 |
| Temas de FC |
M.S. Camacho Lovillo*, M.J. Lirola Cruz**
*Hospital Virgen del Rocío de Sevilla. **Instituto Hispalense de Pediatría
| Resumen
Las vasculitis son un grupo de enfermedades heterogéneas caracterizadas por inflamación de los vasos sanguíneos. Su incidencia es baja en la infancia, a excepción de la púrpura de Schölein-Henoch (PSH) y la enfermedad de Kawasaki (EK). Existen ciertos hallazgos clínicos que deben hacer considerar la posibilidad diagnóstica de un cuadro de vasculitis: fiebre prolongada de origen desconocido, lesiones cutáneas sugestivas, neuropatía periférica y/o afectación multisistémica, especialmente renal, pulmonar o cardiovascular, de causa no determinada. Para establecer el diagnóstico, puede ser necesaria la realización de pruebas de imagen y biopsia de los tejidos afectos. |
| Abstract
The vasculitis syndromes are a group of heterogeneous diseases characterized by inflammation of the blood vessels. Its incidence is low in childhood with the exception of Henoch Schönlein purpura (HSP) and Kawasaki disease (KD). In the presence of certain clinical features the possibility of an underlying vasculitic disease must be considered: prolonged fever of unknown origin, suggestive cutaneous lesions, peripheral neuropathy and/or multi-systemic involvement (especially renal, pulmonary or cardiovascular) of unexplained etiology. To establish a correct diagnosis appropriate imaging and biopsy of the affected tissue may be necessary. |
Palabras clave: Vasculitis; Enfermedad de Kawasaki; Púrpura de Schölein-Henoch
Key words: Vasculitis; Kawasaki disease; Henoch-Schölein purpura
Pediatr Integral 2017; XXI (3): 183-195
Púrpura de Shönlein-Henoch, enfermedad de Kawasaki y otras vasculitis
Introducción
Las vasculitis son un grupo heterogéneo de enfermedades que se caracterizan por la inflamación de la pared de los vasos sanguíneos. Las características clínicas dependerán del tamaño, tipo y localización de los vasos afectados. La incidencia estimada de las vasculitis pediátricas se sitúa en 50 casos por cada 100.000 niños por año. Aunque la incidencia global es baja en niños, cierto tipo de vasculitis ocurren casi exclusivamente en la edad pediátrica, como la vasculitis IgA/púrpura de Shönlein-Henoch y la enfermedad de Kawasaki.
Debido a la naturaleza heterogénea de las vasculitis y al conocimiento limitado de sus causas, es difícil establecer subgrupos adecuados. Dado que el diagnóstico no depende de un solo test patognomónico, hablaremos de criterios de clasificación y no de criterios diagnósticos(1).
En la Conferencia de Consenso de Chapel Hill de 2012, se actualizaron las definiciones de vasculitis utilizando el mejor conocimiento sobre la etiopatogenia y hallazgos clínicos de los diferentes tipos. Esto llevó a que algunos de los epónimos fueran reemplazados por nombres más descriptivos y a incluir nuevas categorías (Tabla I)(2).
En 2008, en Ankara, se validaron los criterios de clasificación de PSH, panarteritis nodosa, arteritis de Takayasu y granulomatosis de Wegener(2).
Los hallazgos clínicos y de laboratorio que deben hacer sospechar la existencia de una vasculitis se reflejan en la tabla II.

Las pruebas de imagen son útiles, sobre todo en las vasculitis de medianos y grandes vasos, siendo en la mayoría de casos, necesaria la biopsia de tejidos afectos. Debido a la afectación multisistémica y a la baja incidencia, el diagnóstico es con frecuencia difícil y consecuentemente tardío, lo cual suele asociarse a una importante morbi-mortalidad. El limitado número de estudios específicos en niños condiciona que muchos aspectos sobre su manejo se extrapolen a partir de la experiencia publicada en adultos(1).
Purpura de Shönlein-Henoch
Introducción
La vasculitis IgA (VIgA) es la nueva forma de denominar a la Púrpura de Shönlein-Henoch (PSH) y es la vasculitis sistémica más frecuente en Pediatría.
En la Conferencia de Consenso de Chapel Hill de 2012, las vasculitis de pequeños vasos (VPV) quedaron clasificadas en 2 grandes grupos en función del proceso etiológico subyacente: las vasculitis asociadas a ANCA y las asociadas a inmunocomplejos (IC)(1,2).
Las VPV asociadas a IC se caracterizan por el depósito de inmunoglobulinas y/o factores del complemento predominantemente en las paredes de los pequeños vasos. La VIgA/ PSH es la forma más frecuente en Pediatría, siendo el resto raras en este grupo de edad, afecta a pequeños vasos con depósitos inmunes de IgA1. La IgA es la inmunoglobulina más importante de la inmunidad de mucosa y su glicosilación es fundamental para el aclaramiento de las moléculas IgA1. Desde que se conoce que una glicosilación anómala en la región pesada de la IgA1 lleva a la acumulación de grandes inmunocomplejos, a la activación de la vía alternativa del complemento y reclutamiento de células inflamatorias, el término descriptivo “Vasculitis IgA” ha sustituido al de “Púrpura de Shönlein-Henoch”.
Epidemiologia
La VIgA/PSH es más frecuente durante la infancia (3-15 años) y su incidencia oscila entre los 10 y 20 casos por cada 100.000 niños.
La VIgA/PSH puede aparecer en todos los grupos de edad, siendo más frecuente durante la infancia, ocurriendo el 50% de los casos en menores de 5 años y el 75-90%, en menores de 10 años. Los hallazgos clínicos son, con frecuencia, atípicos en las edades extremas y de mayor gravedad en el adulto.
La incidencia oscila entre los 10 y 20 casos por cada 100.000 niños menores de 17 años, pudiendo alcanzar los 70,3 casos/100.000 en el grupo de edad comprendido entre los 4 y 7 años. La distribución según el sexo es similar, aunque con predominio en varones en algunas series (1,5-2:1). Los afroamericanos rara vez se afectan y es algo más frecuente en niños con ascendente asiático. La enfermedad es más frecuente en invierno y otoño, lo que hace probable la implicación de determinados procesos infecciosos en su patogénesis(3).
Etiopatogenia
La PSH es una vasculitis mediada inmunológicamente, probablemente resultado de la formación de complejos inmunes en respuesta a determinados estímulos antigénicos en personas genéticamente susceptibles.
La patogénesis de la enfermedad continúa siendo desconocida, aunque se le supone una base genética sobre la que actúan factores ambientales desencadenantes. Su predominio estacional y la evidencia clínica apoyan la hipótesis de la participación en su etiología de determinados componentes infecciosos (parvovirus, VHB, VHC, adenovirus, Estreptococo de grupo A betahemolítico, Staphylococcus aureus, Mycoplasma)(4).
Predisposición genética
La mayoría de los casos de VIgA/PSH son esporádicos, sin embargo, se han descrito determinadas asociaciones intrafamiliares. Se postula la participación de los genes del complejo mayor de histocompatibilidad en la patogénesis de la enfermedad. En este sentido, se ha relacionado en varios trabajos, ciertos polimorfismos del HLA-DRB1 y HLA-B*41:02 con la susceptibilidad de presentar una VIgA/PSH y con su gravedad. El HLA B35 y DQA1 se han relacionado con el riesgo de padecer una nefritis en pacientes con VIgA/PSH. El impacto de la expresión de los genes del sistema renina-angiotensina en la presentación y desarrollo de una VIgA/PSH se ha estudiado en varios trabajos. Igualmente, se han encontrado asociaciones entre la VIgA/PSH y los genes que codifican moléculas relacionadas con la inflamación (citoquinas y moléculas de adhesión)(4). La VIgA/PSH aparece hasta en el 7% de los pacientes con fiebre mediterránea familiar (FMF), enfermedad autoinflamatoria determinada genéticamente. Estudios procedentes de Israel y Turquía muestran un aumento significativo de la prevalencia de mutaciones que afectan al gen MEFV, causa de la FMF, en los niños con VIgA/PSH en comparación con la población general. La glicosilación aberrante de la región bisagra de la IgA1 se ha descrito como factor de riesgo para desarrollar una VIgA/PSH y nefritis por IgA (IgAN). Basándose en esto, se han estudiado los polimorfismos del gen C1GALT1, que codifica a la enzima β1,3-galactosiltransferasa, que juega un papel importante en la glicosilación de esta región bisagra.
Glicosilación aberrante de la IgA1
Existen 2 subclases de IgA, la IgA1 y la IgA2. La IgA1 es la subclase predominante (80-90% de la IgA sérica), contiene una región bisagra con múltiples lugares de glicosilación. En la VIgA/PSH, existe una glicosilación aberrante de esta región que predispone a la formación de inmunocomplejos.
Se ha documentado la existencia de niveles séricos elevados de IgA1, complejos inmunes que contienen IgA1 (de pequeño peso molecular), IgA- ANCA, IgA- FR en los pacientes con VIgA/PSH. En aquellos que, además, presentan nefritis (VIgA/PSHN), se detectan complejos inmunes circulantes IgA1-IgG de gran masa molecular. Así mismo, los niveles séricos de IgA1 con defecto de galactosa (Gd-IgA1) son significativamente más elevados en los pacientes con VIgA/PSHN que en los controles sanos y en los pacientes con VIgA/PSH sin nefritis(5).
Citoquinas en la VIgA/PSH
Durante los últimos años, muchos estudios han implicado a diferentes citoquinas proinflamatorias en la patogénesis de la enfermedad (TNF-α, IL-1-β, IL-2, IL-6, IL-8, TGF-β y VEGF). Son segregadas por las células endoteliales vasculares e inician y propagan la respuesta inflamatoria.
Complemento en la VIgA/PSH
Se piensa que la activación del complemento es un importante factor para el daño tisular en la VIgA/PSH. El aclaramiento defectuoso de IC que contienen IgA por el sistema de complemento, juega un papel en la patogenia de la nefritis por IgA y la VIgA/PSH. Los depósitos granulares patognomónicos de IgA y C3 en el mesangio en la VIgA/PSHN son indistinguibles de los que se observan en la nefropatía por IgA.
Daño endotelial y anomalías en la coagulación en la VIgA/PSH
La circulación de IC y alteraciones en la hemostasia pueden provocar daño vascular en la VIgA/PSH. En algunos estudios, se ha relacionado la gravedad de la enfermedad con los niveles séricos de dímeros-D, complejo trombina-antitrombina, fragmentos de protrombina y factor de Von Willebrand, indicando estos hallazgos, probablemente, una reacción local en el vaso sanguíneo inflamado, más que una activación sistémica de la coagulación e hiperfibrinolisis. Una disminución marcada de la actividad del factor XIII podría dar lugar a complicaciones severas, tales como hemorragia intracraneal o hemorragia pulmonar, presumiblemente debido a una degradación específica del factor XIII por las enzimas proteolíticas liberadas por células inflamatorias, con defecto local de la hemostasia. La concentración y actividad del factor XIII podría constituirse como indicador pronóstico en la VIgA/PSH.
Clínica
La VIgA/PSH es una vasculitis sistémica con afectación multiorgánica. La presentación clásica incluye: púrpura palpable, artralgia/artritis, dolor abdominal y enfermedad renal (hematuria/proteinuria) (Tabla III).

Manifestaciones cutáneas
La lesión cutánea característica es la púrpura palpable, pudiendo presentar el paciente desde petequias a grandes equimosis, precediéndose con frecuencia de un exantema maculopapular eritematoso o urticarial. Aparecen de forma simétrica en las zonas declives (miembros inferiores y nalgas) (Figs. 1 y 2), aunque también pueden encontrarse en los brazos, cara, orejas y espalda.

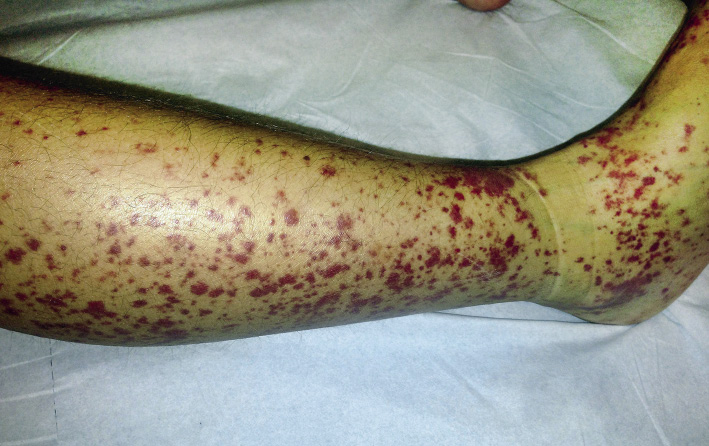
Figura 1 y 2. Exantema característico de púrpura de Scölein Henoch: púrpura palpable en miembros inferiores.
Al inicio del cuadro, y sobre todo en niños pequeños, puede acompañarse de edema de cuero cabelludo, cara, manos, pies y escroto. Las lesiones ampollosas o hemorrágicas y necróticas son raras en los niños (2%), ocurriendo hasta en el 60% del paciente adulto. La recurrencia de la púrpura, que podría estar relacionada con una afectación renal más severa, se observa en un 25% de los pacientes(3).
La biopsia cutánea, si se realizase ante una presentación atípica y dudas diagnósticas, mostraría una vasculitis leucocitoclástica de pequeños vasos con depósitos de IgA e infiltración de neutrófilos y células mononucleares perivasculares.
Manifestaciones digestivas
Se describen en el 50-75% de los pacientes, siendo el primer síntoma de la enfermedad en el 14-36% de los casos. Se producen como consecuencia del edema y la hemorragia secundaria a la vasculitis de la pared intestinal, con mayor afectación del intestino proximal. El síntoma más frecuente es el dolor abdominal, generalmente se trata de un dolor cólico leve-moderado que puede acompañarse de vómitos; en algunos casos, el dolor puede ser muy intenso y limitante. Se encuentra sangre oculta en heces en el 56% de los pacientes, aunque la hemorragia intestinal masiva es rara (2%). La invaginación es la complicación gastrointestinal más frecuente, limitándose al intestino delgado en el 60% de los casos, siendo la incidencia de esta complicación muy variable, hasta en un 2,3-3,5% en algunas series. La pancreatitis, el hidrops vesicular, la enteropatía pierde-proteína y la perforación intestinal son complicaciones raras, pero también descritas. La enteropatía pierde proteínas podría sospecharse ante la presencia de hipoproteinemia en ausencia de proteinuria y/o disfunción hepática severa, pudiéndose confirmar mediante la cuantificación de alfa1-antitripsina en heces(3).
En un trabajo reciente, encuentran manifestaciones digestivas de la enfermedad en el 71% de los pacientes, sin tener ninguna manifestación cutánea el 7,6% de los mismos, llegándose al diagnóstico de VIgA/PSH tras endoscopia oral y biopsia(5). En este mismo trabajo, encuentran que en los pacientes con manifestaciones digestivas, los niveles de dímeros-D y de productos de degradación del fibrinógeno (PDF) están consistentemente más elevados que los marcadores inflamatorios (leucocitos, neutrófilos, VSG, PCR), pudiéndose utilizar estos como marcadores de afectación gastrointestinal en la fase aguda de la VIgA/PSH. La determinación de calprotectina fecal podría ser un marcador útil de afectación gastrointestinal en la VIgA/PSH.
Manifestaciones articulares
La artritis o artralgia puede ser el primer síntoma de la enfermedad en el 15-25% de los pacientes, encontrándose algún grado de afectación articular en el 82% de los mismos. Característicamente, la inflamación es poliarticular, dolorosa, sin eritema ni calor, pero con limitación, afectando con mayor frecuencia a las grandes articulaciones de miembros inferiores. Son transitorias y se resuelven en pocos días sin dejar deformidad(3,6).
Manifestaciones renales
Un 30-50% de los pacientes con VIgA/PSH desarrollarán una glomerulonefritis (PSHN), pudiendo hacerse crónica y dar lugar a un daño renal permanente, motivo por el que es el factor pronóstico a largo plazo más importante de la enfermedad. Se manifestará con: hematuria microscópica/macroscópica, proteinuria, síndrome nefrótico/nefrítico, fracaso renal e hipertensión. En la mayor parte de los pacientes, la afectación es leve y autolimitada. Los niños que no presentan alteraciones urinarias durante los 6 primeros meses de la enfermedad, no desarrollarán una disfunción renal en el seguimiento a largo plazo. El 20% de los pacientes con PSHN (7% de todos los casos de PSH) desarrollarán un síndrome nefrítico o nefrótico. La afectación renal se producirá durante las primeras 4 semanas de la enfermedad en el 75-80% de los pacientes, durante las primeras 6 semanas en el 91% y en los primeros 6 meses en el 97% de los casos, por lo que se aconseja realizar un seguimiento mediante uroanálisis durante, al menos, los seis primeros meses desde el inicio de la enfermedad. El tiempo en que se va a instaurar el daño renal es impredecible, pudiéndose desarrollar incluso años después de alteraciones urinarias menores.
La severidad de los daños histológicos renales, también se correlaciona con el pronóstico renal a largo plazo y, en consecuencia, puede condicionar la toma de decisiones terapéuticas, por lo que se aconseja la realización de una biopsia renal en los casos de VIgA/PSHN más severas(3,7).
La clasificación, desde el punto de vista anatomopatológico de la VIgA/PSHN, está basada preferentemente en la gravedad de las lesiones proliferativas y están definidas según la clasificación para PSH, acordada por el Estudio Internacional de Enfermedades Renales en el Niño (clasificación de Haas).
Manifestaciones neurológicas
Son raras, aunque la cefalea seguida de una ligera encefalopatía con mínimos cambios en el estado mental, tales como: labilidad emocional, apatía e hiperactividad, podría ser más frecuente de lo que se pensaba. Podemos encontrar alteraciones electroencefalográficas y convulsiones. Se han descrito casos de: hematoma subdural, hemorragia subaracnoidea, hemorragia cerebelosa, sangrado intraparenquimatoso e infarto(3).
Manifestaciones pulmonares
La afectación pulmonar es rara. Aunque ocurre con mayor frecuencia en el adulto, se han descrito casos aislados en los niños de hemorragia difusa alveolar, neumonía intersticial y fibrosis intersticial(3).
Manifestaciones urológicas
Las manifestaciones escrotales y testiculares asociadas a la VIgA/PSH son relativamente comunes en los niños, en forma de: escroto agudo, epididimitis, orquitis y complicaciones del cordón espermático (hematoma y edema). Aunque la realización de una eco-doppler normalmente nos permite establecer un diagnóstico, puede llegar a ser necesaria la exploración quirúrgica para descartar la existencia de torsión testicular. De forma poco frecuente, puede existir afectación ureteral durante la fase aguda de la enfermedad o tras resolución de la misma, en forma de obstrucción ureteral o ureteritis. La obstrucción puede ser uni o bilateral, parcial o total, secundaria a una vasculitis periureteral que puede desencadenar una isquemia ureteral. Los síntomas de ureteritis típicamente aparecen 1-2 meses después de la fase aguda de la enfermedad, en ocasiones, asociada a nefritis(8).
Diagnóstico
El diagnóstico de la enfermedad es fundamentalmente clínico. Las diferentes pruebas complementarias irán encaminadas a descartar otras patologías y a conocer el alcance de la enfermedad.
No existen pruebas de laboratorio específicas para el diagnóstico de la enfermedad, por lo que nos basaremos fundamentalmente en los hallazgos clínicos, precisándose, en ocasiones, hallazgos anatomopatológicos. La investigación irá encaminada a descartar otros posibles diagnósticos y a conocer la extensión de la afectación orgánica.
En el estudio inicial, podremos encontrar anemia, leucocitosis con neutrofilia y un discreto aumento de la VSG y de la PCR; en algunos casos, una función renal y/o hepática alterada; en pacientes con proteinuria importante, podemos encontrar hipoalbuminemia. La trombocitosis se ha asociado con enfermedad más severa. Los estudios estándares de coagulación son habitualmente normales, aunque la actividad del factor XIII se encuentra disminuida en relación con enfermedad más severa; no se aconseja su determinación rutinaria. La IgA se encuentra elevada en la mitad de los pacientes y no se correlaciona con la severidad de la enfermedad; pueden encontrarse IC circulantes de IgA. El significado de la presencia de IgA ANCA en la VIgA/PSH, está aún por dilucidar. En general, el estudio básico inmunológico suele ser normal, encontrándose en alguna ocasión, niveles descendidos de C3 y C4. Podremos necesitar diferentes pruebas de imagen para conocer el alcance de la enfermedad gastrointestinal y urológica. En casos de dudas diagnósticas o buscando conocer el alcance de la enfermedad renal, se realizará biopsia cutánea y/o renal.
Tratamiento
Dada la tendencia a la resolución espontánea de la enfermedad, el tratamiento, en la mayor parte de los casos, será conservador, con medidas de sostén, reposo y analgesia.
En caso de manifestaciones cutáneas severas, puede ser necesario el uso de corticoides orales, habiéndose utilizado con éxito agentes ahorradores de corticoides, como la colchicina y dapsona.
La artritis responde generalmente a antiinflamatorios no esteroideos, aunque existen datos sobre una respuesta más rápida y acortamiento de la duración de los síntomas con dosis bajas de corticoides orales.
El uso de prednisolona a 1-2 mg/kg (máximo, 60 mg) se podría considerar en niños con VIgA/PSH y dolor abdominal moderado-severo, una vez descartada patología abdominal potencialmente quirúrgica, como la invaginación y la perforación intestinal. En caso de vasculitis gastrointestinal muy severa (enteropatía pierde-proteínas y la hemorragia gastrointestinal severa), se ha descrito el éxito del tratamiento con: infusión de gammaglobulinas, pulsos de metilprednisolona, plasmaféresis e incluso bolos de ciclofosfamida. El dolor abdominal persistente o crónico es poco común, pero parece responder a metotrexate o micofenolato mofetilo, aunque deben ser valorados sus efectos secundarios gastrointestinales(3,9).
El tratamiento de la VIgA/PSHN sigue siendo controvertido. La toma de decisiones terapéuticas es difícil dada la alta proporción de pacientes con pronóstico favorable y el curso clínico impredecible de pacientes individuales. En algunos estudios retrospectivos, el inicio tardío del tratamiento se asociaba a un peor pronóstico; por lo que, a pesar de la posibilidad de una remisión espontanea, podría ser aconsejable tratar a los pacientes severamente afectados tan pronto como sea posible. El abordaje terapéutico actual se basa en la fisiopatología supuesta y en las series de casos publicados(7).
Los efectos antihipertensivos y renoprotectores de los inhibidores de la enzima convertidora de la angiotensina (IECA) o de los antagonistas del receptor de la angiotensina II (ARA II) están bien documentados en el adulto con hipertensión y/o insuficiencia renal crónica. También, se ha constatado su eficacia en un estudio prospectivo realizado en niños con IgAN. Los corticoides orales o intravenosos forman parte de la mayoría de los regímenes terapéuticos y existe alguna evidencia sobre su efecto beneficioso a largo plazo en adultos con IgAN. En un estudio reciente, se evaluaron de forma retrospectiva a 142 niños con VIgA/PSHN a los que se le realizó biopsia renal. Concluyen que el tratamiento precoz con IECA y ARA-II (tan pronto como la PSHN sea diagnosticada) se relaciona con la existencia de menor proteinuria, al menos, a medio plazo, y que el tratamiento precoz con pulsos de metilprednisolona parece disminuir el riesgo de secuelas(10). De forma análoga, a los pacientes con glomerulonefritis rápidamente progresivas de diferente etiología, la ciclofosfamida se ha utilizado en aquellos pacientes con VIgA/PSHN con manifestaciones más severas. Otras terapias inmunosupresoras, tales como: azatioprina, micofenolato mofetilo, ciclosporina A o rituximab, han sido beneficiosas en casos individuales o pequeñas series de pacientes. En algunos, la plasmaféresis precoz ha sido beneficiosa, incluso sin terapia inmunosupresora adicional(9,10).
Pronóstico
La VIgA/PSH es generalmente una enfermedad autolimitada (en 2-4 semanas), aunque hasta el 33% de los pacientes pueden presentar síntomas recurrentes (entre 1 y 6 episodios). Estas recurrencias suelen acontecer durante los 2-3 primeros meses, aunque se describen recaídas que sobrepasan los 18 meses del inicio de la enfermedad. Normalmente, los síntomas son similares a los del debut y parece ser que aquellos pacientes con afectación renal pueden recaer con mayor facilidad.
El pronóstico a largo plazo de los niños con VIgA/PSH se relaciona predominantemente con la existencia de enfermedad renal. Aunque ningún hallazgo es absolutamente predictivo, muchos estudios coinciden en que la presencia de síndrome nefrítico/nefrótico, la disminución de la actividad del factor XIII, la hipertensión, el desarrollo del fallo renal al inicio de la enfermedad y la presencia de esclerosis glomerular/semilunas/afectación tubulointersticial (lesiones histopatológicas clase IV y V), se considerarían como factores de mal pronóstico; de tal manera que, aunque puedan presentar una recuperación inicial, en el seguimiento a largo plazo (más de 20 años en algunos casos), casi la mitad de estos pacientes pueden presentar hipertensión o insuficiencia renal. Aquellas mujeres a las que se les diagnosticó una VIgA/PSH durante la infancia, presentarán con mayor frecuencia hipertensión arterial y proteinuria durante el embarazo. El tratamiento inicial de la VIgA/PSH con corticoides no previene el desarrollo de nefritis. Por todo lo referido anteriormente, dado el potencial riesgo de deterioro renal de los pacientes con historia de VIgA/PSHN, se aconseja su seguimiento de por vida. En aquellos pacientes sin alteraciones del sedimento urinario y con tensiones arteriales normales, el seguimiento se podría abandonar a los 6-12 meses del inicio de la enfermedad o de la última recaída.
Enfermedad de Kawasaki
Introducción
La enfermedad de Kawasaki (EK) es una vasculitis aguda y autolimitada de etiología desconocida. Constituye la causa más frecuente de enfermedad cardiaca adquirida en la infancia.
Fue descrita por primera vez por Tomisaku Kawasaki, en Japón en 1967. Desde entonces, su incidencia ha ido en aumento, describiéndose en todos los grupos raciales y étnicos. Su importancia se debe a que el 15-25% de los niños no tratados desarrollan anomalías coronarias (AC) que puede conducir a infarto de miocardio, muerte súbita o enfermedad isquémica cardiaca. El tratamiento va dirigido a reducir la inflamación y prevenir el desarrollo de AC(11).
Epidemiología
Afecta a toda la edad pediátrica, aunque con mayor frecuencia a menores de 5 años. La incidencia es más elevada en países asiáticos.
La EK afecta principalmente a niños entre 6 meses y 5 años, existiendo una mayor proporción de varones (1,5:1). La incidencia más alta se ha registrado en Japón (265 casos/100.000 menores de 5 años)(11,12). En nuestro país, se estima que la incidencia anual acumulada es similar a la encontrada en Estados Unidos (20,8/100.000 niños menores de 5 años en 2006). La incidencia es mayor en americanos de origen asiático. En Japón y Corea, la incidencia continúa aumentando, mientras que en Estados Unidos y Canadá permanece estable. Las recurrencias son poco frecuentes (Japón 3% y Estados Unidos 1-2%). Los casos familiares son raros (0,7-2,1%) y el 50% aparece en los 10 días siguientes al primer caso. Existe un predominio estacional que varía de unos países a otros(11,12).
Etiopatogenia
La EK se debe a una respuesta inmunológica inapropiada a uno o más desencadenantes en individuos genéticamente susceptibles(12,13).
La etiología de la EK es aún desconocida, si bien, se piensa que el desencadenante puede ser un agente infeccioso. Sin embargo, las características clínicas pueden ser el resultado de una vía común de inflamación inmunomediada después de una variedad de infecciones, más que un solo patógeno(14). La baja frecuencia en adultos y en los primeros meses de la vida, sugiere un agente que produce inmunidad y del cual están protegidos los lactantes pequeños por el paso de anticuerpos maternos. La hipótesis que relaciona la EK con determinadas toxinas bacterianas estreptocócicas o estafilocócicas que actuarían como superantígenos es, hoy día, controvertida. Los estudios actuales señalan que el desencadenante podría ser un virus y que la puerta de entrada sería el tracto respiratorio(12,13).
La alta incidencia en sujetos de origen asiático y el aumento del riesgo en familiares de pacientes, sugieren la importancia de los factores genéticos en la patogénesis de la EK. Actualmente, existen múltiples estudios de asociación del genoma completo (GWAS) encaminados a identificar marcadores genéticos de susceptibilidad de la enfermedad, de severidad y de resistencia al tratamiento (CTL-4, caspasa 3, linfocito Kinasa B,FCGR2A, IL-10, IL-1B, CD40, PD-1, ORAI1 e inositol 1,4,5 trifosfato3kinasaC[ITPKC]) y HLA clase II(HLA-DQB2) y HLA-DOB(15).
Estudios recientes, que analizan las variaciones estacionales y epidemias, apoyan la hipótesis de que el desencadenante de EK es transportado por vientos de la troposfera y que las provincias del noreste de China sirven como fuente para la epidemia anual de EK en Japón, Hawaii y sur de California(12).
Una etiología autoinmune primaria es improbable, debido a la naturaleza autolimitada y no recurrente de EK. Se postula que en la respuesta aguda de EK intervienen tanto la inmunidad innata como la adaptativa, siendo la inmunidad innata importante en el desarrollo de AC(12).
El paso que conduce a la arteritis coronaria aún no está del todo aclarado. En el epitelio bronquial, se produce un infiltrado primero de neutrófilos, seguido de monocitos/macrófagos CD68, linfocitos CD8 y células plasmáticas Ig A. El trigger infeccioso fagocitado por los macrófagos pasaría a través del torrente sanguíneo al tejido coronario donde produciría un aumento de citocinas y enzimas, como CD40L, VGEF (factor de crecimiento del endotelio vascular), MCP-1, TNF α, IL 1 e IL 6 y metaloproteinasas, que contribuirían al daño vascular. La inflamación acaba produciendo una destrucción de la íntima y la formación de aneurismas. Las células T reguladoras (Tregs) están disminuidas en sangre periférica en EK aguda y aumentados los Th17(16). Existen evidencias de la activación de la vía de IL-1 y del predominio de citocinas de los ejes IL-6/Th17 y IL-12/interferon gamma(12,13).
Clínica
En ausencia de una prueba diagnóstica específica o características clínicas patognomónicas, se han establecido unos criterios clínicos para ayudar al diagnóstico de EK (Tabla IV)(11).

Es característico que todas las manifestaciones clínicas no se presenten a la vez en el tiempo; por lo que, a veces, es necesario esperar varios días antes de hacer el diagnóstico(11).
Es importante excluir otras enfermedades con manifestaciones clínicas similares. El polimorfismo de los signos y síntomas obliga a considerar un amplio diagnóstico diferencial, que incluye procesos infecciosos, alérgicos o tóxicos y reumatológicos (Tabla V)(14).

El término de Kawasaki incompleto se refiere a pacientes que, aunque no cumplen suficientes criterios, pueden ser diagnosticados de EK. Es más frecuente en menores de 1 año y mayores de 9. En 2004, la Asociación Americana de Cardiología ha desarrollado un algoritmo de actuación para la EK incompleta, basada en opiniones de expertos, que utiliza los datos clínicos, analíticos y ecocardiográficos para mejorar el diagnóstico que, en muchos casos, resulta difícil (Algoritmo 1). Debe considerarse el diagnóstico de EK en todos los niños con fiebre inexplicable de 5 o más días de duración, con 2 o 3 criterios clínicos principales y en cualquier niño menor de 6 meses con fiebre de más de 7 días de duración con hallazgos de laboratorio compatibles con inflamación y sin causa explicable de la fiebre(11).
Manifestaciones clínicas principales
La fiebre suele ser elevada, en picos y, a menudo, resistente a antitérmicos. Los cambios en las extremidades son característicos. En la fase aguda, suele aparecer eritema de palmas y plantas. Pueden estar, además, edematosas y el niño evita coger objetos y la deambulación. La descamación se inicia en la región periungueal a las 2 o 3 semanas del comienzo de la fiebre y puede extenderse a palmas y plantas (Fig. 3).
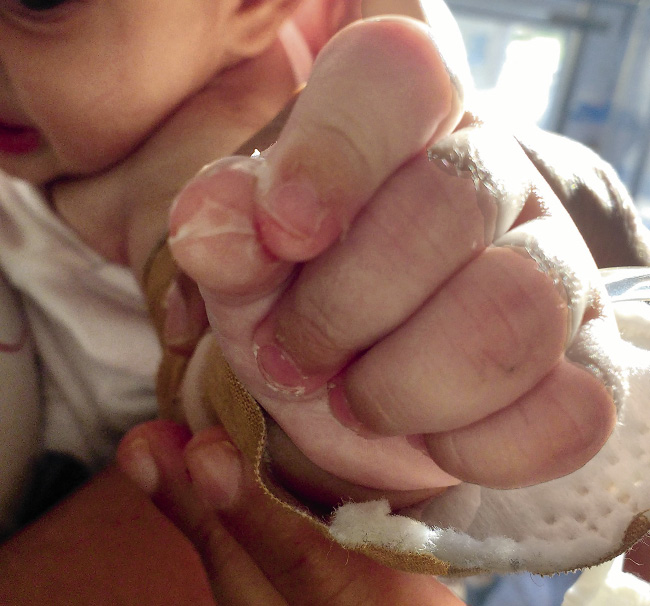
Figura 3. Descamación ungueal en manos en fase subaguda.
1-2 meses tras el inicio de la fiebre, pueden aparecer las líneas de Beau (surcos transversales en las uñas). A los 5 días del inicio de la fiebre, suele aparecer un rash eritematoso. Puede presentarse de muchas formas, aunque la más frecuente es una erupción maculopapular difusa inespecífica (Fig. 4).

Figura 4. Exantema inespecífico maculopapular. Edema en dorso de pies.
A veces, se manifiesta, como: urticaria, rash escarlatiniforme, eritrodermia, similar al eritema multiforme o, menos frecuente, erupción micropustular. No se han descrito las formas bullosas, vesiculares y petequias. Suele localizarse en tronco y extremidades, acentuándose en la región perineal, donde puede aparecer una descamación temprana (Fig. 5).
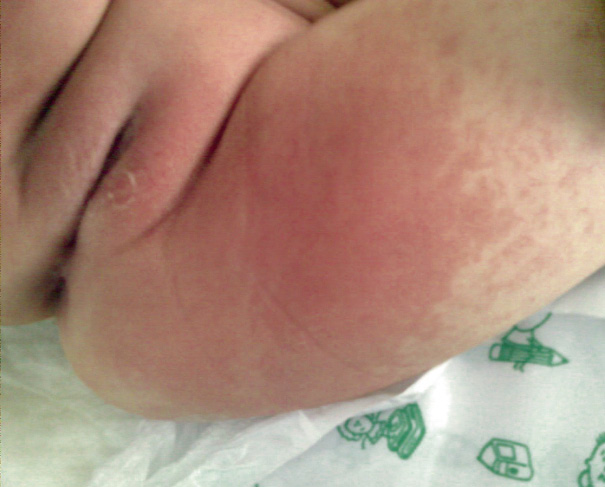
Figura 5. Exantema perineal con descamación precoz.
La inyección conjuntival bilateral suele aparecer poco después del inicio de la fiebre. Afecta de forma característica a la conjuntiva bulbar y no se asocia a exudado, edema conjuntival o ulceración corneal. Normalmente, no es dolorosa. Los cambios en los labios y la cavidad oral incluyen: eritema (Fig. 6), sequedad, fisuras, descamación, grietas y sangrado de labios, lengua aframbuesada indistinguible de la escarlatina, y eritema difuso de la mucosa orofaríngea.
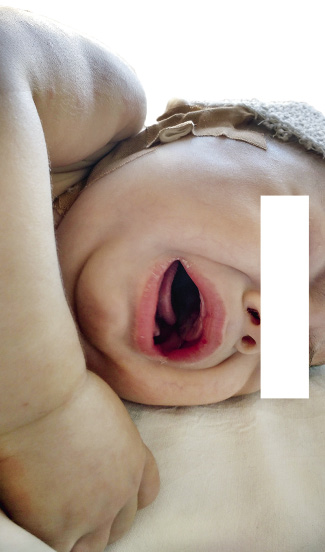
Figura 6. Lactante irritable con labios rojos y edema en manos.
No suelen verse úlceras orales y exudado faríngeo. La linfadenopatía cervical es la menos frecuente de todas las características clínicas principales. Suele ser unilateral y localizada en el triángulo cervical anterior. Para que sea criterio de la EK debe haber una o más linfadenopatías mayores de 1,5 cm de diámetro. Suelen ser firmes, no fluctuantes y sin eritema en la piel. Puede iniciarse como una adenopatía cervical confundiéndose con una adenitis bacteriana o con un edema retrofaríngeo. Pueden aparecer otras manifestaciones descritas en la tabla VI(11).

Son formas poco frecuentes de presentación, pero de especial gravedad, el shock y el síndrome de activación macrofágica(15).
Manifestaciones clínicas cardiovasculares asociadas
En los primeros 10 días no suelen detectarse aneurismas coronarios, pero puede apreciarse mediante ecocardiografía un aumento de la brillantez que rodea la luz arterial o ectasias. Esta lesión precoz puede resolverse o evolucionar hacia aneurismas. También se puede observar disminución de la función ventricular, regurgitación valvular o derrame pericárdico. Los aneurismas se suelen detectar en la fase subaguda (4-6 semanas de enfermedad)(11).
Hallazgos de laboratorio
Las pruebas de laboratorio no son específicas, pero pueden contribuir al diagnóstico (Tabla VI). Suelen normalizarse de 6 a 10 semanas después del inicio de la enfermedad. El biomarcador N-terminal tipo pro-B péptido natriurético (NT-pro BNP) se correlaciona con marcadores de inflamación, estrés oxidativo y disfunción diastólica cardiaca. Algunos estudios sugieren que se eleva en pacientes con EK en comparación con controles febriles y que podría servir como dato analítico suplementario para diferenciar la EK de otros procesos febriles; para facilitar el diagnóstico de EK incompleto e incluso podría ser marcador de riesgo de desarrollo de AC(11).
Tratamiento
El tratamiento con IGIV, dentro de los 10 primeros días del comienzo de la enfermedad, disminuye la incidencia de AC desde un 20-25% a menos de un 5%.
Ya que no se ha podido establecer un escore de riesgo, en población no asiática, que nos permita distinguir pacientes con mayor probabilidad de desarrollar AC, todos los pacientes diagnosticados de EK deben ser tratados con IGIV(15).
La IGIV se utiliza a dosis elevadas (2 g/kg) en una sola infusión. Su mecanismo de acción no es del todo conocido. Parece tener un efecto antiinflamatorio, disminuyendo la producción de citocinas proinflamatorias y aumentando la producción de antagonista del receptor de IL1(12). Es un producto seguro, con efectos secundarios poco frecuentes (reacción infusional, anafilaxia, urticaria, meningitis aséptica, anemia hemolítica autoinmune, tromboembolismo y fallo renal agudo). Aunque solo se han demostrado los beneficios para pacientes tratados en los primeros 10 días de la enfermedad, se recomienda su empleo en niños con evidencia de inflamación persistente (persistencia de fiebre o elevación de reactantes de fase aguda) o alteraciones coronarias, que son diagnosticadas después de esta fecha. Tras administración de IGIV, se recomienda retrasar 11 meses las vacunas que contengan sarampión o varicela, ya que pueden disminuir la respuesta vacunal(16).
La aspirina se emplea por sus efectos antiinflamatorios (a dosis alta) y antitrombóticos (dosis bajas). Aunque no está demostrado que reduzca la incidencia de dilatación coronaria, si parece disminuir la incidencia de infartos miocárdicos fatales. La dosis a emplear es controvertida. Dosis bajas de 30-50 mg/kg/día en 4 dosis, no han demostrado ser menos eficaces en la prevención de AC que las dosis altas de 80-100 mg/kg/día durante la fase aguda, y son mejor toleradas(15). Tras 48-72 horas afebril, la dosis de aspirina se puede reducir a dosis antitrombóticas (3-5 mg/kg/día). El uso de ibuprofeno concomitante antagoniza el efecto antitrombótico de la aspirina. Los pacientes que reciben salicilatos de forma crónica deben vacunarse anualmente de la gripe y estar vacunados de la varicela(11,16).
Enfermedad de Kawasaki refractaria
Entre 11,6 y 38,3% de pacientes tratados inicialmente con IGIV y aspirina a dosis alta, tienen fiebre persistente 24-48 horas tras la primera dosis de IGIV o bien recurre a las 36 horas o más después de completar la infusión de IGIV. Estos pacientes tienen mayor riesgo de desarrollo de AC. La progresión a aneurismas, a pesar de un diagnóstico y un tratamiento adecuado, ha impulsado los estudios hacia tratamientos más efectivos(9,11,12,15).
La utilización de corticoides es controvertida, si bien, cada vez más estudios apoyan su empleo. En el meta-análisis de Chen, et al. y el estudio RAISE concluyeron que la combinación de corticoides con IGIV, como tratamiento inicial en EK grave, reduce el riesgo de desarrollo de AC sin aumentar los efectos secundarios(17,18). Los expertos recomiendan valorar el uso de corticoides en: pacientes con EK refractaria; pacientes con enfermedad severa (menores de 1 año, marcadores de inflamación severa, como PCR elevada persistente, disfunción hepática, hipoalbuminemia y anemia; y características de linfohistiocitosis hemofagocítica o shock); y pacientes con afectación coronaria en la primera semana de la enfermedad. No está establecida la dosis, duración y vía óptima de administración(15). Un meta-análisis reciente apoya que el tratamiento con corticoides de inicio, añadido a IGIV disminuye el riesgo de AC y que, sobre todo, se benefician los pacientes con mayor riesgo de resistencia a IGIV(19). Es importante que estos pacientes con más riesgo reciban un tratamiento a tiempo y más agresivo.
Los escores de riesgo de resistencia a IGIV utilizados en población japonesa tienen una baja sensibilidad en otras poblaciones(15). Anemia severa, trombocitopenia, hipoalbuminemia, elevación de alanino aminotransferasa (ALT) por encima de 200 U/L y PCR y VSG muy elevadas o persistentemente aumentadas, se correlacionan con el riesgo de desarrollar AC(15). Se identifican, además, como factores de riesgo de resistencia a IGIV: la elevación de bilirrubina, polimorfonucleares y NT- pro BNP y la disminución del sodio en sangre(20). También, son factores de riesgo de aparición de AC, las edades extremas y la larga duración de la fiebre antes del tratamiento. Los menores de 6 meses y mayores de 8 años suelen tener una presentación atípica, una mayor demora diagnóstica y, probablemente, una mayor vulnerabilidad genética(11).
En la EK refractaria, la mayoría de los expertos recomiendan retratamiento con IGIV y/o corticoides por el elevado riesgo de desarrollo de AC, previa reevaluación clínica, por si existe un diagnóstico alternativo(15). Se puede considerar el uso de anti TNFα. El tratamiento con infliximab (anticuerpo monoclonal anti TNFα) en EK refractaria, puede ser tan seguro y efectivo como la IGIV en cuanto a duración de síntomas, si bien, se requieren más estudios que definan su papel como primera línea de tratamiento. En caso de fracaso a la segunda dosis de IGIV, corticoides y anti TNFα, se pueden considerar ciclofosfamida, ciclosporina o plasmaferesis(15).
Actualmente, está en marcha un estudio con anakinra (antagonista del receptor de IL-1), como tratamiento de EK refractaria, y otro para evaluar la eficacia de ciclosporina combinada con IGIV, como tratamiento de EK severa en Japón(9). El abciximab (anticuerpo monoclonal trombolítico) se ha empleado en pacientes con aneurismas gigantes en la fase subaguda(11). Se está llevando a cabo un estudio con anakinra y otro con atorvastin (estatina) en pacientes con EK y aneurismas coronarios(12).
Pronóstico
Muchos casos de infartos en personas jóvenes (3ª-4ª década), son atribuidos a una infradiagnosticada EK en la infancia.
El pronóstico de los pacientes sin AC o mínima dilatación a las 6 semanas del inicio de la enfermedad, es normalmente bueno(15). En los pacientes en los que persiste el aneurisma más allá de 6 semanas, precisan un seguimiento cardiovascular hasta la edad adulta, aunque haya una resolución ecocardiográfica, ya que la remodelación se realiza por fibrosis y proliferación del tejido endotelial(12). La mortalidad (0,17% en Estados Unidos) se relaciona, siempre, con la persistencia de secuelas cardiacas. La causa más frecuente de muerte es el infarto de miocardio por trombosis de aneurismas, que suele suceder durante el primer año de enfermedad(11). En varones japoneses con secuelas cardiacas por EK existe un aumento de 1,86 en la tasa estandarizada de mortalidad(14).
En Estados Unidos el tamaño del aneurisma por ecocardiografía se determina en base al tamaño de la luz interna del segmento proximal coronario en relación a la superficie corporal, y se expresa en desviaciones estándares de la media. Se considera aneurisma pequeño si z score ≥ 2 y <5, mediano si ≥ 5 y <10 y gigante si z score ≥ 10 o > de 8 mm(12). El 50-70% de los aneurismas coronarios se resuelven en 1-2 años, si bien, los aneurismas gigantes no se resuelven totalmente(15).
El seguimiento evolutivo de pacientes con EK se basa en la estratificación de riesgo relativo de isquemia miocárdica. Se suele recomendar estudio ecocardiográfico al diagnóstico, a las 2-3 y a las 6-8 semanas del comienzo de la enfermedad y, en función de los hallazgos, se establecen los niveles de riesgo:
• Nivel de riesgo I: pacientes sin AC en ninguno de los estadios. Pueden suspender tratamiento con aspirina a las 6-8 semanas de inicio de la enfermedad.
• Nivel de riesgo II: pacientes con ectasia transitoria de la arteria coronaria en la ecocardiografía (desaparece durante la enfermedad aguda) que pueden recibir recomendaciones similares a las del nivel I y continuar reevaluación cardiológica cada 3-5 años.
• Nivel de riesgo III, IV y V: pacientes con aneurismas que precisan mantener tratamiento con aspirina, con anticoagulación si son aneurismas gigantes, restricción de actividad física y controles cardiológicos periódicos. La angio resonancia magnética y el angio tomografía computada se utilizan cada vez con más frecuencia en el seguimiento de pacientes con AC(11,12).
El riesgo cardiovascular a largo plazo en pacientes con EK sin AC es desconocido; si bien, parece que puede ser un factor de riesgo para el desarrollo de arteriosclerosis precoz en la edad adulta debido a una disfunción endotelial, por lo que se deben generalizar a todos ellos las recomendaciones sobre factores de riesgo cardiovascular: dieta saludable, ejercicio moderado, peso adecuado, control de tensión arterial y evitar consumo y exposición al tabaco.
Otras vasculitis
Vasculitis asociadas a ANCA
Son raras en los niños, con una incidencia de 2,4 casos por millón y año. La poliangeítis con granulomatosis (PAG), antes denominada granulomatosis de Wegener (GW), es la más frecuente. Para diagnosticarla deben de estar presentes, al menos, 3 de las 6 características: afectación renal (hematuria, proteinuria, glomerulonefritis pauciinmune necrotizante), granulomas necrotizantes en las biopsias, afectación de vía aérea superior, afectación laringotraqueobronquial, radiografía o TAC pulmonares patológicos y ANCA positivos. La poliangeítis microscópica (PAM) se define como: una vasculitis necrotizante de pequeños/medianos vasos que se asocia frecuentemente a GN pauci-inmune y ANCA (90%). La poliangeítis granulomatosa eosinofílica (PGE), antes denominada síndrome de Churg-Strauss (SCS), se define como: una vasculitis necrotizante de pequeño/mediano vaso que a menudo afecta al tracto respiratorio superior, junto con inflamación granulomatosa rica en eosinófilos. Se caracteriza por un inicio tardío de asma y frecuentes rinitis-sinusitis no infecciosas.
Vasculitis de mediano vaso
Panarteritis nodosa (PAN): Los criterios de clasificación para los niños son: evidencia histológica de vasculitis necrotizante en arterias de mediano-pequeño tamaño o anomalías angiográficas (angiografía convencional si la angioRMN es negativa) más uno de los siguientes: afectación cutánea, mialgias o sensibilidad muscular, hipertensión, neuropatía periférica o afectación renal.
Vasculitis de grandes vasos (VGV)
Arteritis de Takayasu (TA): Afecta a la aorta y sus ramas principales. Tiene una incidencia mayor en el este de Asia. Para su clasificación se requerirá: demostración en una angiografía (convencional, TAC o RM) de un aneurisma/dilatación, estenosis, oclusión, engrosamiento de la pared arterial y, al menos, uno de los siguientes: falta de pulso o claudicación; soplos; discrepancia de la tensión arterial en los cuatro miembros mayor de 10 mmHg; hipertensión arterial (>p95 según edad); y elevación de reactantes de fase aguda. El síntoma más frecuente en los niños, es la hipertensión arterial seguida de las manifestaciones músculo-esqueléticas.
Vasculitis monogénicas
Recientemente, se han descrito enfermedades monogénicas autoinflamatorias con un importante componente de vasculitis.
• DADA2: la deficiencia de adenosina deaminasa tipo 2 es una enfermedad autosómica recesiva, causada por una mutación del gen CECR1, que se asemeja a la PAN. Los hallazgos clínicos cardinales son: livedo racemosa, afectación neurológica con propensión a los infartos lacunares, neuropatía periférica vasculítica, isquemia digital y ulceraciones cutáneas e inflamación sistémica.
• CANDLE: dermatosis neutrofílica atípica crónica con lipodistrofia y temperatura elevada. Es un síndrome autoinflamatorio asociado al proteasoma. En estadios precoces en la histología, se puede encontrar vasculitis leucocitoclástica/neutrofílica.
• SAVI: estimulador de los genes de interferón (STING) asociados a vasculitis de la infancia. Surge de una mutación del gen TMEM173. Se presenta en edades tempranas con un exantema vasculítico que afecta a mejillas, nariz y zonas acras con ulceraciones crónicas y enfermedad pulmonar intersticial progresiva asociada a hipertensión pulmonar(1,9).
Bibliografía
Los asteriscos reflejan el interés del artículo según los autores.
1.*** Batu ED, Bilginer Y. Classification of Vasculitis in Childhood. Ann Paediatr Rheum. 2016; 5: 1-10.
2. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013; 65: 1-11.
3. Trnka P. Henoch–Schönlein purpura in children. J Paediatr Child Health. 2013; 49(12): 995-1003.
4. Park SJ, Suh JS, Lee JH, Lee JW, Kim SH, Han KH, et al. Advances in our understanding of the pathogenesis of Henoch-Schönlein purpura and the implications for improving its diagnosis. Expert Rev Clin Immunol. 2013; 9(12): 1223-38.
5. Hong J, Yang HR. Laboratory markers indicating gastrointestinal involvement of henoch-schönlein purpura in children. Pediatr Gastroenterol Hepatol Nutr. 2015; 18(1): 39-47.
6. Wang X, Zhu Y, Gao L, Wei S, Zhen Y, Ma Q. Henoch-Schönlein purpura with joint involvement: Analysis of 71 cases. Pediatr Rheumatol Online J. 2016; 14(1): 20.
7.** Pohl M. Henoch–Schönlein purpura nephritis. Pediatr Nephrol. 2015; 30: 245-52.
8. Dalpiaz A, Schwamb R, Miao Y, Gonka J, Walzter W, Khan SA. Urological Manifestations of Henoch-Schonlein Purpura: A Review. Curr Urol. 2015; 8(2): 66-73.
9.*** Eleftheriou D, Brogan PA. Therapeutic advances in the treatment of vasculitis. Pediatr Rheumatol Online J. 2016; 14(1): 26.
10.** Tudorache E, Azema C, Hogan J, Wannous H, Aoun B, Decramer S, et al. Even mild cases of paediatric Henoch-Schönlein-Purpura nephritis show significant long-term proteinuria. Acta Paediatr. 2015; 104(8): 843-8.
11.*** Newburger J, Takahashi M, Gerber M, Gewitz M, Tani L, Burns J, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: Statement for Health Professionals From the Committee on Rheumatic Fever, A Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004; 114; 1708-33.
12.** Newburger JW, Takahashi M, Burns JC. Kawasaki disease. J Am Coll Cardiol. 2016; 67: 1738-49.
13. Wang CL, Wu YT, Liu CA, Kuo HC, Yang KD. Kawasaki Disease Infection, Immunity and Genetics. Pediatr Infect Dis J. 2005; 24: 998-1004.
14. Cohen E, Sundel R. Kawasaki Disease at 50 Years. JAMA Pediatr. 2016; 170(11): 1093-109.
15.** Leftheriou D, Levin M, Shingadia D, Tulloh R, Klein NJ, Brogan PA. Management of Kawasaki disease. Arch Dis Child. 2014; 99(1): 74-83.
16.** Esposito S, Bianchini S, Dellepiane RM, Principi N. Vaccines and Kawasaki disease. Expert Rev Vaccines. 2016; 15(3): 417-24.
17. Chen S, Dong Y, Yin Y, Krucoff MW. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart. 2013; 99: 76-82.
18. Kobayashi T, Saji T, Otan T, Takeuchi K, Nakamura T, Arakawa H, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label,blinded-endpoints trial. Lancet. 2012; 379: 1613–20.
19.** Chen S, Dong Y, Kiuchi MG, Wang J, Li R, Ling Z, et al. Coronary Artery Complication in Kawasaki Disease and the Importance of Early Intervention: A Systematic Review and Meta-analysis. JAMA Pediatr. 2016.
20. Baek JY, Song MS. Meta-analysis of factors predicting resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. Korean J Pediatr. 2016; 59(2): 80-90.
21. Camacho MS, Lirola MJ. Púrpura de Schönlein-Henoch, enfermedad de Kawasaki y otras vasculitis. Pediatr Integral. 2013; XVII(1): 34-46.
Bibliografía recomendada
– Batu ED, Bilginer Y. Classification of Vasculitis in Childhood. Ann Paediatr Rheum. 2016; 5: 1-10.
Muy interesante artículo de revisión en el que se hace una puesta al día de la clasificación de las vasculitis pediátricas.
– Eleftheriou D, Brogan PA. Therapeutic advances in the treatment of vasculitis. Pediatr Rheumatol Online J. 2016; 14(1): 26.
Se hace un repaso sobre las últimas novedades en los tratamientos de las principales vasculitis en la infancia, y una actualización sobre enfermedades autoinflamatorias con manifestaciones vasculíticas descritas muy recientemente.
– Newburger J, Takahashi M, Gerber M, Gewitz M, Tani L, Burns J, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: Statement for Health Professionals From the Committee on Rheumatic Fever, A Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004; 114; 1708-33.
Completa y estupenda revisión de EK con un algoritmo de manejo de EK incompleto, muy útil para la práctica diaria.
– Leftheriou D, Levin M, Shingadia D, Tulloh R, Klein NJ, Brogan PA. Management of Kawasaki disease. Arch Dis Child. 2014; 99(1): 74-83.
Contiene un algoritmo muy útil, con las recomendaciones de tratamiento de la enfermedad de Kawasaki refractaria.
– Esposito S, Bianchini S, Dellepiane RM, Principi N. Vaccines and Kawasaki disease. Expert Rev Vaccines. 2016; 15(3): 417-24.
Interesante artículo, en el que se explican todos los aspectos relacionados con las vacunas y la enfermedad de Kawasaki.
| Caso clínico |
|
Lactante de 5 meses que acude por fiebre de 72 horas de evolución. Exantema maculopapuloso generalizado que ha aparecido en las últimas 24 horas. Rechazo parcial de las tomas. No antecedentes personales ni familiares de interés. Exploración Destaca la irritabilidad. Presenta lesiones maculopapulosas generalizadas. No se encuentran hallazgos patológicos en el resto de la exploración. Pruebas complementarias al ingreso Hemograma: leucocitos: 17.480/uL, 48% de PMN, Hb: 10,8 g/dL, plaquetas: 406.000/uL. Coagulación normal. Bioquímica: normal, excepto proteínas totales: 3,7 g/dL. PCR: 88 mg/L. Orina: leucocitos +. LCR: 26 células, 58% PMN, glucosa y proteínas normales. Evolución Tras 48 horas de antibioterapia empírica, persiste la fiebre y el exantema. Aparece hiperemia conjuntival bilateral y edema de manos y pies. Se repite la analítica en la que la Hb ha descendido a 7,9 g/dl y la PCR se ha elevado a 172 mg/l. El urocultivo, cultivo de LCR y hemocultivo son negativos.
|

