 |
| Regreso a las bases |
E. Panizo Morgado*, J.A. Páramo Fernández**
*Servicio de Pediatría. Clínica Universidad de Navarra. **Director de la Unidad de Hemostasia y Trombosis. Clínica Universidad de Navarra y del Laboratorio de Hematología
| Resumen
La hemostasia comprende un complejo sistema de reacciones en cadena, sinérgicas y coordinadas, cuya finalidad última es mantener la sangre fluida en el interior de los vasos sanguíneos. Para ello, existe un delicado equilibrio entre los factores procoagulantes y anticoagulantes. Disponemos de una amplia variedad de pruebas analíticas que exploran el sistema hemostático en sus distintas fases (hemostasia primaria, hemostasia secundaria y fibrinólisis). Para poder solicitarlas con criterio y saber interpretarlas, es preciso tener unas nociones básicas de la fisiología de la hemostasia. En el presente artículo, se explican las bases fisiológicas de la coagulación, haciendo hincapié en las peculiaridades de la “hemostasia del desarrollo” del niño y se exponen las pruebas de estudio disponibles, sus indicaciones y su interpretación. |
| Abstract
Hemostasis is a complex system of coordinated and synergistic chain reactions. Its ultimate purpose is to keep the blood flowing within the blood vessels. Thus, there is a delicate balance between procoagulant and anticoagulant factors. A wide variety of coagulation tests exploring the hemostatic system in its diverse phases (primary hemostasis, secondary hemostasis and fibrinolysis) are available. In order to be able to request them judiciously and to interpret them knowledgeably, it is necessary to possess some basic notions of the physiology of hemostasis. In this article, the physiological bases of coagulation are explained, making emphasis in the “developmental hemostasis” of childhood. The available coagulation tests are also reviewed here, with their indications and interpretations. |
Palabras clave: Hemostasia; Fisiología de la coagulación sanguínea; Pruebas de coagulación sanguínea; Niño.
Key words: Hemostasis; Blood coagulation physiology; Blood coagulation tests; Child.
Pediatr Integral XXV (5): 265.e1 – 265.e11
Interpretación de las pruebas de coagulación
Introducción(1)
La hemostasia es un complejo sistema de mecanismos procoagulantes y anticoagulantes que, en última instancia, permiten que la sangre permanezca líquida cuando circula en el interior de vasos sanguíneos. Cuando ocurre una lesión endotelial, el sistema hemostático evita el sangrado excesivo, al promover la formación de coágulos, pero también impide la activación excesiva de la coagulación, limitando su formación en el sitio de la lesión. Este equilibrio hemostático depende de muchos parámetros, siendo los elementos más relevantes: plaquetas, factores de coagulación, inhibidores de la coagulación y endotelio vascular.
Existen diversas determinaciones analíticas que nos permiten estudiar la hemostasia, pero para su correcta solicitud e interpretación, es necesario tener un conocimiento básico de los procesos fisiológicos que subyacen en los fenómenos hemorrágicos.
Fisiología de la hemostasia(2-4)
A nivel didáctico, podemos dividir la hemostasia en 2 fases fundamentales (Fig. 1):
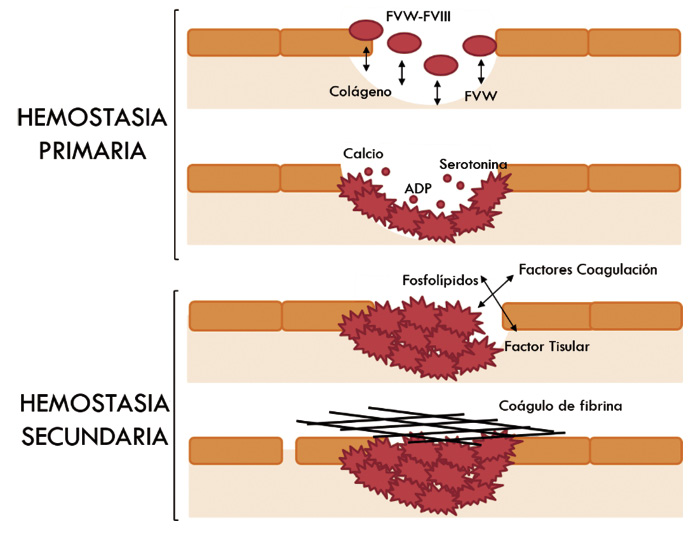
Figura 1. Esquematización de los procesos más relevantes de la hemostasia primaria y secundaria. Hemostasia primaria: tras la lesión vascular se produce la interacción entre las plaquetas, las proteínas estructurales del subendotelio y el factor de von Willebrand (FVW). Las plaquetas se adhieren sobre estas proteínas, se activan, liberan el contenido de sus gránulos e interaccionan entre ellas para formar agregados. Hemostasia secundaria: el factor tisular expresado en el tejido dañado y la superficie de fosfolípidos en las plaquetas activadas iniciarán los mecanismos de la coagulación que aseguran la formación de fibrina.
1. Hemostasia primaria, donde participa principalmente el llamado “componente celular” (plaquetas y endotelio). Comprende la interacción entre el vaso lesionado y las plaquetas, y culmina con la formación del “tapón plaquetario”.
2. Hemostasia secundaria o coagulación, donde interviene el “componente plasmático” (proteínas procoagulantes, anticoagulantes y sistema fibrinolítico). El proceso final es la formación de fibrina, lo que consolidará el tapón hemostático inicial.
Todo el mecanismo hemostático tiene un sistema de regulación, donde, entre otros factores, destacan los anticoagulantes naturales y el sistema fibrinolítico.
Aunque, a continuación, se vayan a repasar los componentes de la hemostasia por separado, es importante recordar que el proceso in vivo no acontece en compartimentos independientes, sino que todos los procesos son concomitantes y sinérgicos, en una serie de reacciones perfectamente coordinadas.
Hemostasia primaria (el compartimento celular)
Tras una lesión vascular, se sucede una vasoconstricción, por la cual el flujo de sangre queda en contacto con la matriz subendotelial, donde hay factor von Willebrand (FVW), sintetizado por las células endoteliales, y otras proteínas estructurales como el colágeno. También, el FVW circulante se depositará sobre la superficie dañada. Las plaquetas poseen receptores para detectar y contactar con estas proteínas. Tras el primer reconocimiento, las plaquetas se adhieren al colágeno expuesto a través de la glicoproteína VI (GPVI) y al FVW a través del complejo glicoproteíco Ib-IX-V (GPIb-IX-V). Posteriormente, se activan y experimentan un cambio conformacional con emisión de pseudópodos. Además, liberan el contenido de sus gránulos e interaccionan entre ellas por medio del fibrinógeno, a través de la integrina GPαIIbβ3 (conocida también como GPIIb/IIIa), para formar agregados que facilitarán la formación del denominado tapón hemostático.
Durante el proceso de activación de las plaquetas, los fosfolípidos aniónicos de la membrana se exponen al exterior. Las estructuras vasculares expuestas, el factor tisular (FT) expresado en el tejido dañado y la superficie de fosfolípidos en las plaquetas activadas iniciarán los mecanismos de la coagulación.
Hemostasia secundaria (coagulación)
La hemostasia secundaria es el proceso por el que se activa la cascada de la coagulación, dando lugar a la fibrina estable. Los factores de la coagulación circulan en el plasma como proteínas precursoras inactivas (zimógenos) que se convierten en enzimas proteolíticas al quedar expuesto su centro activo, por la acción de otra enzima proteolítica. Una vez activadas, actúan de igual manera sobre la siguiente proteína de la cascada en una reacción en cadena. Tradicionalmente, se ha dividido la coagulación en dos vías: intrínseca y extrínseca, que confluyen en la vía común. Esta separación tiene un sentido didáctico y es útil para interpretar pruebas de coagulación in vitro (Fig. 2), pero no se corresponde con los fenómenos in vivo, ya que no contemplan el componente celular.
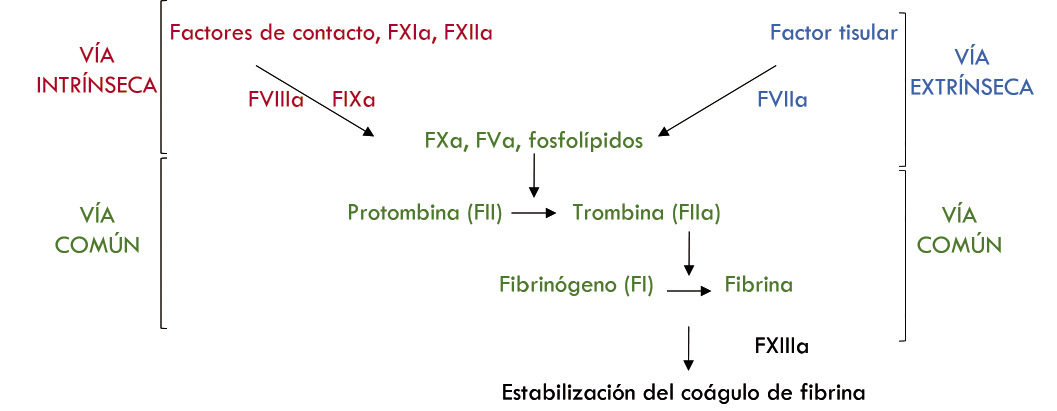
Figura 2. Representación esquemática del modelo clásico de la coagulación. La vía intrínseca (en rojo) se evalúa mediante el tiempo de tromboplastina parcial activado (TTPA). Esta vía se inicia al contacto con el vidrio: se activa el factor XII, que junto con sus cofactores, kininógeno de alto peso molecular y kalicreína (los factores de contacto), activan el factor XI, activando de forma sucesiva al resto de los factores. La vía extrínseca (en azul) se evalúa por el tiempo de protrombina (TP). El proceso se inicia al añadir al plasma la “tromboplastina tisular” (equivalente del factor tisular FT) que, a su vez, activa el factor VII, que activa al factor X, llegando a la vía común (en verde), donde confluyen las 2 vías descritas. El tiempo de trombina (TT) evalúa el paso final de la vía común, la conversión de fibrinógeno en fibrina, tras la adición de trombina exógena. La fibrina se reticula mediante la acción del factor XIII, lo que hace que el coágulo de fibrina final sea insoluble. Esta última función no es probada por el PT, TTPA o TT.
El endotelio sano es una superficie tromborresistente. Actualmente, se acepta que el evento fisiológico principal en el inicio de la coagulación es la interacción entre el factor VII activado (FVIIa) y el factor tisular expuesto tras la lesión vascular. El producto inicial del complejo FT-FVIIa es la conversión del FX en FXa (factor X activado), que puede generar pequeñas cantidades de trombina (fase de iniciación). Esta pequeña cantidad inicial de trombina generada activa el FXI de forma retroalimentada, lo que conduce a la amplificación de la generación de trombina, mediante una serie de reacciones en cadena que incluyen la activación del FV, FVIII, FXI y las plaquetas, que van a exponer los fosfolípidos aniónicos en su superficie, para apoyar el ensamblaje de los complejos enzimáticos multicomponente. Por tanto, la pequeña cantidad inicial de trombina generada ceba la cascada de coagulación y activa las plaquetas, lo que luego conduce a la generación explosiva de trombina. Finalmente, la trombina generada actúa sobre la molécula de fibrinógeno y, tras un proceso de proteólisis, da lugar a la generación de los monómeros de fibrina (fibrinopéptidos A y B). Estos monómeros polimerizan de forma instantánea y son estabilizados por el FXIII, previamente activado por la trombina (Fig. 3).

Figura 3.Modelo celular de la coagulación con tres fases: inicio, amplificación y propagación. Se inicia en el interior de los vasos sanguíneos, cuando las células endoteliales, que expresan el factor tisular (FT), activan al FVII. El complejo FVIIa-FT genera pequeñas cantidades de trombina (iniciación) que, a su vez, activa los factores V, VIII y XI y a las plaquetas (amplificación). Estas últimas cambian su configuración y estructura, exponiendo fosfolípidos cargados negativamente, que van a ser el de anclaje de los factores de la coagulación, dando lugar a la “gran explosión de trombina” y, finamente, al paso de fibrinógeno a fibrina. Se recogen también los principales reguladores de la coagulación.
Mecanismos limitantes de la coagulación
Existen dos mecanismos principales responsables de regular la coagulación: los inhibidores naturales y el sistema fibrinolítico (Fig. 3).
Anticoagulantes naturales
Se pueden dividir en dos grupos:
• Inhibidores de las serín proteasas o “serpinas”: inhiben los factores activados. Estos son: antitrombina, cofactor II de la heparina, inhibidor de la vía de factor tisular (TFPI), proteína C activada, inhibidor de C1 esterasa o α1-antitripsina.
La antitrombina es el principal inhibidor de la trombina y del FXa, aunque también puede inhibir otros factores. Su actividad se ve muy incrementada en presencia de heparina. In vivo, esta función la cumplen los proteoglucanos tipo heparina, presentes en el endotelio vascular y necesarios para el reconocimiento de la antitrombina. La unión de la antitrombina a la heparina o a los proteoglucanos tipo heparina, produce un cambio en su conformación que libera su centro activo.
• Reguladores de los cofactores, como el sistema de la proteína C (proteína C, proteína S y trombomodulina). Este sistema regula al FVa y al FVIIIa, esenciales para mantener la formación de trombina. La proteína C se activa en las superficies vasculares mediante el complejo que forman la trombina con una proteína endotelial llamada trombomodulina. Tras esta activación, la proteína S se une sobre la superficie del endotelio o de la membrana plaquetaria e inactiva proteolíticamente a los cofactores FVa y FVIIIa. Por tanto, la trombina, en contacto con el endotelio sano, no actúa ya como factor procoagulante, sino que actuará como anticoagulante.
La fibrinolisis
El sistema fibrinolítico constituye el mecanismo de defensa final de la hemostasia, que consiste en eliminar el trombo formado, una vez que el vaso se ha reparado (Fig. 4).
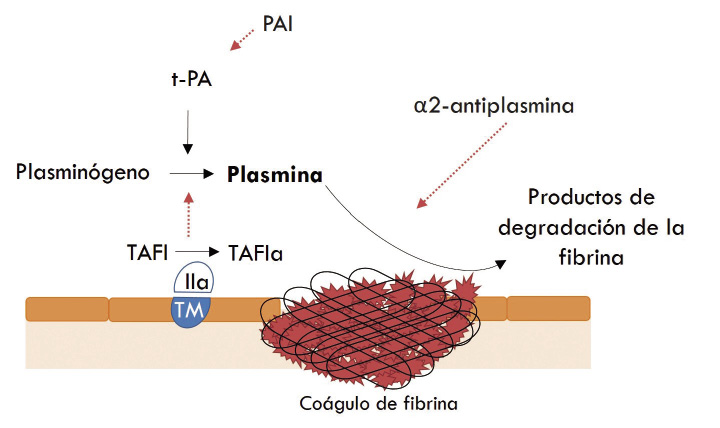
Figura 4. Representación esquemática de la fibrinólisis. Flechas continuas: activación. Flechas discontinuas: inhibición. PAI: Inhibidor del activador tisular del plasminógeno. t-PA: activador tisular del plasminógeno. TAFI: inhibidor de la fibrinólisis activable por trombina. TM: Trombomodulina.
El elemento principal de este sistema es el plasminógeno, que debe transformarse en su forma activa, la plasmina. El principal activador del plasminógeno es el activador tisular del plasminógeno (t-PA), producido por el endotelio y otros tejidos. Existen otros activadores del plasminógeno, como la urokinasa o la estreptokinasa.
Cuando la plasmina se une a la fibrina rompe la forma polimérica de esta, quedando los productos de degradación de fibrina (p. ej.: dímero D), pero también actúa sobre el fibrinógeno y sobre otros factores de la coagulación (p. ej.: V y VIII).
A su vez, este sistema tiene sus propios factores limitantes: los inhibidores del activador del plasminógeno (PAI-1 y PAI-2), la α-2 antiplasmina y el inhibidor de la fibrinólisis activable por trombina (TAFI).
Un nuevo concepto en hemostasia: innmunohemostasia(5)
El sistema hemostático actúa en concierto con la cascada inflamatoria, creando un ciclo hemostasia-inflamación, en el que cada uno de los procesos promueve la activación del otro, siguiendo un sistema de retroalimentación positivo. La comunicación entre ambos se produce a nivel de todos los componentes del sistema hemostático, incluyendo: células endoteliales, plaquetas, proteínas de la coagulación, sistemas anticoagulantes naturales y actividad fibrinolítica.
Durante la respuesta inflamatoria, diversos mediadores, en particular citocinas, juegan un papel central, afectando el sistema hemostático a través de la disfunción endotelial, aumento de la reactividad plaquetar, activación de la cascada de la coagulación, disminución de la función de los sistemas anticoagulantes naturales y supresión de la actividad fibrinolítica. La interacción entre hemostasia e inflamación explica la tendencia protrombótica, lo que se conoce como inmunotrombosis.
Proceso madurativo de la hemostasia en el niño(1,6-8)
El sistema hemostático en el niño cambia y madura a lo largo del tiempo, desde la vida fetal hasta la vida adulta, aunque a partir de los 6 meses se puede considerar que el sistema está plenamente desarrollado. Esto hace que se observen grandes diferencias entre los valores analíticos de niños y adultos, que reflejan los cambios fisiológicos que acontecen en la llamada “hemostasia del desarrollo”. En consecuencia, el conocimiento de esta variabilidad en los rangos de normalidad es fundamental para evitar la clasificación errónea de niños con defectos de factores o inhibidores del sistema de coagulación.
Hoy en día sabemos que, si bien, todos los componentes del sistema hemostático están presentes al nacer, la síntesis de factores de coagulación por parte del feto comienza durante la quinta semana de gestación para el fibrinógeno y, más tardíamente, para el resto de factores. También, se conoce que existen diferencias importantes entre los recién nacidos prematuros, a término, los niños mayores y los adultos, que afectan a todo el sistema hemostático. En general, al nacimiento y en los primeros meses de vida, los niveles plasmáticos de los factores de la coagulación se encuentran disminuidos (alrededor de la mitad de los valores adultos), estando especialmente bajos en los recién nacidos pretérminos; las plaquetas, aunque suelen ser normales en cifras, presentan una marcada hiporreactividad; los niveles de FVIII, por el contrario, suelen estar más elevados y los inhibidores naturales suelen estar descendidos. Las diferencias más destacables se recogen de forma resumida en la figura 5.
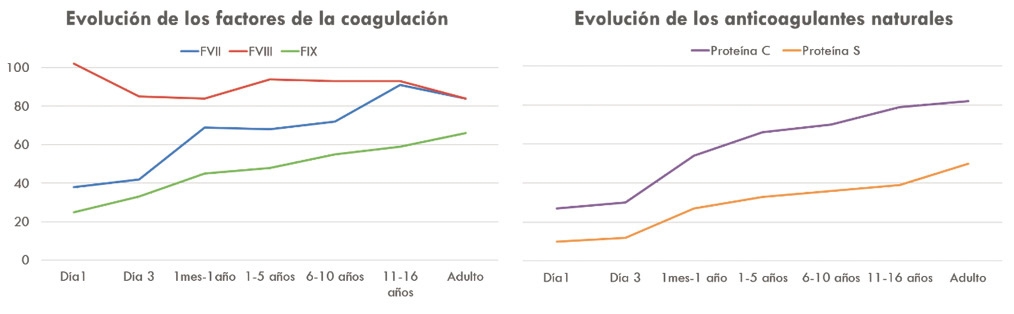
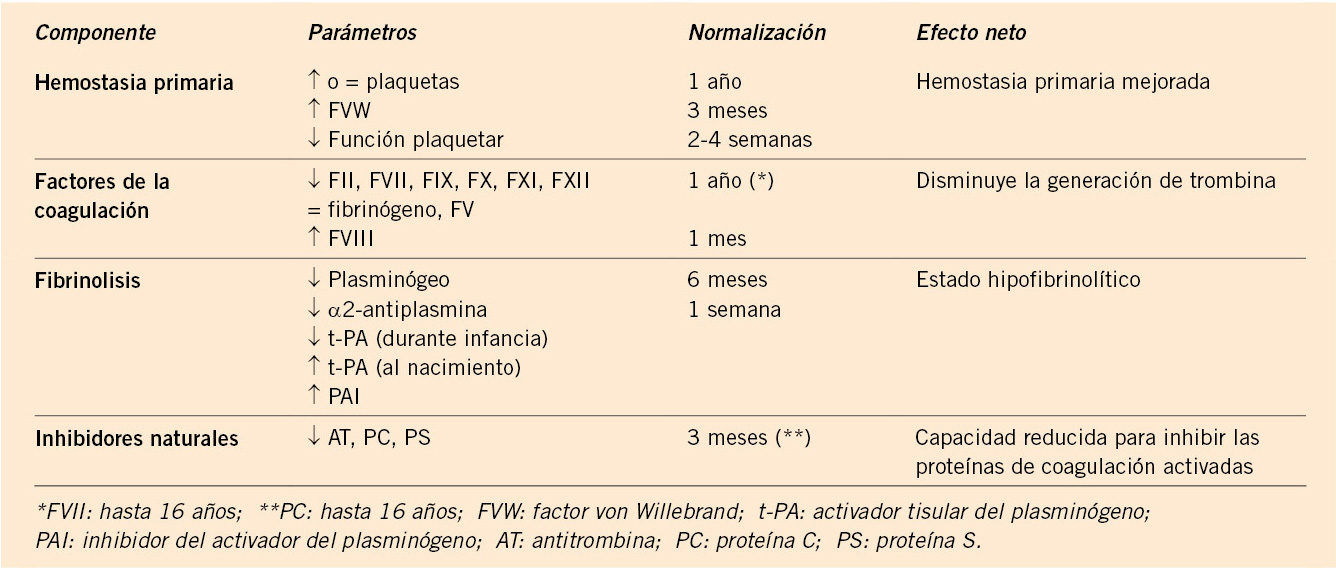
Figura 5. Datos extraídos de Attard C. et al. Developmental hemostasis: Age-specific differences in the levels of hemostatic proteins.
A pesar de todas las diferencias, los niños sanos tienen una hemostasia equilibrada y no tienen una mayor tendencia al sangrado ni a la trombosis. Sin embargo, cuando los niños enferman, este equilibrio puede perderse, favoreciendo la aparición de fenómenos hemorrágicos o trombóticos según el sentido del desequilibrio.
Tal y como se expone en la tabla I, los valores adultos se alcanzan entre los pocos meses de edad y hasta por encima de los 16 años, para parámetros específicos como el factor de coagulación VII o la proteína C. Esto, unido a la variabilidad inter-laboratorio condicionada por el empleo de distintos reactivos y/o instrumentos de medida, ha condicionado que el Comité Científico de Estandarización de la Sociedad Internacional de Trombosis y Hemostasia (ISTH) recomiende que cada laboratorio defina sus propios rangos de normalidad ajustados a la edad del paciente y utilizando sus propias condiciones técnicas.

Métodos de estudio de la hemostasia(3,4,9-11)
Para aproximarnos al estudio de la coagulación, disponemos de una serie de ensayos que nos permiten conocer qué parte del sistema hemostático puede estar o no afecto. Siguiendo el modelo didáctico anteriormente expuesto, valoraremos estas pruebas de acuerdo a la fase de la hemostasia que correspondan.
Aunque cada centro debe disponer de sus propios valores de referencia, se adjunta en la tabla II, unos rangos de normalidad que pueden ser orientativos para los distintos parámetros que vamos a comentar.

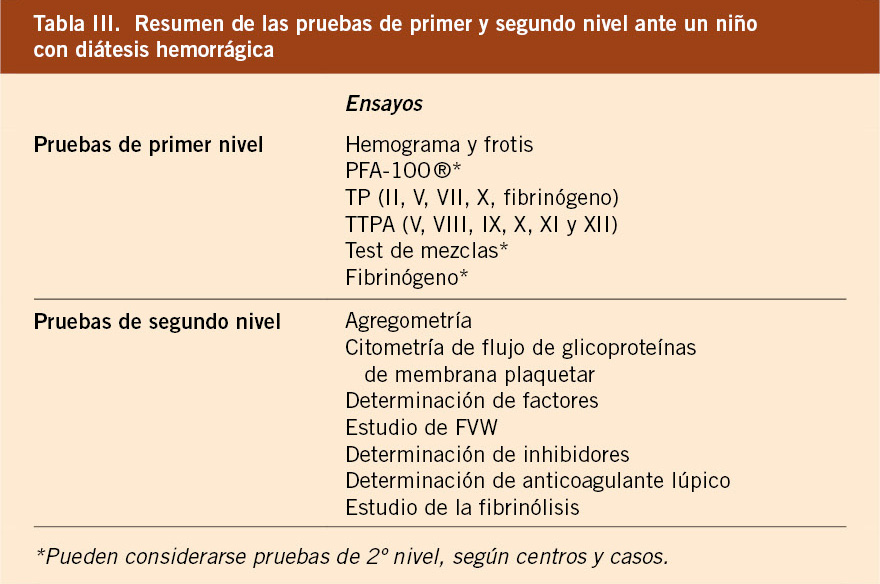
Para facilitar la aplicabilidad de estos estudios en la práctica clínica, los vamos a clasificar en pruebas de primer y segundo nivel en función de su complejidad, disponibilidad y especificidad (aunque los límites de los niveles, como se verá más adelante, dependerán de cada caso particular y de cada centro) (Tabla III).
Estudio de la hemostasia primaria
Estudiaremos los “componentes celulares”, fundamentalmente las plaquetas y el FVW.
Exploración de las plaquetas
Pruebas de primer nivel
• Hemograma: en el hemograma se contabiliza el número de plaquetas. Los valores se expresan en plaquetas x 109/L y la cifra normal oscila entre 150-400 x 109/L, aunque se han descrito cifras inferiores y superiores en sujetos sanos. La vida media es de 8 a 11 días. Un número de plaquetas inferior a 100 x 109/L se denomina trombocitopenia y un valor superior a 400 x 109/L trombocitosis.
Además del recuento, existen otros parámetros que pueden orientar el diagnóstico de una trombocitopenia, si bien, su utilidad es algo controvertida:
– Volumen plaquetar medio (VPM): es el valor medio del volumen de las plaquetas. Se expresa en fl, siendo normal valores de 9 ± 2 fl. Estará aumentado en presencia de plaquetas jóvenes (trombocitopenia inmune, trombocitopenia en recuperación, etc.) y en algunas trombopatías (síndrome de Bernard-Soulier, May-Hegglin, macrotrombopenia familiar). Estará disminuido de forma característica en el síndrome de Wiskott-Aldrich.
– Índice de dispersión de plaquetas o distribución de tamaño de plaquetas (PDW): se expresa en porcentaje y corresponde a la anisocitosis plaquetar. Los valores normales son: 45 ± 20%. Aumenta en las trombocitopenias en recuperación, en las trombocitosis y en algunas hemopatías.
– Plaquetocrito (PCT): es el porcentaje del volumen total de las células sanguíneas que ocupan las plaquetas. En condiciones fisiológicas, la cantidad de plaquetas en la sangre se mantiene en un estado de equilibrio por regeneración y eliminación. El rango normal es 0,12-0,36%.
• Morfología plaquetaria (morfología o frotis de sangre periférica): la observación al microscopio de las plaquetas corroborará el resultado dado por el autoanalizador, siendo muy útil para identificar agregados plaquetarios no cuantificados correctamente, lo que da lugar a falsas plaquetopenias o pseudotrombocitopenia. El EDTA, presente en el anticoagulante utilizado para las muestras de sangre que se emplean en los analizadores automáticos, puede causar este tipo de aglutinación. En esos casos, la observación al microscopio y la realización del recuento plaquetario en sangre citratada, pueden ayudarnos a determinar la cifra real de plaquetas.
La valoración del resto de series en la extensión de sangre periférica puede ayudar también en el estudio de otras causas, como los fenómenos microangiopáticos (se observarán esquistocitos) o las hemopatías malignas.
• Test de obturación plaquetaria o analizador de la función plaquetaria (PFA-100®): es una prueba que evalúa de forma global el funcionamiento plaquetario y que prácticamente ha sustituido al clásico tiempo de sangrado o tiempo de hemorragia (tiempo realizado in vivo que consiste en registrar el tiempo en que cesa el sangrado tras haber practicado una incisión). El analizador de función plaquetaria (PFA-100®) es una prueba rápida y sencilla. Mide el tiempo que tarda en obturarse la abertura central de una membrana recubierta con colágeno y ADP (COL-ADP) o colágeno y epinefrina (COL-EPI) al pasar por ella sangre citratada. Los estímulos químicos (COL-ADP y COL-ADP) en presencia de unas condiciones de flujo estandarizadas dan como resultado la adhesión, activación y agregación de las plaquetas, que crearán un tapón plaquetario estable en la apertura. El tiempo necesario para obtener la oclusión completa de la apertura se registra como tiempo de cierre. Los rangos de referencia medias de los niños sanos son muy similares a las de los adultos sanos, pero más largos que los de los recién nacidos sanos y oscilan, entre 85-120 s, para los cartuchos de COL-ADP; y entre 110-160 s, para los de COL-EPI. En la tabla IV, se presentan los distintos patrones obtenidos con esta prueba y la orientación diagnóstica.

Aunque su utilidad como estudio inicial de rutina es controvertida, dada su simplicidad y disponibilidad, en el caso de que la clínica del paciente sugiera un trastorno de la hemostasia primaria (trombopatía o enfermedad de von Willebrand), se puede realizar como una prueba de primer nivel. No obstante, es importante tener en cuenta sus limitaciones en caso de trombocitopenias por debajo de 100 x 109/L y en el caso de ingesta de AINES reciente.
Pruebas de segundo nivel
• Estudios de agregación plaquetaria: la agregometría es la técnica clásica para el diagnóstico de las alteraciones de la función plaquetaria. La agregometría óptica es una técnica laboriosa que requiere un personal entrenado para su realización e interpretación. Por ese motivo, se han implementado sistemas semiautomáticos, pero aún se requiere avanzar en su desarrollo para su generalización. En los estudios de agregación, las plaquetas son expuestas a agentes agregantes como: ADP, epinefrina, colágeno o ácido araquidónico. El patrón agregométrico obtenido permitirá clasificar el defecto funcional: déficit o anomalía de las glicoproteínas, defectos de almacenamiento o secreción, ya sea por defectos cuantitativos de los gránulos o alteraciones del metabolismo del ácido araquidónico. Por ejemplo, en el caso de la enfermedad de Glanzman (ausencia del complejo GPIIb-IIIa), la agregación plaquetaria estará disminuida frente a todos los agentes agregantes con respuesta conservada a la ristocetina; o en el caso del síndrome de Bernard-Soulier (plaquetas deficientes en glicoproteínas del complejo GPIb-IX-V), la respuesta a la ristocetina estará ausente, pero la agregación al resto de agonistas plaquetarios estará conservada.
• Otras pruebas para estudio plaquetario: citometría de flujo (valora la presencia de las glicoproteínas de membrana), estudio de retracción del coágulo, microscopía electrónica o de fluorescencia (estudia la morfología plaquetaria y los gránulos de las plaquetas), etc. Todas estas exploraciones son laboriosas y se realizan en exclusivamente en laboratorios altamente especializados.
Estudio de enfermedad de von Willebrand
El FVW es una proteína que participa en la hemostasia primaria, contribuyendo a la adhesión plaquetaria ante un endotelio lesionado. En plasma, es el transportador del factor VIII coagulante. Se realizará el estudio de la enfermedad de von Willebrand (EVW) en presencia de un cuadro hemorrágico sugerente, con un estudio básico de la coagulación (TTPA) también alargado, si bien un test normal no excluiría definitivamente la EVW.
Además de los estudios de TTPA (alargado), FVIII (disminuido) y tiempo de obturación (alargado con epinefrina y/o ADP), contamos con estudios específicos de FVW:
• FVW antigénico (FVW:Ag): mide la cantidad de FVW disponible en circulación.
• FVW cofactor de ristocetina (FVW:RCo): se basa en la capacidad del FVW del plasma de unirse a plaquetas normales en presencia de ristocetina. Estudia la funcionalidad del FVW.
• Estructura multimérica del FVW: es una técnica compleja que se realiza únicamente en laboratorios especializados de coagulopatías congénitas y no es imprescindible para el diagnóstico.
En la tabla V, se recoge la orientación diagnóstica según los resultados de las pruebas.

Estudio de la coagulación
Estudios de primer nivel
• Tiempo de protrombina (TP): esta prueba valora la vía extrínseca y la vía común de la coagulación. Consiste en inducir la coagulación mediante la combinación de: plasma citratado, factor tisular (FT), fosfolípidos y calcio, para posteriormente detectar ópticamente el coágulo formado por el autoanalizador. Estudia la integridad de los factores vitamina K dependientes (II, VII, IX y X), el V y fibrinógeno, y se utiliza para monitorizar el tratamiento con antagonistas de la vitamina K. Los resultados se suelen proporcionar como segundos y porcentaje de la actividad (%). Este tiempo también se emplea para el control del tratamiento anticoagulante oral, expresado como INR (razón normalizada internacional), para lo que se tiene en cuenta el índice de sensibilidad internacional (ISI) de cada reactivo: INR = (TP paciente/TP normal)ISI.
• Tiempo de tromboplastina parcial activada (TTPA): esta prueba sirve para monitorizar los niveles de factores de la coagulación de la vía extrínseca y común, así como fármacos antitrombóticos, tipo heparinas e inhibidores directos de la trombina, y detección de anticoagulante lúpico. Será la prueba principal para el despistaje inicial de pacientes hemofílicos (déficit de factor VIII o IX). Se utiliza como reactivo un activador compuesto por fosfolípidos (tromboplastina parcial, de ahí el nombre) y una sustancia cargada negativamente como caolín/sílica, que activa la fase de contacto y la generación del factor XIIa. El TTPA es sensible a las deficiencias de los factores II, V, VIII, IX, X, XI, XII y fibrinógeno. Es menos sensible que el TP a deficiencias dentro de la vía común (p. ej.: II, V, y fibrinógeno) y no se ve afectado por alteraciones en los factores VII y XIII. Por el contrario, aunque los niveles altos de un solo factor (p. ej.: factor VIII) pueden acortar el TTPA, sigue siendo controvertido si existe una asociación entre un TTPA acortado y un estado de hipercoagulabilidad.
Si bien, el PT y el TTPA proporcionan una evaluación general de la formación de coágulos, no proporcionan información sobre la reticulación de la fibrina o la disolución del coágulo y, por lo tanto, serán insensibles a las anomalías de la función del factor XIII o la fibrinólisis anormal, que tendrán que estudiarse de forma independiente.
• Fibrinógeno: la mayoría de los laboratorios miden el fibrinógeno funcional con el método de Clauss, basado en la coagulación de un plasma citratado tras la adición de una concentración elevada de trombina. El autoanalizador proporciona los resultados directamente que se expresan en mg/dL. De forma menos habitual, también se pueden determinar los niveles de fibrinógeno mediante técnicas inmunológicas, de utilidad para el despistaje de disfibrinogenemias.
En la mayoría de los casos de trastornos del fibrinógeno clínicamente significativos, tanto el TP como el TTPA se prolongan, por lo que, en ocasiones, esta prueba se omite en los cribados iniciales. No obstante, es importante recordar que esta prueba es más sensible que el TP / TTPA para detectar trastornos del fibrinógeno, por lo que nosotros sí la recomendamos de manera habitual.
• Test de mezclas: aunque es una prueba que no se realiza de forma rutinaria en algunos laboratorios, es una prueba relativamente sencilla de realizar y que puede aportar mucha información acerca de la etiología de las alteraciones del laboratorio que podemos encontrar. En nuestro laboratorio, se solicita y realiza cuando alguno de los tiempos anteriormente descritos (TP o TTPA) resulta prolongado. Se realiza incubando el plasma del paciente con plasma normal (generalmente a una concentración 1:1) a 37ºC durante 30 minutos. En la muestra resultante, se determina de nuevo el TTPA o el TP. Si se ha corregido tras la incubación, sugiere déficit de alguno de los factores de la coagulación evaluados con esa prueba. Si persiste la alteración tras la incubación, es posible que exista un inhibidor.
Pruebas de segundo nivel
• Dosificación de factores: se solicitará la determinación de los factores de coagulación cuando algunas de las pruebas anteriormente descritas sugieran la deficiencia de uno o varios de ellos. Es importante conocer los factores que estudian cada una de las pruebas globales, para poder dirigir el estudio de la forma más rentable posible.
En la tabla I, se recogen los valores de referencia de los distintos factores según la edad. Se puede apreciar que, mientras algunos factores como el FV o el FVIII se mantienen estables durante prácticamente todo el desarrollo, otros factores tienen niveles muy diferentes al valor normal del adulto. Es importante conocer estos cambios y repetir las determinaciones en aquellos niños que presenten déficits leves de algún factor, ya que es posible que no estemos ante déficits reales, sino ante variaciones fisiológicas del desarrollo.
• Tiempo de trombina/tiempo de reptilase: mide el tiempo de formación de fibrina en presencia de trombina humana o bobina. Esta prueba se emplea para la evaluación de la hemostasia en pacientes que reciben tratamiento anticoagulante (heparina o inhibidores de la trombina), deficiencias congénitas o adquiridas en la formación y polimerización de la fibrina, y paraproteinemias y disfibrinogenemias.
Dado que esta prueba es sensible a la presencia de heparina en la muestra y a niveles reducidos de fibrinógeno, en caso de duda, se puede emplear el tiempo de reptilase, cuya normalidad confirma la presencia de heparina, mientras que el alargamiento indica alteraciones del fibrinógeno. La reptilasa es un enzima similar a la trombina, pero procedente de veneno de serpiente.
• Detección y cuantificación de inhibidores en hemostasia: los inhibidores de la hemostasia son inmunoglobulinas que interfieren en la coagulación. Se pueden distinguir 2 clases de inhibidores:
– Anticoagulante lúpico: prolonga los tiempos de coagulación y suele asociarse con trombosis en adultos. En el paciente pediátrico estos inhibidores suelen ser transitorios y asintomáticos, en el seno de procesos infecciosos. Lo detectaremos ante TTPA prolongados que no corrigen tras la adición de plasma normal.
– Inhibidores específicos de factores concretos de la coagulación, cuya presencia se asocia generalmente a hemorragias. Aunque el más frecuente es contra el factor VIII, también se han descrito inhibidores contra el factor V, XI XII, XIII, vitamina K dependientes y FVW. La prueba de cribado es el test de mezclas y la cuantificación de los mismos se realiza mediante estudios funcionales. Se basan en el descenso de la actividad del factor correspondiente tras incubación del plasma problema con plasma normal. Los resultados se expresan en unidades Bethesda: 1 unidad Bethesda es la cantidad de inhibidor que resulta en un 50% de la actividad residual.
Métodos de estudio de fibrinolisis
Dímero D (DD): el DD es el resultado de la degradación de la fibrina polimerizada por acción de la plasmina. Existen múltiples métodos disponibles para su realización, teniendo cada ensayo rangos de referencia diferentes. El punto de corte habitual para sujetos sanos es de 0,5 µg/ml (500 ng/ml). Su principal utilidad reside en el elevado valor predictivo negativo que presenta en casos de sospecha de tromboembolismo venoso. Sin embargo, es importante recordar que sus niveles aumentan en múltiples circunstancias protrombóticas: CID, inflamación, malignidad, traumatismo, embarazo, cirugía, enfermedad hepática, patologías cardíacas, edad, etc. Por el contrario, encontraremos valores disminuidos de DD en pacientes con tratamiento anticoagulante (eficacia del tratamiento) y en sujetos con deficiencias de factor XIII (presentan alteración de la polimerización de la fibrina).
Aunque disponemos de los ensayos necesarios para poder estudiar la fibrinólisis en mayor profundidad (determinación de plasminógeno, et-PA, α-2 anti-plasmina, PAI1, TAFI, etc.), al tratarse de patologías tan poco frecuentes y que solo se estudian en laboratorios especializados, no profundizaremos en su análisis.
Pruebas globales de hemostasia
Tromboelastometría: es un método para medir las propiedades viscoelásticas durante la formación de un coágulo de sangre y puede ayudar a mejorar el pronóstico de los pacientes, guiando la terapia hemostática y evitando transfusiones innecesarias, sobre todo, en situaciones de hemorragia masiva o cirugías de elevado riesgo hemorrágico. El tromboelastómetro (ROTEM) proporciona curvas sobre el inicio, la firmeza del coágulo y la lisis, de forma rápida (Fig. 6).
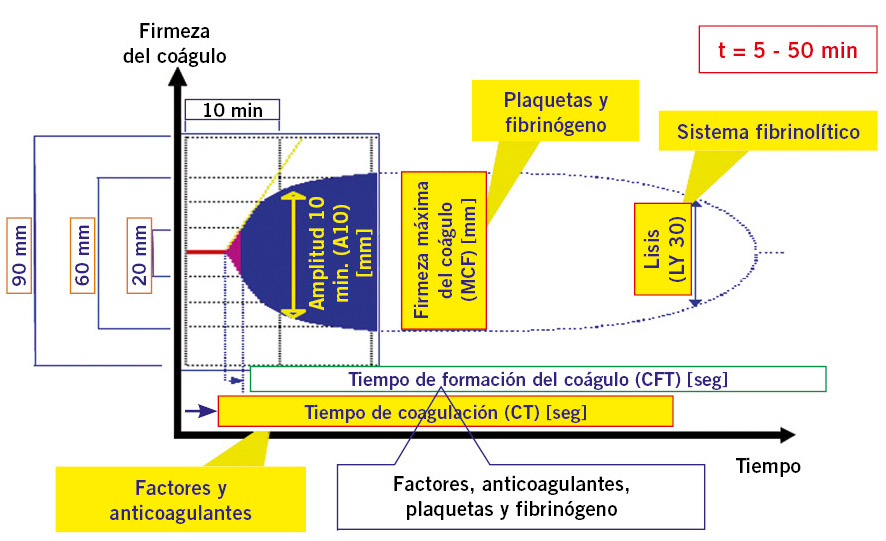
Figura 6. Perfil de coagulación obtenido con test viscoelásticos (ROTEM). En esta figura se puede observar, tanto el trazado del tromboelastograma como todos los parámetros que de él podemos obtener.
Estas pruebas pueden ser de interés en población pediátrica, para decidir sobre la administración de hemoderivados en hemorragia masiva secundaria a trauma o cirugía cardiaca, por ejemplo(12).
Pruebas de laboratorio útiles para el control y monitorización de fármacos anticoagulantes
Las pruebas de valoración de la hemostasia secundaria sirven también para monitorizar la terapia anticoagulante empleada en el tratamiento de la trombosis arterial y venosa. Dada la gran variabilidad de fármacos anticoagulantes disponibles en la actualidad, se resumen a continuación, los test más empleados para su control.
• Monitorización del tratamiento con heparina no fraccionada: el TTPA, al explorar la vía intrínseca, es la prueba de elección para su monitorización. Se expresa como ratio en comparación a un TTPA normal y el rango terapéutico comprende 1,5-2,5.
• Monitorización del tratamiento con heparina de bajo peso molecular (HBPM): se emplea la determinación de la actividad anti-Xa (Anti-Xa). Debe determinarse a las 4-6 h tras la administración de la HBPM. Los niveles terapéuticos oscilan entre 0,5-1 U/mL.
• Monitorización del tratamiento con antivitaminas K: se emplea el TP, expresado en forma de ratio normalizado o INR. Los valores terapéuticos se encuentran entre 2-3 (en pacientes con válvulas mecánicas el rango es mayor, 2,5-3,5).
• Monitorización de los anticoagulantes orales directos:
- Inhibidores del factor Xa (rivaroxabán, apixabán y edoxabán): se empleará la antividad anti-Xa utilizando los calibradores adecuados.
- Inhibidores directos de la trombina (dabigatran): el TTPA es útil como medida cualitativa de la presencia de dabigatrán, pero no sirve para su cuantificación. Para ello utilizaremos: tiempo de trombina diluido o el tiempo de ecarina, ambas variaciones del tiempo de trombina.
Variables preanalíticas a considerar en el paciente pediátrico(9,11)
Las pruebas de hemostasia son particularmente susceptibles a la fase preanalítica, ya que multitud de elementos pueden alterar sus resultados. A continuación, se exponen algunas recomendaciones para evitar errores:
• Se aconseja la extracción de muestra en ayunas de 4 h.
• No se debe tomar ningún medicamento que afecte la función plaquetaria el día de la prueba. En el caso de la aspirina y los AINEs, se deben evitar, al menos, 1 semana.
• Se desaconseja fumar, ingerir cafeína, tomar remedios a base de hierbas o ajos e ingerir alcohol, antes de las pruebas.
• La sangre debe obtenerse mediante punción venosa directa realizada por personal experimentado, utilizando preferiblemente agujas de calibre 19 a 21G (las muestras obtenidas de accesos venosos permanentes pueden estar contaminadas con heparina o líquidos intravenosos lo que falseará los resultados).
• Se deben desechar los primeros 1-2 ml y muestras con hemólisis o coágulos visibles.
• Se debe emplear sangre citratada (una proporción citrato por nueve de sangre; proporciones distintas pueden alargar falsamente los resultados, por exceso de anticoagulante o provocar la coagulación de la muestra, por defecto).
• La sangre extraída debe mezclarse suavemente mediante inversión (3-4 veces), enviarse al laboratorio sin demora (evitando tubos neumáticos) y analizarse dentro de las 2 horas posteriores a la extracción, o en las 4 horas siguientes si se mantiene fría. Las muestras de plasma deben congelarse si no se analizan en ese plazo.
• Los resultados de coagulación se ven afectados por poliglobulias (ocasionará una reducción en la proporción del citrato) y el PFA-100 por la trombocitopenia y el hematocrito bajo.
Bibliografía
1. Van Ommen CH, Sol JJ. Developmental Hemostasis and Management of Central Venous Catheter Thrombosis in Neonates. Semin Thromb Hemost. 2016; 42: 752-9.
2. Cervera Bravo A, Álvarez Román MT. Fisiopatología y trastornos de la coagulación hereditarios más frecuentes. Pediatr Integr. 2016; 5: 318-30.
3. Jiménez Yuste V. Atlas de hemofilia. Salermo: Momento Médico; 2013.
4. González Porras J, Páramo Fernández JA, Mateo Arranz J. Hemostasia y trombosis. Manual práctico. Arán Ediciones; 2018.
5. Delabranche X, Helms J, Meziani F. Immunohaemostasis: a new view on haemostasis during sepsis. Ann Intensive Care. 2017; 7: 117.
6. Toulon P. Developmental hemostasis: laboratory and clinical implications. Int J Lab Hematol. 2016; 38: 66-77.
7. Lippi G, Franchini M, Montagnana M, Guidi GC. Coagulation testing in pediatric patients: The young are not just miniature adults. Semin Thromb Hemost. 2007; 33: 816-20.
8. Breakey VR. Reference Ranges for Common Tests of Bleeding and Clotting. In: SickKids Handbook of Pediatric Thrombosis and Hemostasis. 2013. p. 232-4.
9. Melo Valls M, Murciano Carrillo T. Interpretación del hemograma y pruebas de coagulación. Pediatr Integr. 2012; XVI(5): 413.e1-413.e6.
10. Revel-Vilk S. Clinical and laboratory assessment of the bleeding pediatric patient. Semin Thromb Hemost. 2011; 37: 756-62.
11. O´Brien S, Mahoney D. Approach to the child with bleeding symptoms. UpToDate. Versión 2021. Actualizado el 19/09/2019. Consultado: abril 2021. Disponible en: http://www.uptodate.com/contents/approach-to-the-child-with-bleeding-symptoms.
12. Cunningham AJ, Condron M, Schreiber MA, Azarow K, Hamilton NA, Downie K, et al. Rotational thromboelastometry predicts transfusion and disability in pediatric trauma. J Trauma Acute Care Surg. 2020; 88: 134-40.


 Anemia. Classification and diagnosis
Anemia. Classification and diagnosis