 |
| Temas de FC |
A. Cervera Bravo, F. Muñoz Bermudo
Adjuntos del Servicio de Pediatría. Hospital Universitario de Móstoles. Madrid
| Resumen
La púrpura es una lesión hemorrágica de piel o mucosas secundaria a alteraciones de la integridad vascular, de las plaquetas o de la coagulación. Se muestra cómo realizar el diagnóstico diferencial de un niño con clínica de púrpura. Se describen las dos enfermedades purpúricas más frecuentes, la trombocitopenia inmune primaria (PTI) y la vasculitis por IgA (VIgA) (anteriormente púrpura de Schönlein-Henoch). La PTI es un trastorno benigno de carácter autoinmune que produce principalmente destrucción plaquetar con clínica de hemorragia cutánea acompañada o no de hemorragia mucosa gastrointestinal o génito-urinaria pero puede ser asintomática. En el 70% de los casos, se resuelve de forma espontánea en los primeros 6-12 meses. El diagnóstico es por exclusión de otras patologías por anamnesis, exploración física y pruebas complementarias básicas. El tratamiento va enfocado a corregir la clínica hemorrágica y no la cifra de plaquetas. La VIgA es una vasculitis de origen inmune que afecta al vaso pequeño, con clínica aguda de púrpura cutánea en miembros inferiores sin/con manifestaciones sistémicas asociadas. El pronóstico en la mayoría de los casos es excelente, con resolución espontánea, salvo si hay afectación renal. Se comenta: etiopatogenia, diagnóstico, diagnóstico diferencial, manejo terapéutico y seguimiento según la presentación clínica. |
| Abstract
Purpura refers to a hemorrhagic lesion of the skin or mucosal membranes due to disruption in vascular integrity, or abnormalities in platelets or clotting. |
Palabras clave: Trombocitopenia; Púrpura; Trombocitopenia inmune (PTI); Vasculitis IgA.
Key words: Thrombocytopenia; Purpura; Immune thrombocytopenia (ITP); IgA vasculitis.
Pediatr Integral 2021; XXV (5): 254 – 264
Púrpuras más frecuentes. Trombocitopenia inmune primaria y vasculitis por IgA (púrpura de Schönlein-Henoch)
Introducción
La púrpura es una lesión hemorrágica de piel o mucosas secundaria a la alteración de la integridad vascular o a problemas de la coagulación o de las plaquetas.
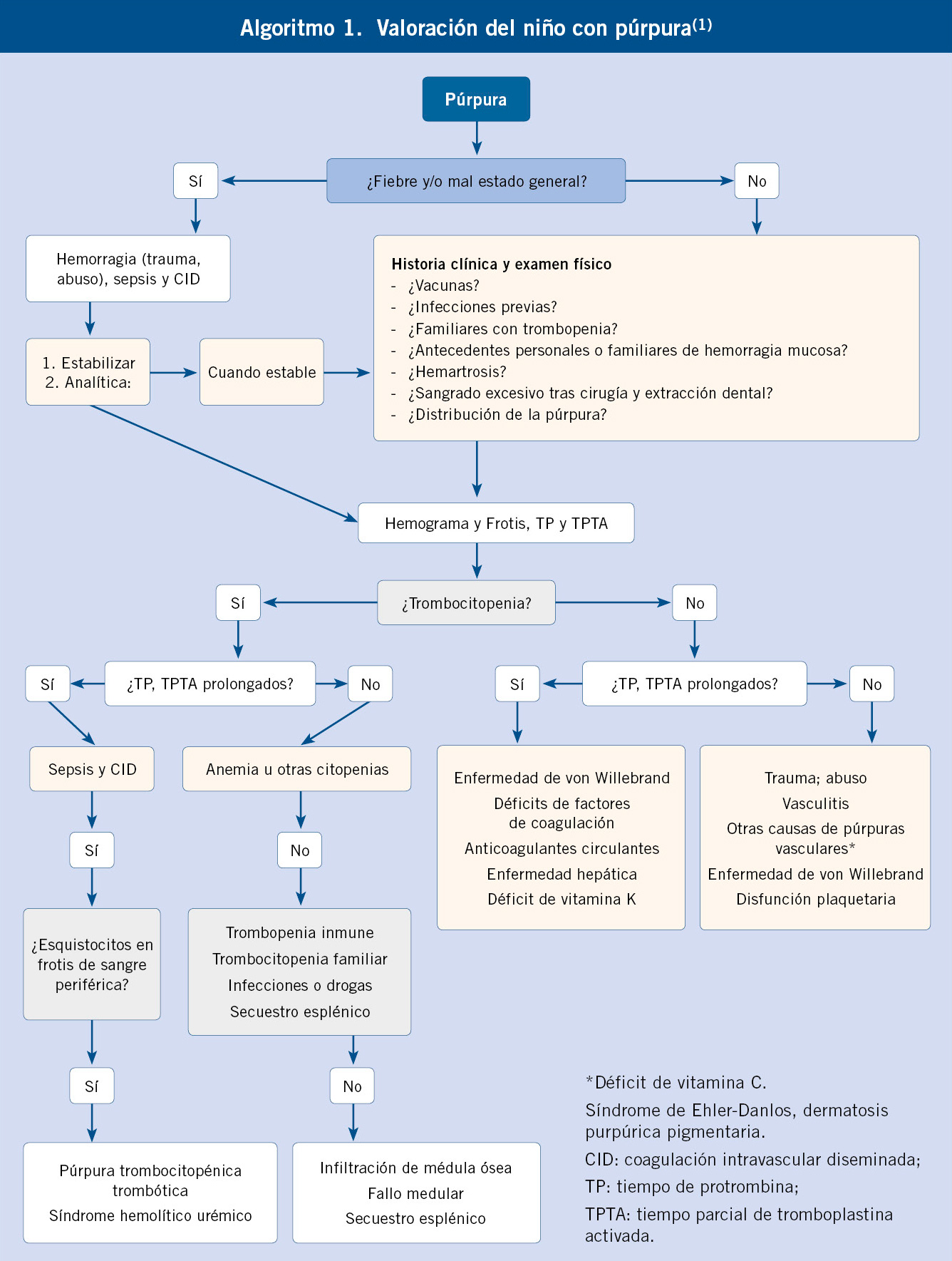
La púrpura se refiere al sangrado cutáneo o en mucosas. Puede ser secundario a causas banales como traumatismos o ser la manifestación de una enfermedad grave. Son lesiones rojo-violáceas que no blanquean con la presión. Según el tamaño, se denominan como petequias (menos de 2-5 mm) o equimosis (>5 mm). A diferencia de ésta, el hematoma se acompaña además de tumefacción. Las causas pueden ser múltiples: por rotura de la integridad vascular (traumatismos, infección, vasculitis, enfermedades del colágeno, escorbuto…), pueden ser debidas a alteraciones de los otros dos componentes de la hemostasia primaria, además de los vasos (las plaquetas o el factor de von Willebrand) o por alteraciones de la hemostasia secundaria (coagulopatías). Los problemas que afectan a la hemostasia primaria se denominan más propiamente, como enfermedades purpúricas. Para el diagnóstico etiológico, véase el algoritmo 1(1).
Excluyendo las causas vasculares, la etiología más frecuente de aparición de púrpura por alteración de la hemostasia primaria es la trombopenia. Por otro lado, ésta puede ser un hallazgo casual en un niño por lo demás asintomático, sin hemorragia cutánea o mucosa. Más raramente, las manifestaciones purpúricas pueden deberse a problemas de la función plaquetar.
La trombopenia aislada, sin afectación de otras series hematológicas, puede deberse a un problema central, con afectación de la megacariopoyesis en la médula ósea, o a un mecanismo periférico (destrucción de plaquetas, secuestro). Además, pueden ser congénitas o adquiridas, representando estas últimas la mayoría de los casos(1) (Tabla I).
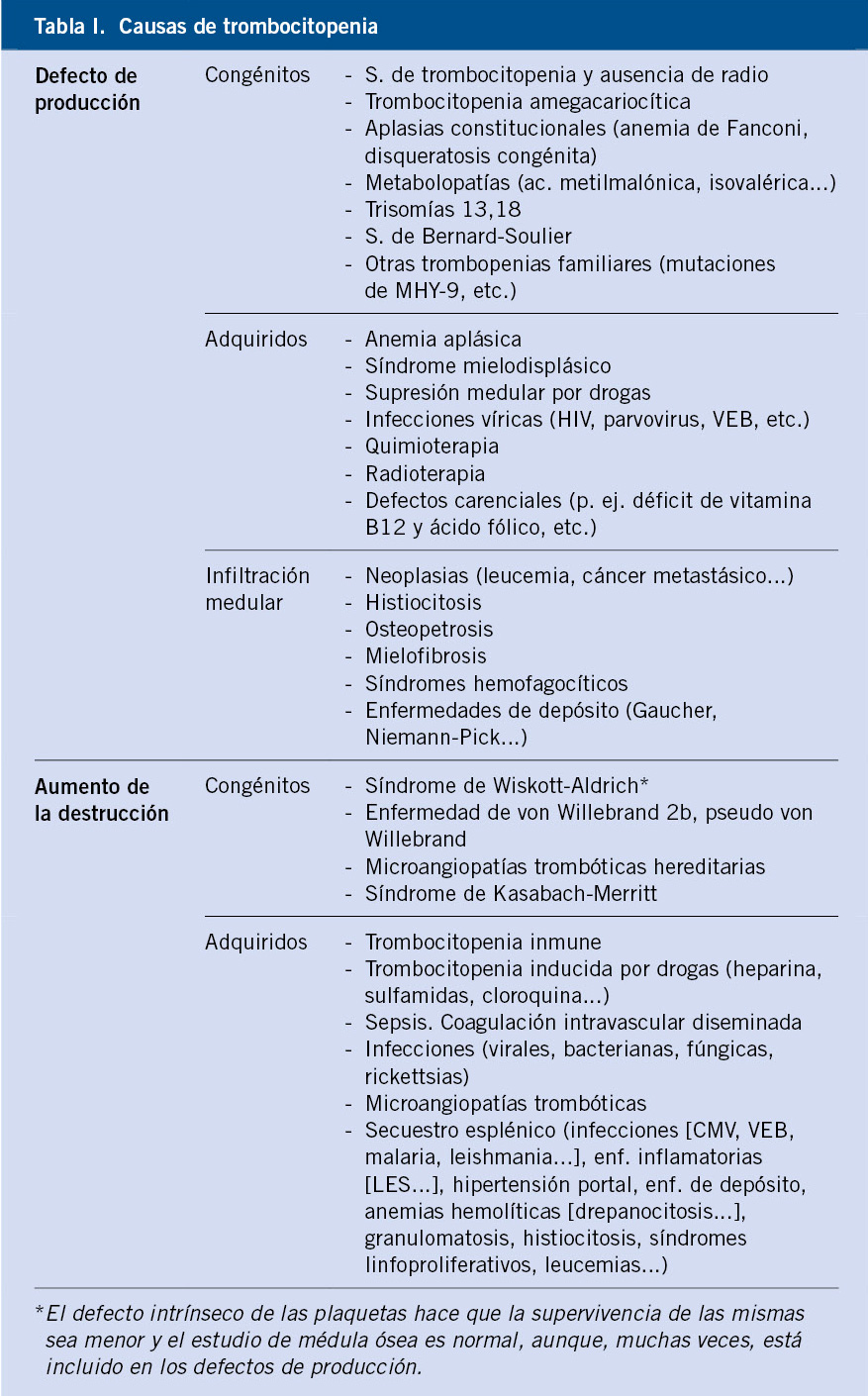
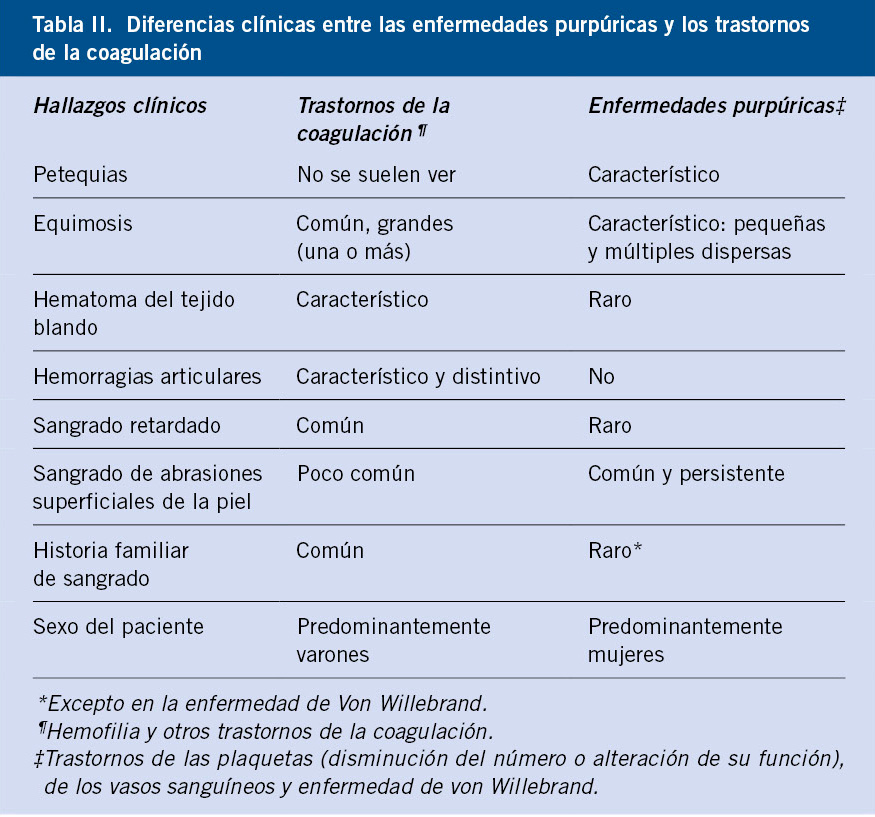
Aunque la cifra normal de plaquetas es >150.000/μL, las manifestaciones de sangrado anormal no aparecen hasta que descienden <30.000/μL a no ser que haya algún traumatismo o cirugía. Cuando existe trombocitopenia de mecanismo periférico, suele haber una respuesta medular con producción de plaquetas jóvenes que son más funcionales, por lo que la presencia de púrpura o sangrado con cifras de plaquetas relativamente altas (>30.000-50.000/μL) debe hacer sospechar un origen central y/o alteraciones en la función plaquetaria. En la tabla II, se muestran las diferencias clínicas entre las enfermedades purpúricas, que afectan a la hemostasia primaria y los trastornos de la coagulación.
Este artículo comenta las dos causas más comunes de púrpura en la infancia: la trombocitopenia inmune que puede ser primaria o secundaria, aunque se centrará en la primaria, más frecuente, y la vasculitis IgA (VIgA) o púrpura de Schönlein-Henoch.
Trombocitopenia inmune
Introducción
La trombopenia inmune es un trastorno adquirido benigno de carácter autoinmune que produce principalmente una destrucción de plaquetas.
La trombocitopenia inmune (o PTI, manteniendo el antiguo acrónimo por su amplia difusión) es un trastorno autoinmune caracterizado por trombopenia (<100.000 plaquetas/µL según consenso internacional de 2009)(2), producido principalmente por la destrucción de plaquetas. Generalmente, es un proceso benigno, adquirido y de origen desconocido que, de forma típica, aparece como un cuadro agudo de púrpura mucocutánea o sangrado menor en un niño sano, asociado a trombopenia, sin otras alteraciones clínicas o analíticas. Un tercio de los pacientes pueden no mostrar clínica hemorrágica alguna, por eso, en el consenso de 2009, se retiró la denominación de “púrpura trombocitopénica”(2). Además, se estandarizó la nomenclatura (véanse las definiciones).
Definiciones
• PTI de reciente diagnóstico: la que lleva menos de 3 meses de evolución desde el diagnóstico.
• PTI persistente: desde los 3 a los 12 meses de duración desde el diagnóstico.
• PTI crónica: más de 12 meses de evolución desde el diagnóstico.
• PTI primaria: cuando no se conoce la causa que la ha originado. Es un diagnóstico de exclusión.
• PTI secundaria: cuando aparece en el curso de enfermedades (infecciones, enfermedades autoinmunes, enfermedades malignas, inmunodeficiencias…).
• PTI refractaria: en paciente esplenectomizado, cuando tiene riesgo o persiste la clínica hemorrágica a pesar del tratamiento. En niños/adolescentes, se intenta evitar la esplenectomía, por lo que algunos autores proponen denominar PTI refractaria a esa edad, cuando hay clínica y/o riesgo de sangrado a pesar del tratamiento, en ausencia de esplenectomía.
Epidemiología
Su incidencia en la edad pediátrica es de unos 5 casos/100.000 personas/año, con un pico de edad entre los 2-5 años y con ligero predominio en varones, salvo en adolescentes, donde predomina el sexo femenino.
Etiopatogenia(3)
El desencadenante suele ser desconocido o tras infecciones o vacunación. La destrucción plaquetar se produce por autoanticuerpos y por disfunción de la inmunidad celular.
Se han descrito múltiples mecanismos en la PTI que dan lugar a la trombocitopenia y que difieren de unos pacientes a otros, justificando la distinta respuesta a los tratamientos. El desencadenante del problema suele ser desconocido, pero, muchas veces, son las infecciones o vacunaciones que, por mimetismo con las plaquetas, dan origen a una respuesta inmune antiplaquetaria que puede mantenerse una vez resuelta la infección. Esa respuesta autoinmune se produce principalmente por una susceptibilidad genética y afecta a la inmunidad innata y a la adaptativa, incluyendo tanto a la respuesta humoral como a la celular. El factor principal es la pérdida de tolerancia de los antígenos plaquetarios de la membrana. Hay varios mecanismos patogénicos fundamentales que dan lugar a la trombocitopenia. El primero es la producción de autoanticuerpos IgG (presentes en el 60% de los pacientes) contra las glicoproteínas (GP) de la membrana plaquetar y de los megacariocitos, especialmente frente a la GPIIb/IIIa. Los fragmentos Fc de esos anticuerpos plaquetarios son reconocidos por los macrófagos, facilitando la fagocitosis plaquetar, especialmente en el bazo. Los megacariocitos dañados aumentan su apoptosis y se altera la producción de plaquetas. Un segundo mecanismo parece producir la destrucción plaquetar y de megacariocitos por linfocitos T citotóxicos auto-reactivos. Además, en algunos casos puede haber una posible destrucción plaquetaria en el hígado(4). Por último, los niveles de trombopoyetina (TPO), principal factor de crecimiento de los megacariocitos, son inapropiadamente bajos. Por otro lado, se ha demostrado una alteración en los mecanismos de regulación inmune (células reguladoras T, linfocitos B y células dentríticas) que no pueden contrarrestar esa respuesta autoinmune.
Clínica
La clínica más frecuente con plaquetas <20.000/μL es el sangrado cutáneo exclusivo, aunque puede haber hemorragia mucocutánea, gastrointestinal o genitourinaria.
En más del 50% de los casos, la clínica suele ser de comienzo agudo, con sangrado mucocutáneo (petequias y equimosis, epistaxis, sangrado gingival, bullas hemorrágicas en labios…) y cifras de plaquetas <20.000/μL, aunque la mayoría tienen exclusivamente sangrado cutáneo(5). Hasta en el 20%, puede haber sangrado más grave, como epistaxis, sangrado gastrointestinal o genitourinario –especialmente menorragias–, que precisan tratamiento médico y/o transfusión(6). La peor complicación es la hemorragia del sistema nervioso central, pero en la edad pediátrica ocurre en menos del 1%. Cuando aparece, la gran mayoría lo hace en la primera semana del diagnóstico, 15% entre los 3-12 meses y un 15% posteriormente(7). Alrededor de un 60% suelen referir historia de infección previa o vacunación(5). En el adolescente, el comienzo es, a veces, indolente, sin sangrado, con cifras de plaquetas >50.000/μL y con mayor riesgo a cronificarse que en niños < 10 años. La mayoría (> 70%), sin embargo, remiten en los primeros 12 meses. No hay correlación exacta entre la cifra de plaquetas y el riesgo de sangrado, aunque éste es mayor si las plaquetas son <10.000/μL, pero hay pacientes con ese grado de trombopenia que apenas sangran. Un número significativo de pacientes refieren fatiga (cansancio, debilidad) que afecta a su calidad de vida(7,8).
Diagnóstico
El diagnóstico es por exclusión, descartando otras patologías por anamnesis, exploración física rigurosa y pruebas básicas de laboratorio.
El diagnóstico es por exclusión, descartando patología medular que se acompaña de otras anomalías en el hemograma (macrocitosis eritrocitaria, otras citopenias, alteraciones en el frotis de sangre periférica), así como otras causas de trombopenia aislada (ver apartado de diagnóstico diferencial). Es importante realizar una buena historia clínica, tanto familiar (posibilidad de trombopenias heredadas), como personal (transfusiones previas, drogas, actividad sexual, inmunizaciones) y un examen físico riguroso, que debe ser normal, salvo por las manifestaciones hemorrágicas.
Para el diagnóstico inicial, se recomienda realizar solo: hemograma, reticulocitos y frotis (para descartar presencia de células malignas y ver morfología plaquetar como microplaquetas o plaquetas gigantes que orienten a otros diagnósticos), inmunoglobulinas (para descartar inmunodeficiencias frecuentes, como la combinada severa), grupo sanguíneo y test de Coombs directo (para descartar el síndrome de Evans, con anemia hemolítica autoinmune asociada), serologías virales (CMV, VEB, parvovirus B19, VHS), bioquímica básica, estudio de coagulación, test del embarazo en adolescentes con actividad sexual y orina elemental(5,9,11). El aspirado medular solo se recomienda de entrada en casos con clínica atípica(5,9). La remisión clínica y una buena respuesta inmediata a los tratamientos de primera línea, apoyan el diagnóstico. Para los casos refractarios, persistentes o crónicos, que podrían deberse a otras patologías, hay que realizar estudios de segundo nivel para reafirmar el diagnóstico: estudios de autoinmunidad, perfil tiroideo y Acs. antitiroideos, subpoblaciones linfocitarias, inmunoglobulinas y complemento, descartar infección viral crónica (PCR para CMV, parvovirus B19) y realizar estudio de médula ósea(5,11). No está clara la indicación de detectar y erradicar el Helicobacter pylori en la edad pediátrica. En los casos crónicos y, especialmente, en adolescentes de sexo femenino o con evolución tórpida, se recomienda repetir los estudios de inmunidad y autoinmunidad cuando presenten clínica sugestiva o, al menos, una vez al año(11). Los anticuerpos antiplaquetarios no parecen muy útiles, pues pueden ser positivos en casos de trombopenias inmunes y no inmunes y pueden dar falsos negativos(5).
Diagnóstico diferencial
Dado que la PTI es un diagnóstico de exclusión, hay que descartar patología medular (síndromes mielodisplásicos, aplasia medular, anemia megaloblástica, leucemias, etc.) con un hemograma normal y por el estudio de médula ósea cuando esté indicado(5,11). Una trombopenia aislada, estable, de larga evolución y que no responde al tratamiento habitual, debe hacer sospechar una trombopenia heredada. Podemos ver trombopenias aisladas asociadas a formas congénitas, a lupus eritematoso sistémico (LES), infecciones, medicamentos, enfermedades de depósito, inmunodeficiencias, etc. (Tabla I). La presencia de esplenomegalia, debe hacernos descartar infecciones (VEB, CMV…), inmunodeficiencias (p. ej.: síndrome linfoproliferativo autoinmune o ALPS), enfermedades malignas o de depósito (Gaucher, etc.). Por debajo del año, debemos pensar en trombopenias congénitas o PTI secundaria a inmunodeficiencias(5,11). En adolescentes, especialmente en mujeres, hay que considerar que puede haber una enfermedad autoinmune de base (LES).
Tratamiento
El tratamiento se centra en el control de la clínica hemorrágica y no en la corrección de la cifra de plaquetas.
Debe realizarse en función de la clínica de sangrado y no por la cifra de plaquetas. En cualquier caso, se recomienda la restricción de la actividad física que conlleve riesgo de traumatismos con trombopenias <30.000/μL. Las guías actuales de tratamiento de PTI, tanto americanas como el protocolo de la Sociedad Española de Hematología y Oncología Pediátricas (SEHOP), recalcan la importancia de la clínica: en los pacientes asintomáticos o con sangrado cutáneo exclusivo, sin factores de riesgo, la observación es la primera opción(5,9,10). En estos casos, hay que valorar además el entorno social, distancia al hospital y otros factores de riesgo (señalados en el Algoritmo 2)(5,9,10). En la PTI de diagnóstico reciente que precise tratamiento, se emplea la prednisona en ciclos cortos, solo si hay sangrado cutáneo-mucoso (dosis de 4 mg/kg/día los primeros 4 días y 2 mg/kg/día 3 días más). Si hay sangrado activo, se recomienda la inmunoglobulina intravenosa (IGIV) por su efecto más rápido, a una dosis de 0,8-1 g/kg/dosis, que se puede repetir a las 72 horas en función de la respuesta (Algoritmo 2). Para los casos persistentes con plaquetas <30.000/μL, se emplean también los corticoides y/o IGIV como tratamiento de primera línea en los episodios de sangrado en espera de la remisión. En la PTI crónica, donde la remisión espontánea es mucho más baja e impredecible, el tratamiento debe emplearse no solo en función del riesgo de sangrado, sino también considerando la calidad de vida, especialmente en el adolescente, por lo que habrá que tener en cuenta su actividad física y estilo de vida a la hora de decidir el manejo más adecuado. Recientemente se ha aprobado en niños > 1 año con PTI crónica refractaria, el empleo de agonistas del receptor de la trombopoyetina (ar-TPO), el romiplostin y el eltrombopag (este último aprobado si más de 6 meses de evolución). Ambos tratamientos han mostrado una eficacia a largo plazo del 40-50%, disminuyendo la necesidad de esplenectomía(9,11). En algunos casos, se ha podido suspender el tratamiento sin recaídas posteriores, bien por la evolución natural de la enfermedad, o bien porque los ar-TPO podrían cambiar su curso natural modulando la respuesta inmune(11). La esplenectomía resuelve el problema en el 65-70% de los pacientes, pero aumenta el riesgo de sepsis fulminantes y de trombosis(5,11). Si hay fracaso de los tratamientos de primera (prednisona, IGIV) o segunda línea (dexametasona, ar-TPO), se puede valorar la esplenectomía o el empleo de los inmunosupresores (micofenolato de mofetilo, rituximab u otros) en centros especializados(5,9-11).
Evolución y pronóstico
El pronóstico es bueno, con resolución en el 70% durante el primer año y mejoría progresiva con los años, en la mayoría de los casos crónicos.
El Grupo de Estudio Cooperativo Intercontinental de la PTI muestra que en los niños (<16 años), la PTI regresa de forma espontánea en el 71% a los 12 meses del diagnóstico, la mayoría en los primeros 6 meses, aunque de los que persisten, un 20-25% lo hacen en los siguientes 6 meses(12). Los adolescentes (10-16 años) se cronifican más que los niños, del 30-40% según las series. Posteriormente, puede haber remisiones tardías, un 28% adicional lo hacen a los 24 meses y aun a partir de entonces, la mayoría mejoran clínicamente con el tiempo, alcanzando cifras de plaquetas >30.000/μL de forma estable, sin precisar tratamiento o incluso con remisiones completas hasta cerca del 50% de ellos, años después(7). De los que no se recuperan, un número significativo de pacientes acaban teniendo otra enfermedad de base, por lo que en el seguimiento, es importante siempre reevaluar al paciente.
Vasculitis por IG-A (púrpura de Schönlein-Henoch)
Introducción
La vasculitis por IgA es la vasculitis pediátrica sistémica más frecuente.
La vasculitis por IgA (VIgA), nueva forma de denominar a la púrpura de Schönlein-Henoch, es la vasculitis sistémica más frecuente en la infancia, caracterizada por el fenómeno de leucocitoclasia y el depósito de complejos inmunes de IgA1 en la pared de los pequeños vasos. Clínicamente, se manifiesta como púrpura cutánea no trombopénica, artralgias y artritis, dolor abdominal y afectación renal.
Epidemiología
Es una enfermedad predominantemente pediátrica, más frecuente entre los 3 y 15 años, ocurriendo el 75-90% en menores de 10 años. La incidencia se estima entre los 10-20 casos/100.000 niños menores de 17 años. La distribución por sexo es similar, con discreto predominio en varones 1,5/1. Es más frecuente en niños asiáticos y caucásicos. Predomina en primavera, otoño e invierno, lo que hace probable la implicación de procesos infecciosos en su patogénesis(13,14).
Etiopatogenia(13)
La etiopatogenia no está bien definida; tras un posible desencadenante infeccioso, en personas genéticamente susceptibles, se provoca la formación y depósito de inmunocomplejos de IgA que dañan la pared vascular.
Es una vasculitis mediada inmunológicamente, resultado de la formación de complejos inmunes en respuesta a determinados estímulos antigénicos en personas genéticamente susceptibles. Su predominio estacional y la evidencia clínica apoyan la hipótesis de la participación en su etiología de agentes infecciosos, el estreptococo beta hemolítico del grupo A, se ha aislado hasta en el 36% de los pacientes. También se han identificado: M. pneumoniae, Legionella, Yersinia, H. pylori, CMV, VEB, parvovirus B19 y virus varicela zóster, entre otros. Se ha descrito, también, su asociación con algunas vacunas: sarampión, rubeola, neumococo o hepatitis B, y con algunos fármacos como: betalactámicos, macrólidos y antiinflamatorios no esteroideos (AINES).
Respecto a la predisposición genética, parece haber una mayor susceptibilidad para presentar la enfermedad con antígenos del complejo HLA, como el HLA-DRB1 y HLA-B*41:02. El HLA B35 y DQA1 se han relacionado con el riesgo de padecer nefritis. Se piensa que es una enfermedad mediada por el depósito de inmunocomplejos, caracterizados por la presencia de IgA1 polimérica a nivel de los capilares dérmicos, gastrointestinales y glomerulares, habiéndose documentado la existencia de niveles séricos elevados de IgA1, complejos que contienen IgA1, IgA-ANCA, IgA-FR en los pacientes con VIgA. En pacientes con nefritis, se detectan además inmunocomplejos circulantes IgA1-IgG de gran masa molecular. Así mismo, los niveles séricos de IgA1 con defecto de galactosa (Gd-IgA1) son algo más elevados en los pacientes con nefritis.
Durante los últimos años, muchos estudios han implicado citoquinas proinflamatorias en la patogénesis de la enfermedad (IL-2, IL-6, IL-8, VEGF, TNFalfa).
Manifestaciones clínicas
La afectación es multiorgánica con púrpura cutánea que puede asociarse principalmente a clínica articular, gastrointestinal o renal.
Manifestaciones cutáneas: la lesión cutánea característica es la púrpura palpable, signo de presentación en el 75% de los pacientes, desde petequias a grandes equimosis que pueden convertirse en lesiones necróticas. Aparecen de forma simétrica en zonas declives (miembros inferiores y nalgas) (Fig. 1), pudiendo encontrarse hasta en un tercio de los pacientes, en extremidades superiores, tronco y cara. Las lesiones son, inicialmente, eritematosas y, progresivamente, cambian a color violáceo y marrón. Mejoran con el reposo. Pueden acompañarse de edema subcutáneo en: cuero cabelludo, cara, dorso de manos y pies, periné y escroto.
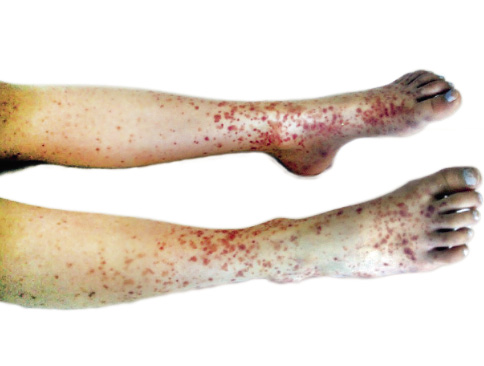
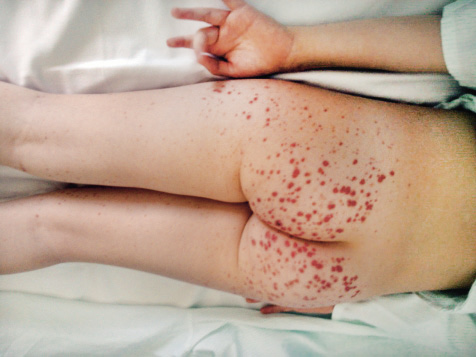
Figura 1. Lesiones purpúricas típicas de la vasculitis por IgA. Fotos de: A. Cervera, del libro Medicina del adolescente, con la autorización de Ergon.
Manifestaciones articulares: la artritis o artralgias pueden ser el primer síntoma de la enfermedad en el 15-20% de los pacientes, y hasta en un 80% de ellos, se presenta algún grado de afectación articular. La inflamación es periarticular, dolorosa, sin eritema ni calor, pero con limitación, afectando con mayor frecuencia a grandes articulaciones de miembros inferiores (rodillas y tobillos). La artritis, generalmente, es oligoarticular, transitoria y se resuelve en pocos días sin dejar deformidad.
Manifestaciones gastrointestinales: se describen en el 50-75% de los niños, siendo el primer síntoma de la enfermedad en el 14-36% de los pacientes. Se producen como consecuencia del edema y la hemorragia secundarios a la vasculitis de la pared intestinal. El síntoma más frecuente es el dolor abdominal cólico. La invaginación es la complicación más frecuente. Otras manifestaciones pueden ser: hemorragia digestiva, úlceras, perforaciones, pancreatitis aguda, afectación hepatobiliar y enteropatía pierde proteínas.
Manifestaciones renales: se producen en el 20-50% de los pacientes y es el factor pronóstico más importante de la enfermedad. Las manifestaciones varían entre: hematuria microscópica/macroscópica, proteinuria, síndrome nefrótico/nefrítico, hipertensión arterial (HTA) y fracaso renal. Raramente, estas manifestaciones preceden a la púrpura, suelen desarrollarse durante el primer mes de la enfermedad en el 75-80% de los pacientes, y en el 97-100% de los casos, en los primeros 6 meses de la presentación de la enfermedad. Se ha descrito algún caso en el que la afectación renal se produjo varios años después. La intensidad de los síntomas renales al inicio de la enfermedad, determina la gravedad de las lesiones glomerulares(13,15).
Otras manifestaciones clínicas mucho menos frecuentes incluyen: neurológicas (cefalea, convulsiones, encefalopatía, hemorragias, neuropatía periférica), pulmonares (hemorragia difusa alveolar, neumonía intersticial, fibrosis intersticial) y urológicas (orquitis, epididimitis, torsión testicular). También se han descrito: miositis, carditis o uveítis anterior y, muy raramente, fenómenos de trombosis arteriovenosa en distintos territorios(15).
Diagnóstico
El diagnóstico es fundamentalmente clínico (Tabla III).

El diagnóstico es clínico y se basa en los criterios del Consenso de Ankara de 2008 (EULAR/PRINTO/PRES)(16) (Tabla III).
No existen estudios de laboratorio específicos, estos irán encaminados a descartar otras patologías y a conocer la afectación orgánica de la enfermedad.
En el estudio inicial, se incluirán: hemograma, coagulación, VSG, PCR, perfil renal, hepático y óseo, análisis de orina y sangre oculta en heces. Si se identifica proteinuria, se determinará en orina de la mañana el índice proteína/creatinina. Si el diagnóstico fuese dudoso, se debe añadir un perfil autoinmune completo, que incluya: ANAS, antiDNAds, ANCA, inmunoglobulinas, C3 y C4.
Se puede encontrar: anemia, leucocitosis, aumento de VSG y PCR y, en algunos casos, una función renal y/o hepática alteradas. El recuento de leucocitos es un factor de riesgo importante para la participación de órganos internos. La trombocitosis se asocia con enfermedad más grave, y hay una correlación positiva entre la PCR y la gravedad de la enfermedad(17).
El estudio de coagulación suele ser normal; puede haber disminución de la actividad del factor XIII. La IgA se encuentra elevada en la mitad de los pacientes y no se correlaciona con la severidad del proceso; pueden detectarse inmunocomplejos circulantes de IgA. El estudio inmunológico suele ser normal, en ocasiones, se detectan niveles bajos de C3 y C4.
Si se sospecha un proceso infeccioso, en función de los síntomas, se deberían realizar: hemocultivo, urocultivo, frotis faríngeo y radiografía de tórax.
La biopsia cutánea, ante una presentación atípica y dudas diagnósticas, pone de manifiesto una vasculitis leucocitoclástica de pequeños vasos con depósitos de complejos inmunes, que contienen IgA, patognomónico de la VIgA o púrpura de Schönlein-Henoch (PSH)(13).
La biopsia renal se realiza en pacientes con afectación renal grave: proteinuria grave durante, al menos, 4 semanas; proteinuria moderada persistente durante, al menos, 3 meses; o deterioro de la función renal (Fig. 2). Se puede encontrar desde una proliferación mesangial leve hasta una glomerulonefritis con formación de semilunas.

Figura 2. Seguimiento de pacientes con nefritis por IgA(18,19).
La ecografía abdominal puede mostrar un engrosamiento de las paredes del intestino delgado y grueso, y ayudará a descartar invaginación intestinal. Se debe realizar ecografía escrotal ante escroto agudo, para descartar una torsión testicular.
En 2019, se han publicado las recomendaciones de diagnóstico y tratamiento para la VIgA/PSH de la iniciativa europea Single Hub and Access point for paediatric Rheumatology in Europe (SHARE), que incluyen 7 recomendaciones para el diagnóstico y 19 para el tratamiento, basadas en evidencia y consenso, con el objetivo de unificar y mejorar la atención de estos pacientes. Para las manifestaciones renales, se definen criterios de gravedad que condicionarán el tratamiento a emplear (Tabla IV)(18).

Diagnóstico diferencial
Se debe diferenciar de otros cuadros de vasculitis, procesos abdominales o enfermedades renales, en casos de presentación cutánea atípica o en función de la presentación clínica.
El diagnóstico se complica en los casos de presentación atípica de la enfermedad, debiéndose considerar otras vasculitis como: granulomatosis con poliangeítis, poliangeítis microscópicas, vasculitis asociada a conectivopatías, vasculitis por hipersensibilidad o crioglobulinémica, entre otras.
El edema agudo hemorrágico del lactante (síndrome de Finkelstein-Seidlmayer) debe ser considerado por su similitud en las manifestaciones cutáneas(13).
Si el paciente presenta síntomas abdominales, se debe diferenciar de otras causas de abdomen agudo, como apendicitis o invaginación. Si presenta síntomas renales, de la glomerulonefritis postestreptocócica, nefritis lúpica o síndrome hemolítico urémico. No se debe olvidar la trombopenia inmune, CID, malos tratos o reacciones de hipersensibilidad, en aquellos niños con predominio de manifestaciones cutáneas (Algoritmo 1).
Tratamiento
El tratamiento en la mayoría de los pacientes es de soporte, salvo en casos de afectación gastrointestinal grave no quirúrgica con corticoides, o de nefritis moderada a grave con corticoides +/– inmunosupresores.
La mayoría de los pacientes con VIgA/PSH se recuperan espontáneamente, por lo que el tratamiento, en la mayoría de los casos, será sintomático, con reposo y analgesia, y siempre que se pueda, se manejará de forma ambulatoria. Serán criterios de ingreso: dolor abdominal grave, hemorragia gastrointestinal, artralgia grave con limitación a la deambulación, afectación renal con hipertensión, síndrome nefrótico o insuficiencia renal, y afectación del estado mental.
Manifestaciones cutáneas: el reposo disminuye la aparición de nuevas lesiones. En las lesiones bullosas, los corticoides pueden ser eficaces. Para la enfermedad crónica cutánea y articular, se utilizan la aspirina y la colchicina.
Manifestaciones articulares: normalmente responde a los AINES o paracetamol. Un estudio randomizado de prednisolona oral frente a placebo mostró muy leve mejoría de la severidad de los síntomas y una tendencia a la menor duración del dolor, pero no significativa(19).
Manifestaciones gastrointestinales: el dolor abdominal, suele resolverse en pocos días sin tratamiento. El uso de prednisolona a 1-2 mg/kg se podría considerar en niños con VIgA y dolor abdominal moderado-grave, una vez descartada la invaginación. En casos de vasculitis grave, se ha descrito respuesta al tratamiento con infusión de gammaglobulina, pulsos de metilprednisolona y plasmaféresis.
Manifestaciones renales: su manejo sigue siendo controvertido. En las nuevas recomendaciones de la iniciativa SHARE(18), se propone el tratamiento con inhibidores de la enzima convertidora de la angiotensina (IECA) o antagonistas del receptor de la angiotensina II (ARA II), para prevenir la lesión glomerular en niños con proteinuria persistente (>3 meses de duración), independientemente de si están recibiendo prednisolona u otro tratamiento inmunosupresor. El tratamiento recomendado con corticoides asociado o no a inmunosupresores según la gravedad de la nefritis, se resume en la figura 3.

Figura 3. Tratamiento de la nefritis de la vasculitis por IgA, según consenso SHARE(18). AZA: azatioprina; MMF: mofetilmicofenolato; CYC: ciclofosfamida; MP: metilprednisolona.
Los corticoides también se proponen para el tratamiento de complicaciones raras como: orquitis, hemorragia pulmonar, vasculitis cerebral u otras manifestaciones graves de vasculitis, que comprometan la función orgánica o la vida(18).
Pronóstico
En la mayoría de los casos, la enfermedad se resuelve en pocas semanas, un tercio presentan recurrencias y, si clínica renal moderada al inicio, hasta un 15% pueden desarrollar HTA o afectación renal a largo plazo.
La VIgA es generalmente una enfermedad autolimitada, que se resuelve en 2-6 semanas, aunque hasta el 33% de los niños tendrán recurrencias (entre 1 y 6 episodios) durante los 2-3 primeros meses, habiéndose descrito recaídas que sobrepasan los 18 meses del inicio de la enfermedad, que parecen más probables entre los niños con afectación renal. Los síntomas de las recaídas son similares al debut, aunque suelen ser cada vez más leves y de menor duración(13).
Las manifestaciones digestivas condicionan la morbilidad en fases iniciales, mientras que la afectación renal es el principal factor pronóstico a largo plazo. El riesgo de nefritis es mayor por encima de los 10 años. Aunque ningún hallazgo es predictivo, la presencia de síndrome nefrítico/nefrótico, la disminución de la actividad del factor XIII, la HTA, el fallo renal al inicio del proceso y la presencia de esclerosis glomerular/semilunas/afectación tubulointersticial, son factores de mal pronóstico: aunque haya una recuperación inicial, en el seguimiento a largo plazo, hasta un 15% de los pacientes pueden desarrollar HTA, proteinuria o disminución del filtrado glomerular. El riesgo de insuficiencia renal terminal es < 1%(13,14).
El seguimiento mínimo de estos pacientes debe ser de 6 meses, incluso algunos autores aconsejan alargarlo hasta los 12 meses del inicio de la enfermedad o de la última recaída, sabiendo que el tiempo en que se va a instaurar el daño renal es impredecible, pudiéndose desarrollar hasta años después (Fig. 2).
Las mujeres con historia de VIgA en la infancia, tienen mayor riesgo de presentar proteinuria e hipertensión arterial durante el embarazo.
Función del pediatra de Atención Primaria
• Reconocer la gravedad o no de un niño con púrpura para su derivación precoz a atención especializada. Se deberá derivar a cualquier niño con púrpura y trombopenia. En la vasculitis por IgA, se podría seguir en primaria si hay afectación cutánea típica sin otras manifestaciones clínicas asociadas o con clínica gastrointestinal o articular leves, la tensión arterial es normal, y tanto la analítica de orina y como la sanguínea son normales (Fig. 2).
• Favorecer la adherencia a los tratamientos en los pacientes que lo precisen, monitorizando los efectos adversos de los tratamientos empleados y colaborando en la realización de los controles analíticos.
• En niños/adolescentes con PTI, se debe evitar el empleo de AINES (ibuprofeno, aspirina). Se puede emplear como analgésico, el paracetamol o el metamizol. Como antiinflamatorio, se podría valorar dar ibuprofeno (en función de la clínica hemorrágica) o emplear inhibidores selectivos de ciclooxigenasa-2 (celecoxib) en >14 años.
• En niños con PTI y clínica hemorrágica, se puede emplear tratamiento adyuvante con antifibrinolíticos, siempre que no tengan hematuria, en donde está contraindicado (p. ej.: ácido tranexámico, el contenido de las ampollas iv puede administrarse por vía oral). En adolescentes con menorragia, el tratamiento hormonal puede ser eficaz.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de las autoras.
1.*** Raffini L, Fleisher GR, Wiley JF. Evaluation of purpura in children. UpToDate. Versión: enero 2021. Actualizado el 31 de julio de 2019. Consultado el 21 de febrero de 2021. Disponible en: www.uptodate.com.
2.** Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura (ITP) of adults and children: report from an International Working Group. Blood. 2009; 113: 2386-93.
3. Audia S, Mahévas M, Samson M, Godeau B, Bonnotte B. Pathogenesis of immune thrombocytopenia. Autoimmun Rev. 2017; 16: 620-32.
4. Li J, van der Wal DE, Zhu G, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun 2015;6:7737.
5.*** Provan D, Arnold DM, Bussell JM, Chong BH, Cooper N, Gernsheimer T, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv 2019;3:3780-817.
6. Neunert C, Noroozi N, Norman G, Buchanan GR, Goy J, Nazi I, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost. 2014; 13: 457-64.
7.** Despotovic JM, Grimes AB. Pediatric ITP: is it different from adult ITP? Hematology Am Soc Hematol Educ Program. 2018; 2018(1): 405-11.
8. Grace RF, Klaassen RJ, Shimano KA, Lambert MP, Grimes A, Bussel JB, et al. Fatigue in children and adolescents with immune thrombocytopenia. Br J Haematol. 2020; 191: 98-106.
9.*** Monteagudo E, Astigarraga I, Cervera A, Dasí MA, Sastre A, Berrueco R, et al (SEHOP). Protocolo de estudio y tratamiento de la P.T.I. (PTI-2018). An Pediatr (Barc). 2019; 91: 127.e1-10.
10.** Neunert C, Terrell DR, Arnold DM, Buchanan G, Cines DB, Cooper N, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv 2019;3:3829-66.
11.** Berrueco R, Dapena JL, Sebastián E, Sastre A. Controversias en el tratamiento de la trombocitopenia inmune pediátrica. An Pediatr (Barc). 2018; 89: 189.e1-8.
12.*** Kühne T, Berchtold W, Michaels LA, Wu R, Donato H, Espina B, et al. Newly diagnosed immune thrombocytopenia in childhood and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011; 96: 1831-7.
13.*** Borlán Fernández S. Vasculitis por IgA (púrpura de Schönlein-Henoch) Protoc Diagn Ter Pediatr. 2020; 2: 225-38. Consultado el 9 de marzo de 2021. Disponible en: https://www.aeped.es/sites/default/files/documentos/20_vasculitis_iga.pdf.
14. Reamy BV, Servey JS, Williams, PM. Henoch-Schönlein Purpura (IgA vasculitis): Rapid Evidence Review. Am Fam Physician. 2020; 102: 229-33
15. Du L, Wang P, Liu C, Li S, Yue S, Yang Y. Multisystemic manifestations of IgA vasculitis. Clinical Rheumatology. 2021; 40: 43-52.
16.** Ozen S, Pistorio A, Lusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheuma Dis. 2010; 69: 798-806.
17. Özdemir ZC, Çetin N, Düzenli Kar Y, Öcal HO, Bilgin M, Bör Ö. Hematologic Indices for Predicting Organ Involvement in Henoch-Schönlein Purpura (IgA vasculitis). J Pediatr Hematol Oncol. 2020; 42: e46-9.
18.*** Ozen S, Marks SD, Brogan P, Groot N, de Graeff N, Avcin T, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatol. 2019; 58: 1607-16.
19. McCarthy HJ, Tizard EJ. Clinical practice: Diagnosis and management of Henoch-Schönlein purpura. Eur J Pediatr. 2010; 169: 643-50.
Bibliografía recomendada
– Raffini L, Fleisher GR, Wiley JF. Evaluation of purpura in children. UpToDate. Versión: enero 2021. Actualizado el 31 de julio de 2019. Consultado el 21 de febrero de 2021. Disponible en: www.uptodate.com.
Aborda, de forma práctica y completa, la evaluación clínica de un niño con púrpura.
– Monteagudo E, Astigarraga I, Cervera A, Dasí MA, Sastre A, Berrueco R, et al (SEHOP). Protocolo de estudio y tratamiento de la P.T.I. (PTI-2018). An Pediatr (Barc). 2019; 91: 127.e1-10.
Es el protocolo nacional vigente del manejo de la PTI, elaborado por el grupo de trabajo de la PTI de la Sociedad Española de Hematología Pediátrica, en función de la evidencia y el consenso.
– Borlán Fernández S. Vasculitis por IgA (púrpura de Schönlein-Henoch) Protoc Diagn Ter Pediatr. 2020; 2: 225-38. Consultado el 9 de marzo de 2021. Disponible en: https://www.aeped.es/sites/default/files/documentos/20_vasculitis_iga.pdf.
Completo y reciente artículo de revisión sobre la vasculitis por IgA.
– Ozen S, Marks SD, Brogan P, Groot N, de Graeff N, Avcin T, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatol. 2019; 58: 1607-16.
Recoge las recomendaciones europeas actuales tras evaluación de la evidencia o por consenso, de cómo se debe realizar el diagnóstico y de cómo tratar la vasculitis por IgA.
| Caso clínico |
|
Un niño de 6 años acude por presentar, desde hace dos días, petequias y equimosis múltiples, y haber sangrado ese día durante un par de minutos por la fosa nasal izquierda. Refiere cuadro catarral febril 10 días antes. A la exploración, presenta buen estado general, numerosas petequias por todo el cuerpo, algunas en mucosa yugal y gingival, tiene algunas ampollas hemorrágicas en los labios y sangre seca en la narina izquierda. No presenta adenopatías periféricas ni hepatoesplenomegalia. El resto de la exploración es normal. Se realiza analítica que muestra los siguientes datos (valores normales): • Hemograma: leucocitos: 6.540/µL (4.000-11.500) (cayados: 0%; segmentados: 35%; linfocitos: 47%; monocitos: 11%; eosinófilos: 7%; basófilos: 0%). Hematíes: 4,5 (x 1012/L) (4-5,3). Hb: 11,8 g/dL (11-14,5). Hcto: 35% (34-42). VCM: 76 fl (75,2-91). HCM: 28 pg (25-33). CHCM: 34 g/dL (32,5-35,5). ADE: 14% (11,5-14). Plaquetas 7.000/µL (150.000-450.000). Frotis de sangre periférica: linfocitos atípicos aislados sin otros hallazgos. • Coagulación: tiempo de protrombina: 13,6 seg (12-15). Actividad: 90% (N>70). INR: 1,05 (0,9-1,25). APTT: 34 seg (29-37). Fibrinógeno: 420 mg/dL (150-450). • Bioquímica: glucosa: 95 mg/dL; urea: 28 mg/dL: ácido úrico: 4 mg/dL; creatinina: 0,4 mg/dL. LDH: 250 (125-220), GOT-AST: 35 UI/L; GPT-ALT: 29 UI/L. • Serología de virus de Ebstein-Barr, CMV, parvovirus B19, virus herpes simple, VIH: negativa. • Orina elemental: pH 6. Densidad: 1.020. Hematíes/Hb: 150/µL. Resto de elementos negativos. Sedimento: 8-10 hematíes/campo.
|


 Anemia. Classification and diagnosis
Anemia. Classification and diagnosis