Beikost sin incidencias. Vacunas al día (incluyendo antineumocócica y meningococo B). No alergias conocidas. Desarrollo psicomotor normal. Desde hace 1 mes ha notado la rodilla derecha inflamada, asociando cojera al levantarse, que mejora progresivamente hasta desaparecer a media mañana. Está mejor por las tardes. Mejoría parcial con AINES. No refiere artritis a otros niveles, aunque si artralgias de dedos de ambas manos desde hace 2-3 años, no sabe si alguna vez con inflamación asociada. No antecedente infeccioso ni traumático. Afebril. Exploración física Color normal. No lesiones cutáneas. No masas ni visceromegalias. No adenopatías. Locomotor: rodilla derecha: tumefacta +++ limitada + dolorosa 0. Articulaciones interfalángicas proximales de dedos de ambas manos: tumefactas + limitadas + dolorosas 0 (Fig. 9).
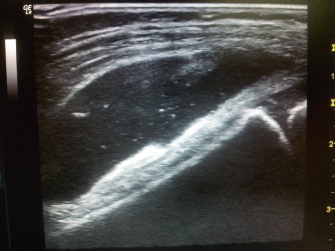
Figura 9. Micrognatia. No otros puntos dolorosos. No dactilitis. No entesitis. Maniobras sacroilíacas negativas. Fuerza conservada. Dismetría 0,5 cm a favor de extremidad inferior derecha. Marcha con cojera derecha intermitente. Exploraciones complementarias • Analítica sangre: hemograma y bioquímica normales (incluyendo transaminasas). PCR: 5 mg/l. VSG: 20 mm. ASLO: 450 UI. ANA: positivos 1/160, FR: negativo, HLA-B27: negativo. • Microbiología: mantoux negativo. Serologías VHB, VHC y VIH negativas. • Ecografía rodilla derecha: moderado derrame articular con leve sinovitis asociada (Fig. 10).
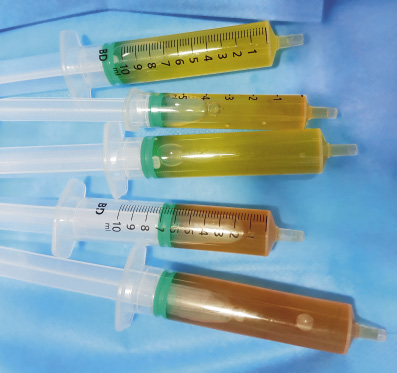
Figura 10. • Artrocentesis rodilla derecha (bajo sedoanalgesia con Kalinox): se extraen unos 40 ml de líquido articular con el aspecto macroscópico que se muestra (Fig. 11).
Figura 11. Leucocitos: 20.000/mm3 (predominio polimorfonucleares), glucosa normal y proteínas aumentadas. Cultivo del líquido articular negativo. Tratamiento Tras resultados microbiológicos que descartan infección latente, con clínica y líquido articular de características inflamatorias, se realizó infiltración de rodilla derecha con acetónido de triamcinolona 40 mg y se inició simultáneamente tratamiento con metotrexato subcutáneo a 15 mg/m2 semanal con suplementos de ácido fólico. Se realizó control analítico al mes del inicio del tratamiento con MTX y, posteriormente cada 3 meses, junto con revisiones en consulta, manteniendo transaminasas normales y reactantes de fase aguda negativos.
|