 |
| Temas de FC |
H. González García, R. Herraiz Cristóbal, J.L. Moreno Carrasco
Unidad de Hemato-Oncología Infantil. Hospital Clínico Universitario de Valladolid. Valladolid
| Resumen
Esta revisión aborda la fisiología de la hemostasia primaria, la cascada de coagulación clásica y el modelo celular de coagulación in vivo, así como los avances recientes en el diagnóstico y el tratamiento de la enfermedad de von Willebrand y la hemofilia. La enfermedad de von Willebrand es el trastorno hemorrágico hereditario más común, tiene un patrón de herencia autosómico dominante y se caracteriza por una disminución cualitativa o cuantitativa de la actividad del factor de von Willebrand y hemorragias mucosas y hemorragias después de cirugía y traumatismos. El diagnóstico se basa en los antecedentes personales o familiares de hemorragias y en las pruebas de laboratorio de las anomalías del factor von Willebrand. Pueden distinguirse varios tipos sobre la base de características fenotípicas que pueden diagnosticarse mediante pruebas de laboratorio específicas. El tratamiento se centra en aumentar los niveles del factor von Willebrand mediante la administración de desmopresina o la infusión de concentrados exógenos que contienen el factor. La hemofilia es una enfermedad genética causada por una deficiencia del factor VIII en la hemofilia A y del factor IX en la hemofilia B, ambas de herencia recesiva y ligadas al sexo. La gravedad clínica está relacionada con la intensidad del déficit de factor. Se analizan el diagnóstico, el diagnóstico diferencial y el tratamiento. |
| Abstract
This review focuses on the physiology of primary hemostasis, the classical coagulation cascade and cellular model of in vivo coagulation, as well as on the recent advances in the diagnosis and treatment of von Willebrand disease and hemophilia. Von Willebrand disease is the most common inherited bleeding disorder, it has a dominant autosomal inheritance pattern, and it is characterized by a qualitative or quantitative decrease of the von Willebrand factor activity and mainly presents with mucosal bleeding and bleeding after surgery and trauma. The diagnosis is based on a personal or family history of bleeding and laboratory evidence of abnormalities in von Willebrand factor. Various types can be distinguished based on phenotypic characteristics that can be diagnosed by means of specific laboratory tests. Treatment is centered on increasing von Willebrand factor levels by administration of desmopressin or infusion of exogenous concentrates containing the factor. Hemophilia is a genetic disease caused by a deficiency of factor VIII in hemophilia A and factor IX in hemophilia B, both of which are recessively inherited and sex-linked. Clinical severity is dependent upon the degree of factor deficiency. Diagnosis, differential diagnosis and treatment are discussed. |
Palabras clave: Hemostasia primaria; Coagulación; Enfermedad de von Willebrand; Hemofilia A; Hemofilia B.
Key words: Primary hemostasis; Coagulation; von Willebrand disease; Hemophilia A; Hemophilia B.
Pediatr Integral 2021; XXV (5): 242 – 253
Enfermedad de von Willebrand y otros trastornos frecuentes de la coagulación
Funcionalismo hemostático en la edad infantil(1-4)
La hemostasia se define como el conjunto de acontecimientos fisiológicos encaminados a detener la hemorragia y la posterior restauración de la circulación.
La hemostasia se inicia para impedir la hemorragia tras una lesión vascular. Secuencialmente, se ponen en marcha la hemostasia primaria (vasoconstricción, adhesión y agregación plaquetaria) y la hemostasia secundaria (coagulación). Finalmente, se produce la regulación del proceso hemostático mediante los mecanismos anticoagulantes y la fibrinólisis.
Hemostasia primaria. La lesión vascular produce de inmediato una vasoconstricción de la zona lesionada que da lugar a una disminución de la superficie dañada. La exposición del subendotelio, desencadena la adhesión de las plaquetas y la agregación plaquetaria con la intervención del factor von Willebrand (FvW), dando lugar al trombo plaquetario (Fig. 1).

Figura 1. Representación esquemática de la hemostasia primaria en un vaso lesionado. El factor von Willebrand ( ) funciona como una glicoproteína adhesiva imprescindible en la interacción entre el endotelio dañado y las plaquetas (
) funciona como una glicoproteína adhesiva imprescindible en la interacción entre el endotelio dañado y las plaquetas ( ), inicialmente a través de la glicoproteína de membrana Ib, produciendo la adhesión plaquetaria. Posteriormente, el colágeno del subendotelio se fija a la glicoproteína VI de membrana plaquetaria, produciendo la activación plaquetaria (
), inicialmente a través de la glicoproteína de membrana Ib, produciendo la adhesión plaquetaria. Posteriormente, el colágeno del subendotelio se fija a la glicoproteína VI de membrana plaquetaria, produciendo la activación plaquetaria ( ), con reclutamiento de nuevas plaquetas y la unión entre ellas, formando puentes entre el fibrinógeno y el complejo glicoproteico de membrana IIb/IIIa: agregación plaquetaria. El factor von Willebrand se produce en el endotelio y en el megacariocito y se encuentra tanto el plasma como en los gránulos alfa de las plaquetas y en el tejido conectivo subendotelial (en los cuerpos de Weibel-Palade). Autor de la figura: H. González.
), con reclutamiento de nuevas plaquetas y la unión entre ellas, formando puentes entre el fibrinógeno y el complejo glicoproteico de membrana IIb/IIIa: agregación plaquetaria. El factor von Willebrand se produce en el endotelio y en el megacariocito y se encuentra tanto el plasma como en los gránulos alfa de las plaquetas y en el tejido conectivo subendotelial (en los cuerpos de Weibel-Palade). Autor de la figura: H. González.
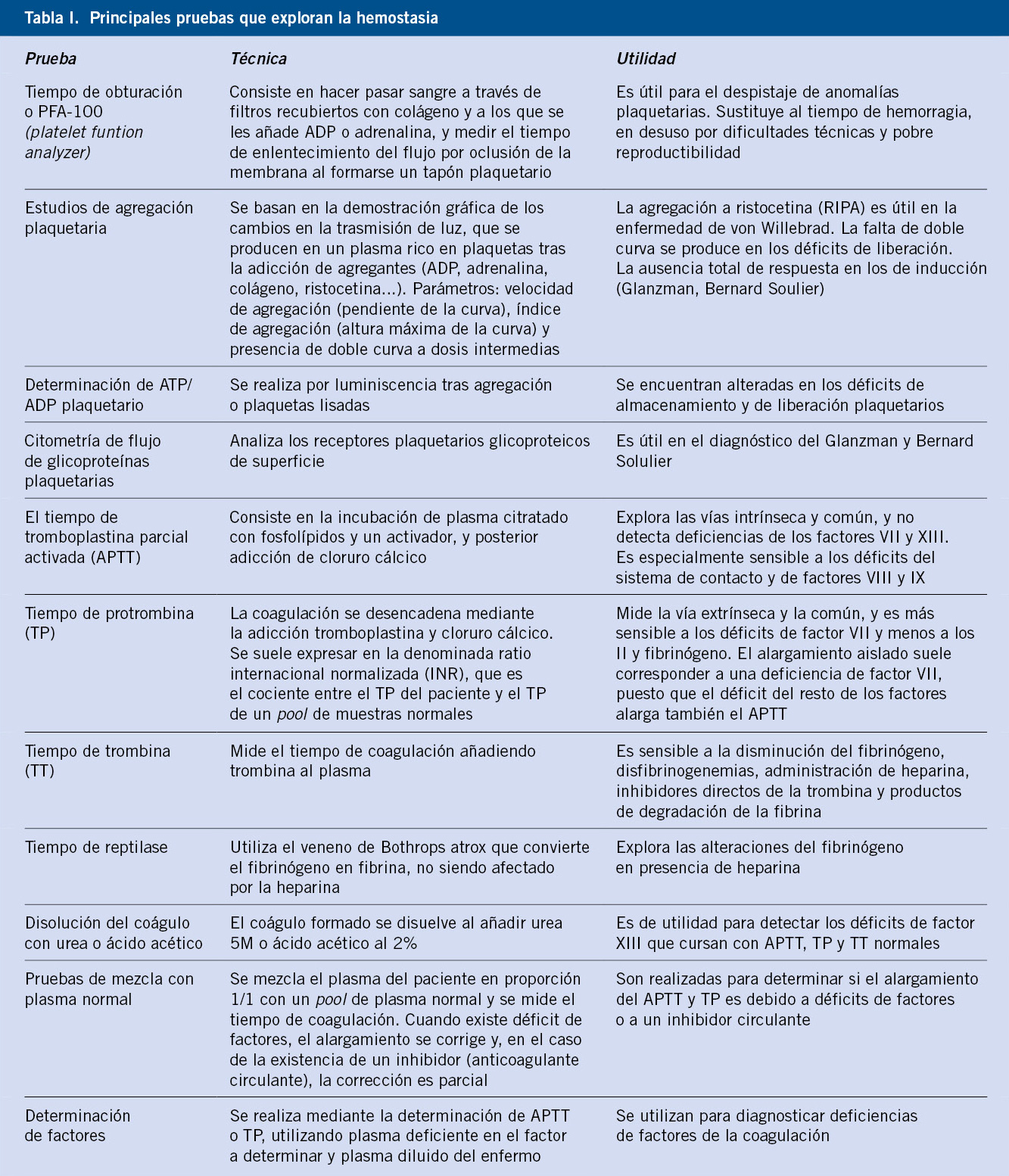
Las pruebas que exploran la hemostasia primaria derivan de la comprobación de que exista un número de plaquetas suficiente (entre 150.000 y 450.000/mm3) y que funcionen adecuadamente(1) (Tabla I).
Hemostasia secundaria (Fig. 2).
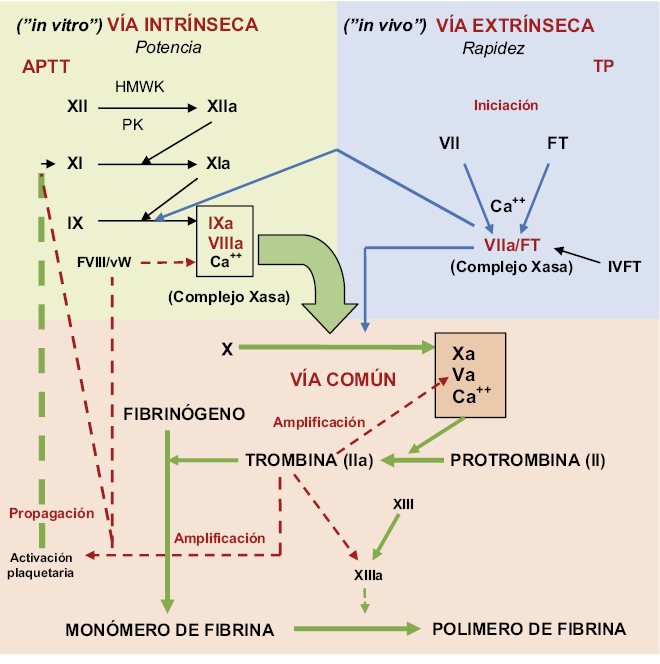
Figura 2. Esquema general de la coagulación, integración del modelo clásico (texto) y el modelo celular. APTT: tiempo de tromboplastina parcial. TP: tiempo de protrombina. HMWK: kininógenos de alto peso molecular. Pk: prekalicreína. FVIII/vW: complejo de unión entre el factor VIII y el factor von Willebrand. En la fase de iniciación sobre la célula lesionada, el factor tisular (FT) actúa sobre el factor VII que en presencia de calcio es activado (VIIa), generando el complejo Xasa. Este activa al factor X, y a la vez activa al factor IX. El factor Xa se une al factor Va y se genera trombina. La cantidad de trombina es pequeña, porque existe un inhibidor de la vía del factor tisular (IVFT), que se libera también de la célula lesionada, este se une al factor X producido en exceso y además inhibe al complejo Xasa, deteniéndose la generación de trombina. En la fase de amplificación, la trombina activa al factor V y también a los factores XI y XIII y activa las plaquetas. También, separa el complejo factor VIII/vW en factor VIIIa para la siguiente fase y FvW para la hemostasia primaria. En la fase de propagación sobre las plaquetas activadas, el factor XI es activado y este activa al factor IX. El factor IX activado se une al factor VIII activado, formándose otro complejo Xasa que es el que sostiene la coagulación, pues este, a diferencia del que se forma en la fase de iniciación, no tiene un inhibidor. A continuación, se activa el factor X que se une al factor Va formando el complejo protrombinasa y se genera gran número de moléculas de trombina. Los factores Va y VIIIa pueden ser inhibidos por la proteína C y el resto de factores por algún inhibidor como la antitrombina, pero esto no sucede en el entorno de la plaqueta activada, donde prima la actividad procoagulante. In vitro, el factor XII se activa iniciando la vía intrínseca que puede ser estudiado por la prueba APTT. La vía extrínseca se estudia mediante la prueba del TP. Autor de la figura: H. González.
El sistema de coagulación plasmática es el responsable del mantenimiento de la fluidez de la sangre, pero cuando es activado, da lugar a la producción del coágulo de fibrina por la conversión del fibrinógeno en el polímero de fibrina, merced a la actuación de un enzima, la trombina, que es capital para el desarrollo de la función hemostática, pues actúa activando las plaquetas y retroalimentando la coagulación. El sistema de coagulación plasmática se encuentra integrado por proenzimas que existen en el plasma de manera inactiva, junto con cationes (calcio) y fosfolípidos de origen celular. Las proenzimas (zimógenos), por la acción de una enzima proteolítica, dejan al descubierto su parte activa, formándose las enzimas proteolíticas que actúan de igual forma sobre la siguiente proteína de la cascada en una reacción en cadena(1,2).
De una forma didáctica (Fig. 2), la coagulación puede ser desencadenada por dos mecanismos, la vía intrínseca y la extrínseca. In vitro, el contacto de la sangre con el tubo de vidrio activa la vía intrínseca mediante la activación del factor XII por el sistema de contacto (kininógenos de alto peso molecular y prekalicreína). El factor XII activado (XIIa) activa, a su vez, al IX (IXa) y este junto con el factor VIII activado, fosfolípidos y calcio, activan al factor X (Xa). In vivo, se activa la vía extrínseca, por el factor tisular que se expone tras la lesión vascular, actuando sobre el factor VII que, en presencia de calcio, es activado (VIIa). El factor VIIa activa al factor X y, a la vez activa al factor IX, con lo que refuerza la acción sobre la vía intrínseca. La vía extrínseca le da a la coagulación plasmática inmediatez de acción y la intrínseca potencia coagulante, pues de una molécula activada inicialmente se generan miles de moléculas de factor Xa. A partir del Xa se establece la vía común de formación de trombina. La trombina, actúa sobre el fibrinógeno polimerizándolo, dando lugar a las redes de fibrina y, a su vez, activa al factor XIII que, actuando sobre la fibrina, estabiliza el coágulo. La trombina formada inicialmente, es la responsable de la activación de los factores VIII y V, piezas claves en los complejos de activación del X y de la protrombina (II); así mismo, activa el factor XI retroalimentando la vía intrínseca e interviene en la agregación irreversible plaquetaria y activa el inhibidor de la fibrinólisis activable por trombina, para que la plasmina no detenga la coagulación(1-3).
Actualmente, se describe el modelo celular de la coagulación como que consta de: una fase de iniciación, por la exposición del factor tisular; una fase de amplificación por la trombina originada en la fase de iniciación; y una fase de propagación sobre las plaquetas activadas. En la figura 2, se representan la integración del modelo clásico y celular de la coagulación. En este modelo celular “in vivo”, el factor XII no es indispensable para la coagulación y de hecho su deficiencia no provoca hemorragia sino trombosis, puesto que activa al factor XI “in vitro”, pero in vivo activa más aún a la plasmina que al factor XI(1-3).
Todos los factores menos el VIII se sintetizan en el hígado y los factores II, VII, IX y X necesitan la actuación de la vitamina K que carboxila el ácido glutámico imprimiéndole mayor carga negativa a las moléculas, ligando con más facilidad calcio y fosfolípidos. Las deficiencias aisladas de factores son poco frecuentes y generalmente son de origen hereditario. Las deficiencias combinadas son más frecuentes y de origen adquirido (CID, hepatopatía, deficiencia de vitamina K, etc…)(1,2).
Una vez formado el coágulo y detenida la hemorragia, el sistema hemostático debe ser inactivado fuera de la lesión vascular para evitar la coagulación masiva de la sangre en los vasos, mediante la activación de anticoagulantes naturales que bloquean el proceso a diferentes niveles anulando acción de agentes procoagulantes (factores VIII y V, factor II, trombina y factor Xa). Los anticoagulantes naturales más importantes son la antitrombina III y la proteína C, y su cofactor la proteína S. Es la propia trombina la que sobre el epitelio vascular sano se une a la trombomodulina activando el sistema proteína C-proteína S, que inhibe a los factores V y VIII activados, convirtiéndose así la trombina sobre el epitelio sano en un factor anticoagulante. La antitrombina III inhibe a la trombina y también a los factores activados IX, X, XI y XII(1-4).
Formado el coágulo y detenida la hemorragia, este se retrae perdiendo agua y volumen, iniciándose su destrucción mediante la fibrinólisis, merced a la formación de plasmina a partir de plasminógeno y de su activador. La plasmina destruye el coágulo produciendo, como elementos residuales, los productos de degradación de la fibrina (PDF, dímero D), que se forman cuando dos moléculas de fibrina son rotas por la plasmina en el D-dominio (Fig. 3).
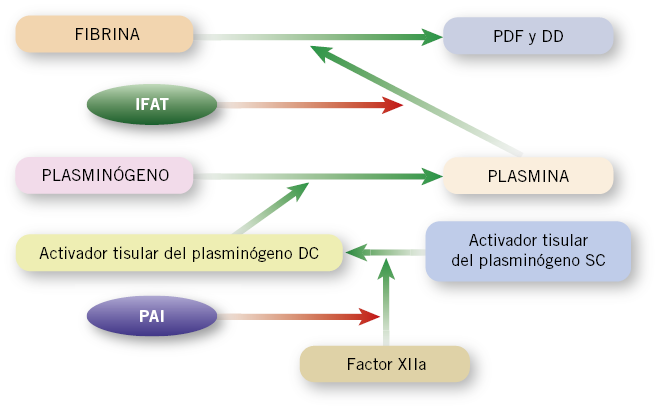
Figura 3. Esquema de la fibrinólisis. Proceso en el que se lisa la fibrina, generándose productos de degradación (PDF) y dímero D (DD). Flechas verdes: estimulación. Flechas rojas: inhibición. La fibrinólisis se inactiva inicialmente por la propia trombina, cuando se inicia el proceso de coagulación por el inhibidor de la fibrinólisis activable por trombina (IFAT), para que la plasmina no detenga la coagulación. Una vez el coágulo ha detenido la hemorragia y, mientras se produce la cicatrización, se inicia la disolución del coágulo por liberación de sustancias, como el activador tisular del plasminógeno. Este, para ser activo, precisa ser de doble cadena (DC), siendo su forma inactiva de simple cadena (SC). El sistema del factor XIIa (que precisa prekalicreína y kininógenos de alto peso molecular) es el responsable de configurar al activador tisular del plasminógeno en doble cadena. El inhibidor del activador del plasminógeno (PAI) regula la actividad fibrinolítica, inhibiendo la formación de dobles cadenas. La deficiencia de factor XII congénita produce, en clínica, trombosis por este mecanismo, aunque alargue el APTT por su efecto sobre la coagulación “in vitro”. Autor de la figura: H. González.
La determinación de dímero-D se realiza mediante inmunoanálisis, utilizando anticuerpos específicos frente a él. El dímero-D se eleva en múltiples situaciones: intervenciones quirúrgicas, grandes hematomas, trombosis venosas y arteriales, sepsis, inflamaciones y coagulación intravascular diseminada (CID)(2-4).
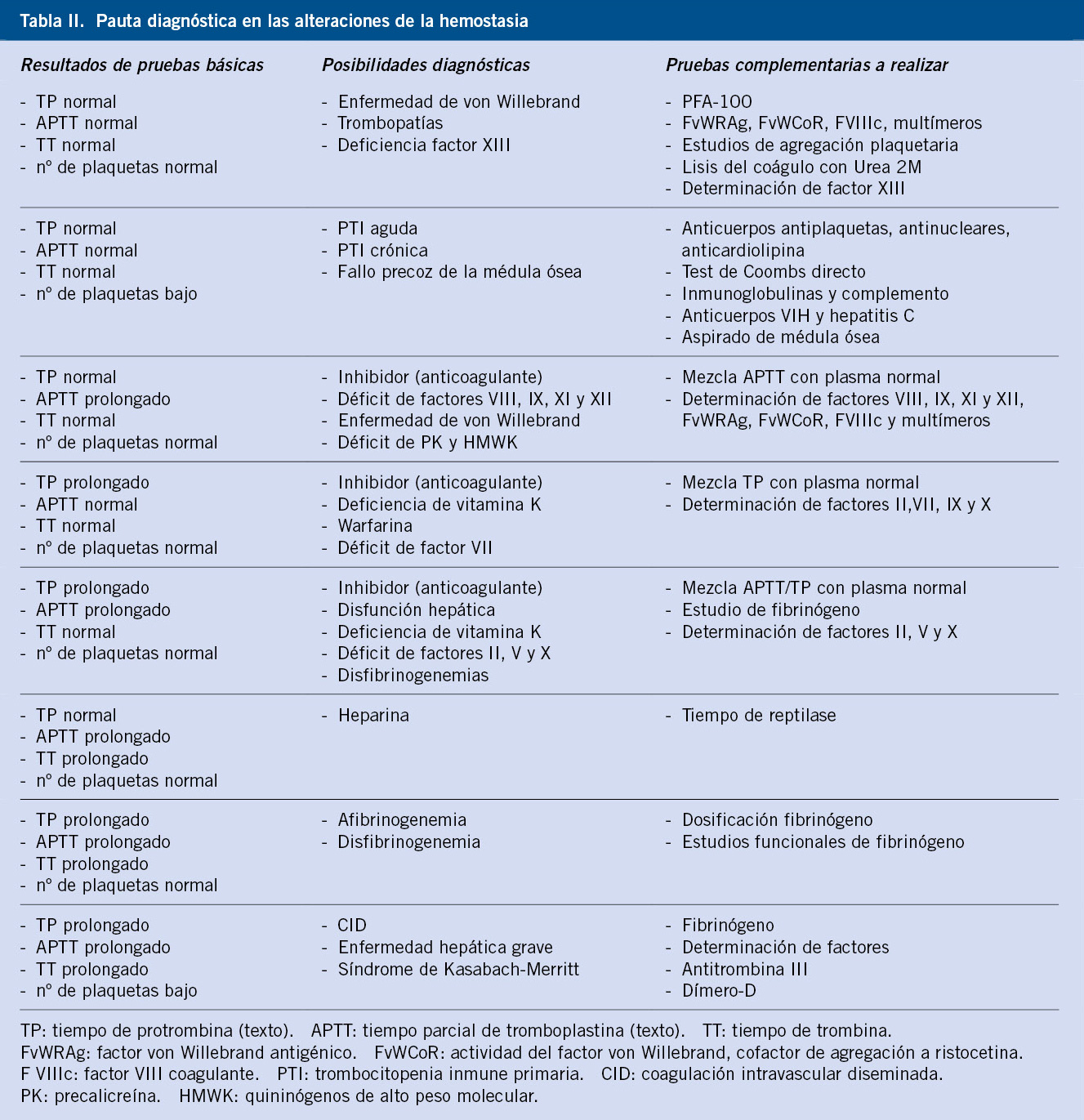
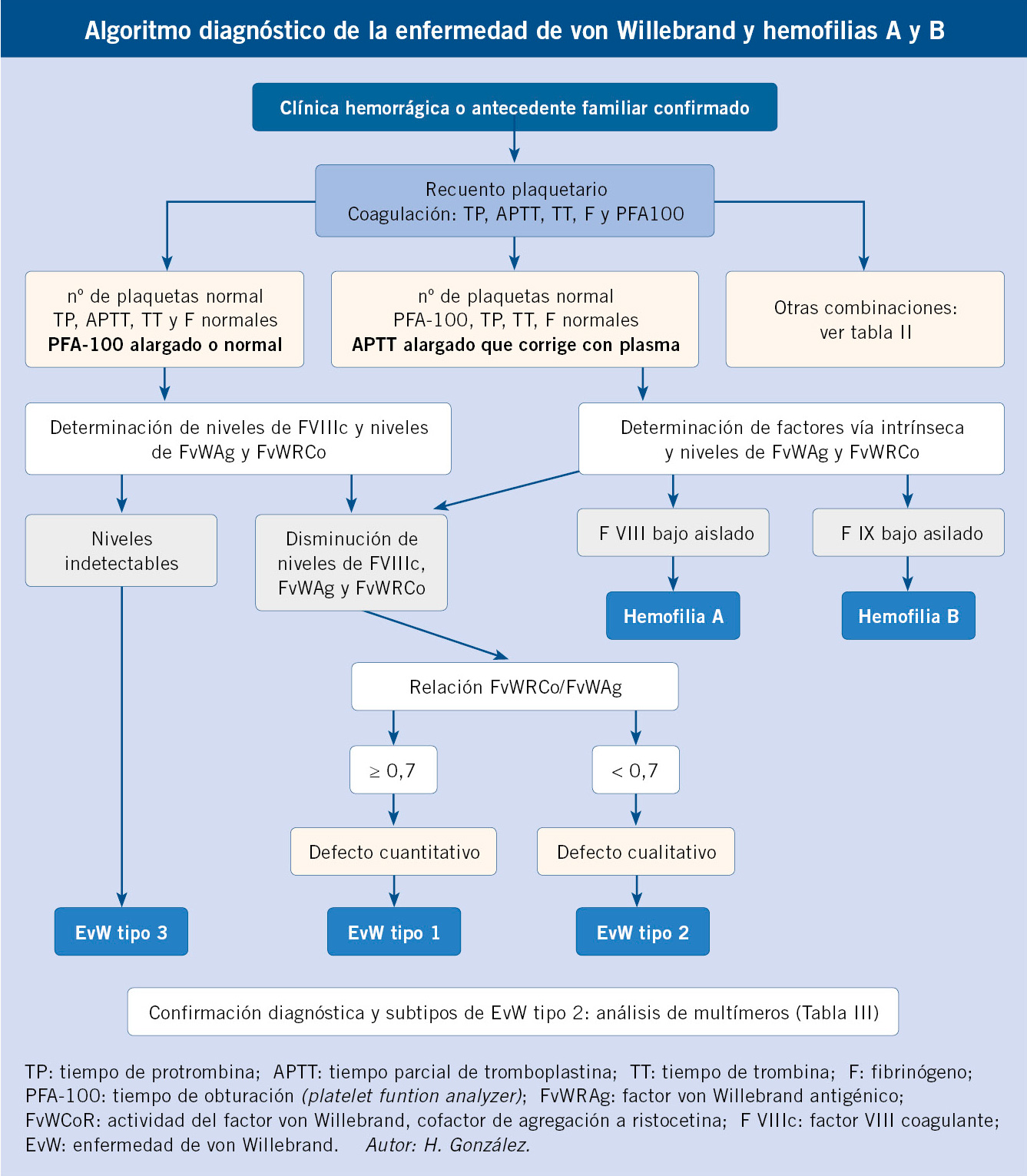
Todas las pruebas que exploran la coagulación plasmática se basan en la observación de la formación de fibrina. En la actualidad, no se usan las pruebas de exploración global (tiempo de coagulación) y sí las pruebas parciales, como el tiempo de tromboplastina parcial activada (APTT), el tiempo de protrombina (TP) y el tiempo de trombina (TT) (Tabla I). En pacientes con clínica de sangrado, dependiendo del resultado de la combinación de las pruebas de hemostasia básicas, se pueden establecer las posibilidades diagnósticas y pruebas complementarias a realizar para un diagnóstico específico (Tabla II)(1).
Enfermedad de von Willebrand
La enfermedad de von Willebrand es la alteración congénita más frecuente de la coagulación.
La alteración cuantitativa o cualitativa del FvW es el origen de la enfermedad de von Willebrand (EvW), que es la causa más frecuente de alteración congénita de la hemostasia, con una frecuencia del 1 por 100 en pruebas de cribado de laboratorio y de 1 cada 1.000-10.000 personas en su forma sintomática. Su transmisión es habitualmente autosómica dominante, aunque hay formas recesivas, así como adquiridas (tumor de Wilms)(5-7).
El FvW es una glicoproteína adhesiva y pieza clave en la interacción entre el endotelio dañado y la plaqueta (adhesión y agregación plaquetarias) y también como transportador del FVIII, al que estabiliza alargando su vida media. Esta molécula se produce en el endotelio y en el megacariocito y se encuentra tanto el plasma como en los gránulos alfa de las plaquetas y en el tejido conectivo subendotelial (Fig. 1). La síntesis está regulada por un gen que se encuentra en el brazo corto del cromosoma 12. La proteína plasmática circula en el plasma con un peso molecular variable que depende de la polimerización de la molécula (entre 500 y 20.000 kDa), siendo los multímeros de alto peso molecular funcionalmente más activos. La vida media es de 8 a 12 horas(5,6). En la figura 4, se muestra el esquema de la formación de la molécula de FvW y los diferentes puntos de unión a estructuras de las que depende su función fisiológica(8,9).
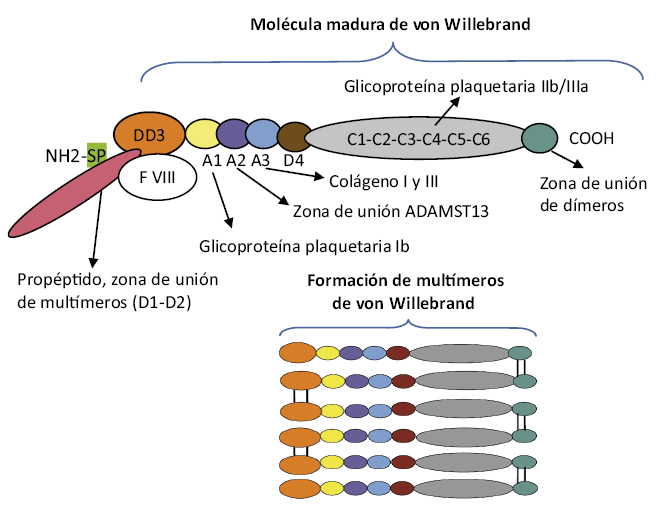
Figura 4. Molécula del factor de Von Willebrand (FvW) y formación de multímeros(8,9,11). El precursor del FvW (pre-pro-FvW) está formado por un péptido señal (SP), un propéptido (zona de unión a multímeros D1-D2) y, finalmente, la molécula madura del FvW. La pérdida del péptido señal origina el pro-FvW. El pro-FvW está organizado en repeticiones de dominios estructurales homólogos (A, C y D), donde se encuentran las diferentes zonas de unión del FvW: al factor VIII a través del DD3; a la glicoproteína plaquetaria Ib a través de la subunidad A1; al sitio de escisión de la proteína ADAMTS 13 en la subunidad A2; al sitio de unión al colágeno I y III gracias a la subunidad A3 y a la importante zona de unión a la glicoproteína plaquetaria IIb/IIIa a través del dominio C4. Para la formación de dímeros y, posteriormente, de multímeros, se requiere un complejo procesamiento. Tras la síntesis del precursor en el retículo endoplásmico, el péptido señal se escinde y el pro-FvW se dimeriza mediante enlaces disulfuro en la región C-terminal en la zona de unión de dímeros. Posteriormente, en el aparato de Golgi se escinden los propéptidos, gracias a lo cual, los dímeros pueden unirse en la región N-terminal (DD3) para la formación de multímeros, que pueden superar los 20.000 kDa. Autores de la figura: R. Herraiz y H. González.
Para una adecuada función del FvW, se precisa de la regulación del tamaño del multímero por ADAMTS 13, que se encuentra en las células endoteliales y se encarga de la escisión de los multímeros ultragrandes de gran afinidad con las plaquetas. Si ADAMTS 13 no actúa, los multímeros de muy alto peso molecular provocan la agregación plaquetaria y formación de trombos (purpura trombótica trombocitopénica)(8,9).
Las pruebas diagnósticas específicas para el diagnóstico de la enfermedad incluyen(5,6,9,10):
• Determinar el factor de von Willebrand antigénico (FvWAg): cantidad circulante de factor de von Willebrand.
• Determinar el factor de von Willebrand cofactor de ristocetina (FvWCoR): cuantifica la actividad del factor. La ristocetina es un antibiótico que consigue cambiar la configuración del FvW para que pueda unirse a las plaquetas a través de la glicoproteína (GP) Ib (GPIb)(5). Consiste en mezclar el plasma del paciente junto con plaquetas fijadas en formol de otro paciente y ristocetina. Las plaquetas se agregan ante mínimas cantidades de FvW del plasma.
• Análisis de los multímeros. Mediante métodos de separación de proteína (electroforesis), se determinan los diferentes tamaños de las moléculas de las subunidades de FvW de acuerdo a su peso molecular.
• Estudio de agregación plaquetaria en presencia de ristocetina (RIPA). Depende de la concentración de ristocetina y de la afinidad de FvW por la GPIb.
Clasificación
La forma más frecuente es la enfermedad de von Willebrand tipo 1, déficit parcial cuantitativo, con antecedentes familiares (herencia dominante).
Existen diversas formas de la enfermedad que obedecen a diferentes mecanismos fisiopatológicos de las alteraciones cuantitativas o cualitativas del FvW(4,10-12) (Tabla III) (Algoritmo).
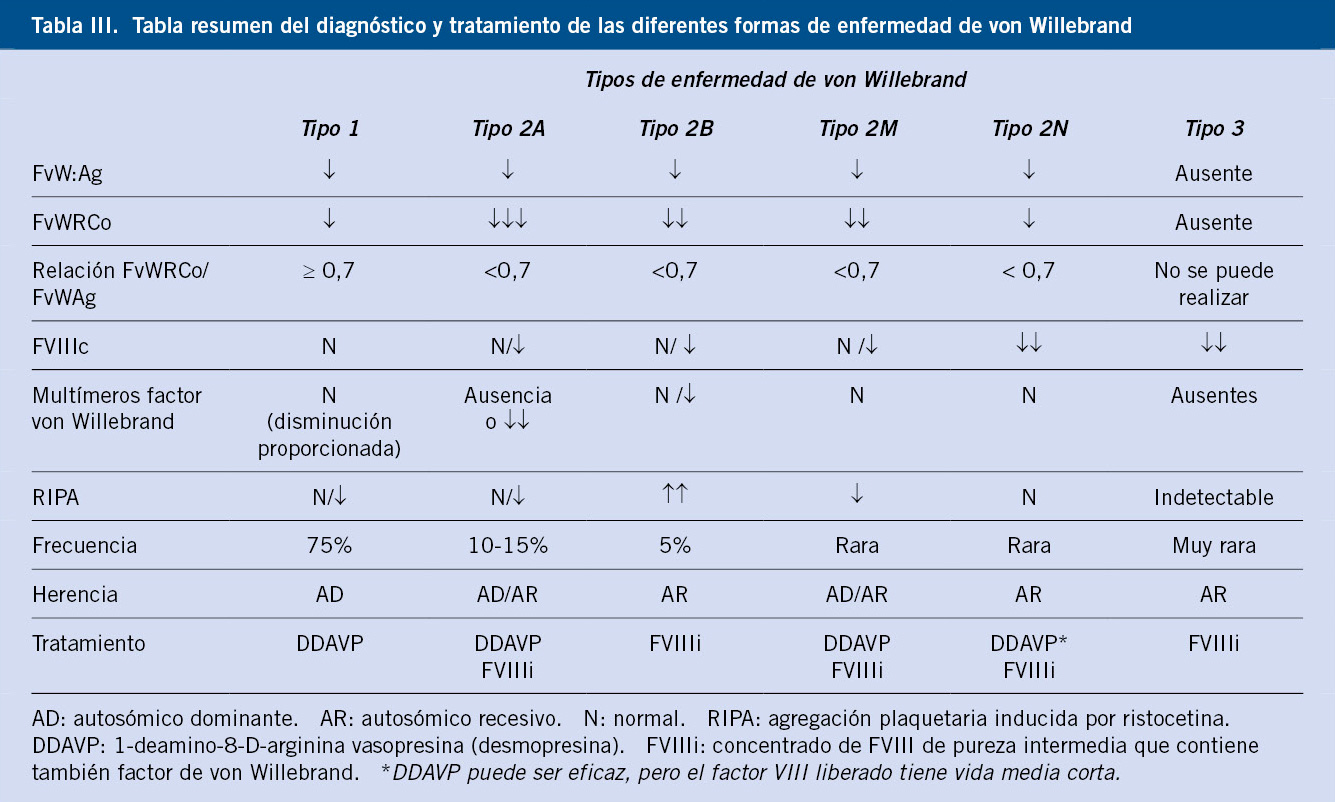
Tipo 1. Defecto cuantitativo (leve, heterocigoto): disminución de cantidad. Es la variante más frecuente (70-75% de los casos). Se trasmite de forma autosómica dominante, aunque con una penetrancia variable. Lo más habitual es que sean pacientes asintomáticos o con síntomas muco-cutáneos leves. El diagnóstico se basa en una prolongación de los tiempos de obturación (PFA-100), aunque, en ocasiones, puede resultar normal, y una disminución proporcionada del FvWAg y del FvWCoR (cociente FvWCoR/FvWAg ≥ a 0,7).
Tipo 2. Defectos cualitativos. Frecuencia del 20-25%. En este caso, se produce una disminución desproporcionada del cociente FvWCoR/FvWAg que será ≤ 0,7. El TTPA se puede encontrar prolongado si existe una disminución del FVIIIc por una anomalía en la molécula transportadora, el FvW, o por una alteración en la zona DD3 del FvW que impida la unión.
• EvW tipo 2A. Frecuencia del 10-15%. Se produce bien por defectos en formación de dímeros o multímeros, o bien por anomalía en la regulación, por presentar una mutación que provoca que la zona A2 del FvW sea más sensible a la acción de la ADAMTS 13, que actúa en exceso, escindiendo los multímeros. En el estudio de multímeros se aprecia la ausencia o disminución importante de los de alto peso molecular. Hay formas de herencia dominante y recesiva.
• EvW tipo 2B. Frecuencia del 5%. Se produce por mutaciones en la zona A1 del FvW que aumentan la capacidad de unión de este a la GPIb. Provoca agregación plaquetaria y fagocitación de multímeros grandes. Se diagnostica, porque hay una hiperagregación plaquetaria con bajas concentraciones de ristocetina (RIPA aumentada). En este caso, puede haber normalidad o ausencia de multímeros de alto peso molecular. La herencia es dominante. Puede cursar con trombocitopenia.
• EvW tipo 2M. Frecuencia muy baja. Se produce una deficiente unión del FvW a la GPIb. Esto origina un defecto de adhesión, ya que impide la unión entre la plaqueta y el FvW. Provoca una alteración similar al déficit de GPIIb/IIIa (enfermedad de Bernard Soulier). El RIPA está disminuido y los multímeros son normales, pero no funcionantes.
• EvW tipo 2N. Frecuencia muy baja. Existe una mal función en la zona DD3, zona de unión entre el factor VIII y su transportador, el FvW. La función hemostática del FVW está conservada (el tiempo de obturación puede ser normal), pero existe una disminución del factor VIII que se cataboliza rápidamente. El diagnóstico de la variante 2N puede realizarse determinando la capacidad de unión del FvW al FVIII(10). Se realiza esta prueba cuando el ratio FVIII/FvW:Ag es <0,5, para diferenciar el tipo 2N de la hemofilia moderada.
Tipo 3. Grave defecto homocigoto o dobles heterocigotos. Frecuencia excepcional. Se trata de una enfermedad más grave que la hemofilia, porque presenta niveles bajos o indetectables de FvW y, consecuentemente, de FVIIIc. Presentan la misma clínica que los pacientes con hemofilia grave, pero asociando el defecto en la agregación plaquetaria.
Clínica
La enfermedad de von Willebrand tipo 1 origina clínica leve, moderada en los tipos 2A, 2B y 2M, y grave en el tipo 3.
En general, la clínica de sangrado suele ser leve en el tipo 1, moderada en los tipos 2A, 2B y 2M, y severa en el tipo 3. El tipo 2N presenta sangrado leve a moderado en tejidos blandos y a nivel intraartícular. El sangrado articular es una complicación frecuente en pacientes con hemofilia, pero no suele ser habitual en los pacientes con EvW, excepto en los pacientes con subtipo 2 N o 3(4,9,11).
No existe correlación entre las mutaciones del gen y la expresividad clínica, ni siquiera entre los miembros de la misma familia, porque hay factores que pueden modular la expresividad clínica. La EvW tipo 1 puede llegar a pasar desapercibida o con clínica leve (hematomas con facilidad, epistaxis prolongadas, hemorragia gingival en la caída de dientes primarios, menorragias), por lo que una forma muy frecuente de presentación es la presencia de sangrados en intervenciones quirúrgicas, incluso con estudio preoperatorio básico de coagulación normal o menstruaciones muy abundantes, con anemia en niñas adolescentes(4,9,11).
Diagnóstico
El diagnóstico precisa de pruebas complementarias que demuestren la disminución de actividad del factor.
El diagnóstico de la EvW puede llegar a ser complejo. Tanto el FvW como el FVIII se comportan como reactantes de fase aguda, por lo que los niveles de ambos pueden encontrarse elevados en caso de estrés, ejercicio, entre otros. Por otro lado, los pacientes con grupo sanguíneo 0 presentan, de forma fisiológica, niveles de FvW un 20-30% más bajos. Además, el umbral del nivel a considerar como patológico es controvertido, aunque hay consenso en considerar como patológicos niveles <30% y niveles bajos, que pueden corresponder, tanto a individuos sanos como enfermos, entre 30-50%(4,11,13).
Para el diagnóstico de la EVW, se requiere la presencia de síntomas de sangrado en el paciente, demostrar la disminución de la actividad del FvW y la presencia de agregación familiar(4,13). Sin embargo, la clínica leve de la EvW tipo I puede pasar desapercibida y, dependiendo de la edad, no es infrecuente que aún no hayan sido sometidos a intervenciones quirúrgicas o extracciones dentarias. Además, los síntomas de sangrado leve son también frecuentes en niños sanos (equimosis en zonas expuestas, epistaxis…). Existen escalas para adultos y adaptadas a niños de clínica de sangrado significativo, asociadas a unas preguntas estandarizadas de las que se obtiene una puntuación que diferencia entre pacientes sanos y enfermos, o con sangrado que requiere estudio(14). Un paciente debe remitirse para estudio especializado, si ha presentado sangrado prolongado tras una cirugía o extracción dental, o la presencia de hematomas musculares o hemartrosis, o ante una historia familiar de una enfermedad hemorrágica(4,11,13). La sistemática diagnóstica se basa en estudios progresivos que, a menudo, requieren de varias determinaciones y que se reflejan en las tablas II y III, y el algoritmo.
En todos los pacientes con EvW, excepto en el tipo 2B o el tipo 3, se debe realizar al diagnóstico el test de desmopresina, que trata de comprobar si hay respuesta a su administración con elevación significativa de los niveles de FvW. La 1-deamino-8-D-arginina-vasopresina (DDAVP o desmopresina) libera, tanto el FvW como el FVIII de su lugar de almacenamiento en el endotelio(4).
Tratamiento
El tratamiento de la enfermedad de von Willebrad se basa en la utilización de desmopresina en casos respondedores y derivados plasmáticos del factor.
En todos los pacientes con EvW, excepto en el tipo 2B o el tipo 3, se puede emplear la desmopresina, si en el test diagnóstico existe respuesta (Tabla IV)(4).
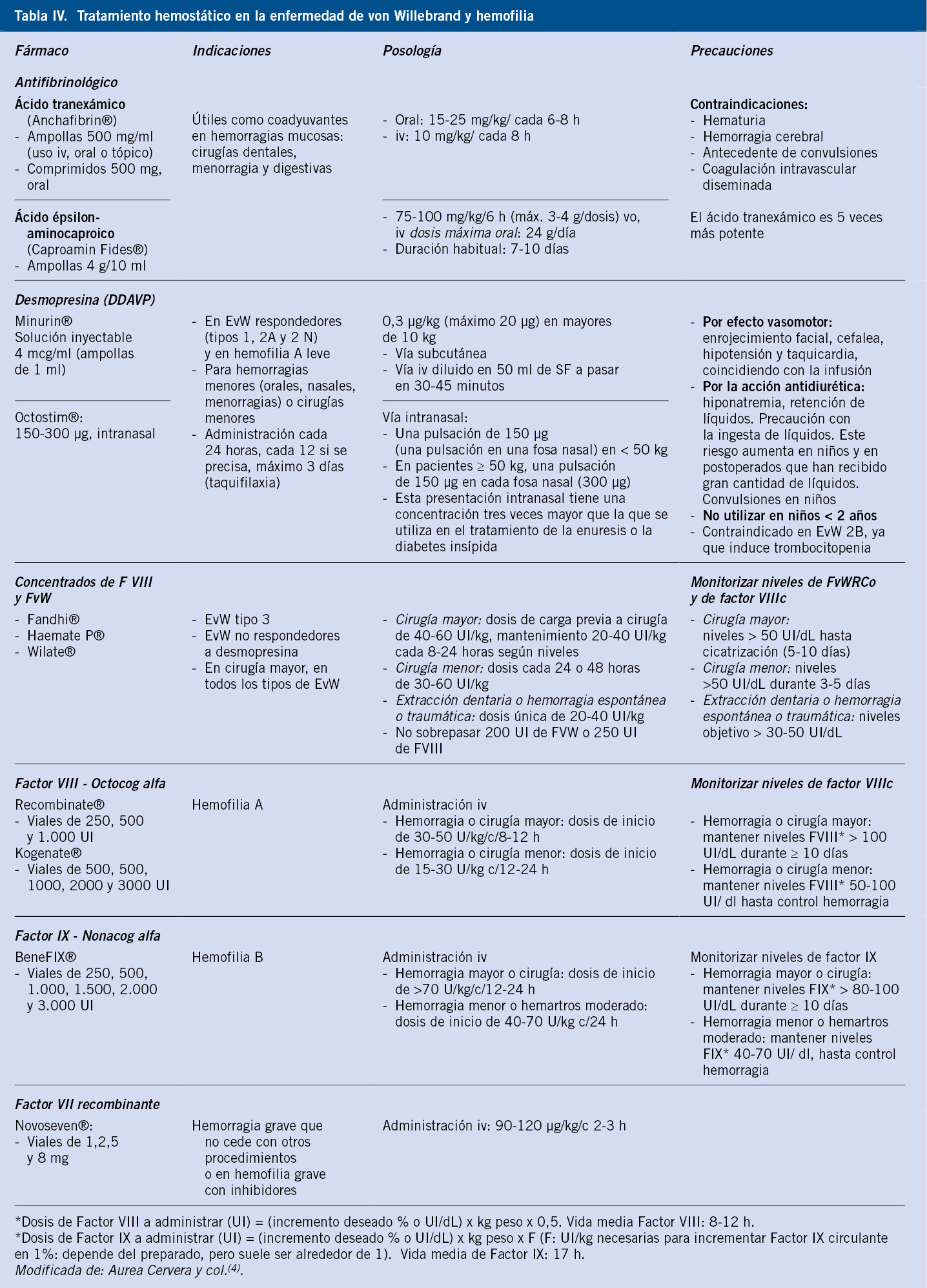
La respuesta puede ser variable de unos pacientes a otros, pero es constante en un mismo enfermo. Produce un incremento de 3 a 5 veces de los niveles basales de FvW y de FVIII hacia los 30-60 minutos y dura entre 6 y 12 horas. La mayor parte de los pacientes con tipo 1 responden y un porcentaje, generalmente los menos graves, de los de tipo 2, aunque el test debe realizarse en todos. En la mayoría de las formas graves del tipo 2, el defecto cualitativo no puede ser compensado por la liberación de más moléculas de FVW defectuoso. En el tipo 2B, la liberación de FvW anormal puede aumentar la aglutinación plaquetaria y acentuar la trombopenia, por lo que no se recomienda, y en el tipo 3 no hay respuesta(15,16). En casos de cirugía mayor, se debe cuantificar la actividad del FVW:RCo y del FVIII, pero no es necesario para episodios de sangrados leves, tras haber comprobado la respuesta. La desmopresina con las dosis repetidas disminuye la acción (taquifilaxia), por lo que no se recomienda más que una vez al día y la duración del tratamiento no durará más de tres días. Las dosis, formas de presentación y efectos secundarios se muestran en la tabla IV. La hiponatremia puede producir convulsiones, por lo que no se recomienda en niños de menos de dos años. La restricción de líquidos disminuye el riesgo de hiponatremia. La formulación intranasal es muy práctica y permite la administración, de forma ambulatoria, antes de procedimientos menores(4,15,16).
En los casos en los que no puede administrarse el DDAVP, o se prevea la necesidad de más de tres días de tratamiento, hay que emplear concentrados plasmáticos que contengan FvW (derivados con FvW y FVIII)(15,16). Las formas de administración, indicaciones y precauciones se exponen en la tabla IV. Para la EvW, en 2015, se ha aprobado el primer recombinante de FvW sin factor VIII asociado(17) (Vonvendi®), con indicación, en la actualidad, para pacientes mayores de 18 años.
Como coadyuvantes para el control de hemorragias mucosas (menorragias, epistaxis, extracción dentaria), pueden usarse antifibrinolíticos (Tabla IV) y para hemorragias de pequeños vasos nasales o bucales agentes tópicos, como trombinas tópicas o selladores de fibrina. No se recomienda administrar anti-inflamatorios no esteroideos (AINES) en la EVW, porque alteran la función plaquetaria(4,16).
Hemofilias A y B
Las hemofilias son enfermedades genéticas por déficit de factor VIII en la hemofilia A y de factor IX en la B.
La hemofilia es una enfermedad genética expresada por un déficit de los factores de coagulación VIII (hemofilia A), IX (hemofilia B) u XI (hemofilia C). La hemofilia A es el tipo más frecuente (1/4.000-5.000 recién nacidos varones, 85% de casos totales), seguida de la hemofilia B (1/30.000-50.000 recién nacidos varones), y la hemofilia C es aún más infrecuente (1/100.000)(18,19). La clasificación de las hemofilias, en estrecha relación con su gravedad clínica y con la edad de aparición, viene determinada por los niveles de actividad del factor en cuestión: leve cuando la actividad del factor es ≥ 5%, moderada entre 1-5% y grave ≤1%. Mientras que el 60-70% de las hemofilias A debutarán como una hemofilia grave, la frecuencia en la hemofilia B es menor (40%)(18).
Esta patología se produce por la alteración en los genes que codifican para la producción de los factores de la coagulación implicados que, en el caso de la hemofilia A y B, se localizan en el cromosoma X (herencia recesiva ligada a X), mientras que en la hemofilia C, se encuentra en el cromosoma 4 (herencia autosómica recesiva). En las hemofilias A y B, las mujeres serán portadoras sanas de la enfermedad, salvo excepciones, mientras que los varones son enfermos. En la hemofilia C, tanto hombres como mujeres pueden padecerla. Hasta un tercio de los casos son esporádicos, producidos por mutaciones de “novo”, sin antecedentes familiares(19).
Clínica
Las manifestaciones clínicas dependen de la gravedad del déficit del factor.
La clínica es idéntica para todas las hemofilias, ya que los factores de coagulación implicados actúan conjuntamente en la vía intrínseca. Las hemofilias leves suelen debutar a edades más tardías, a veces, como hallazgos incidentales en estudios preoperatorios de pacientes asintomáticos, o bien tras traumatismos de gran intensidad o cirugías de alto riesgo hemorrágico(18,19). Las hemofilias moderadas suelen iniciar los síntomas durante los primeros años de vida, siendo las formas más habituales de presentación en forma de equimosis, grandes hematomas frente a mínimos traumatismos o relacionados con la administración de vacunas o fármacos intramusculares, y mediante sangrados leves-moderados (epistaxis, sangrados bucales tras rotura de frenillo o en extracciones dentales…)(20). Las hemofilias graves habitualmente debutan en el primer año de vida (en ocasiones, incluso durante el período neonatal, en forma de hemorragias cerebrales hasta en un 2-5% de casos) y en cualquier localización. Son muy características, por ser más específicas de estos pacientes y por la morbilidad que implican, las hemorragias en articulaciones y los hematomas musculares, los más frecuentes en psoas ilíaco y en antebrazo. La hemartrosis es la localización más frecuente en pacientes ambulatorios, y pueden originarse espontáneamente o ante mínimos traumatismos, siendo las localizaciones más afectadas el tobillo, la rodilla y el codo, por este orden. Además, las hemartrosis de repetición desarrollan mayor susceptibilidad a nuevos sangrados y mayor inflamación que deriva en una sinovitis crónica y, con el tiempo, una artropatía grave irreversible(18,20).
Diagnóstico
El diagnóstico precisa de la demostración de niveles disminuidos del factor afectado.
La sospecha clínica de hemofilia debe realizarse ante cualquier paciente que presente diátesis hemorrágica, sobre todo, en el caso de varones en las hemofilias A y B, y en los casos con antecedentes familiares conocidos de sangrados frecuentes. El diagnóstico se realiza mediante la realización de pruebas de coagulación (TP y TT normales y TTPA alargado, con estudios de FvW dentro de rango normal) y la determinación de niveles de actividad de los distintos factores que confirmará el diagnóstico y determinará la gravedad (Tablas I y II, Algoritmo). Se considera una determinación de factor de coagulación dentro del rango normal el comprendido entre 50-120%, pero los valores de referencia deben ser ajustados a la edad del paciente, pues los menores de 2 años suelen presentar niveles fisiológicos más bajos(18,20). El estudio molecular de las mutaciones de las hemofilias no es necesario para la confirmación diagnóstica, pero, en ocasiones, puede resultar relevante para el estudio de mujeres portadoras y el consejo genético y/o diagnóstico prenatal(19,20).
El diagnóstico diferencial se establece con otras patologías que cursen clínicamente con diátesis hemorrágica: coagulopatías congénitas como la EvW, déficit de otros factores de coagulación como los vitamina-K-dependientes o el factor XIII, con trombocitopenias graves adquiridas o de carácter hereditario como el síndrome de Bernard-Soulier o la trombastenia de Glanzmann(1-4,11) (Tabla II y Algoritmo).
Tratamiento
El tratamiento de la hemofilia grave precisa de la infusión del factor deficiente.
En pacientes hemofílicos A leves/moderados, el uso de desmopresina (DDAVP), vía intravenosa o intranasal, es útil para el manejo de hemorragias menores o cirugías de bajo riesgo hemorrágico, pero no es eficaz para la hemofilia B(4,20,21) (Tabla IV). En el caso de episodios hemorrágicos importantes o la necesidad de realización de cirugías de alto riesgo hemorrágico en pacientes con hemofilia moderada/grave, la terapia sustitutiva con administración de concentrados de factor VIII o IX recombinantes es el tratamiento de elección(20,21). Estos preparados, ampliamente comercializados actualmente, tienen un alto grado de seguridad frente a la transmisión de agentes infecciosos. Su vía de administración es exclusivamente endovenosa y presentan como principal inconveniente una vida media corta, de 10-12 h para el factor VIII y de 17 horas para el factor IX. Según la gravedad de la hemorragia o del tipo de cirugía a realizar, se establecerán diferentes objetivos de hemostasia (Tabla IV). Por otro lado, los avances actuales en el tratamiento de la hemofilia, se centran en el desarrollo de nuevas moléculas o fármacos de administración subcutánea y el uso de la terapia génica como alternativas terapéuticas más efectivas y definitivas(20,21).
Es muy importante en los pacientes hemofílicos, potenciar el ejercicio físico y evitar el sobrepeso, mediante la realización de deportes de intensidad moderada, con el objetivo de fortalecer las articulaciones y mantener un buen tono muscular que ayude a evitar las hemartrosis. Los fármacos AINES están contraindicados, por lo que se usarán paracetamol o metamizol. Se administran las vacunas habituales, siendo preferente la vía subcutánea profunda en lugar de la vía intramuscular, aunque a día de hoy, aún hay autores que la aconsejan, sobre todo, en pacientes en profilaxis, para hacerla coincidir con la administración de una dosis del factor(21).
La profilaxis de los pacientes hemofílicos graves consiste en la administración, a través de una vía central de acceso venoso, de factor recombinante hasta 3-4 veces/semana (para el factor VIII) y 2-3 veces/semana (para el factor IX), convirtiéndolos así en hemofílicos moderados, con la consecuente reducción significativa en el número de episodios hemorrágicos clínicamente relevantes, demostrando su coste-eficacia y evitando el desarrollo de artropatía hemofílica(21,22). Como principal inconveniente, se aprecia el desarrollo de inhibidores en forma de anticuerpos específicos frente a los factores VIII o IX, que puedan hacer ineficaz la administración de los mismos, en un 20-30% de pacientes hemofílicos graves tipo A y en un 5% de los de la hemofilia B(19,22). Para la hemofilia A, se ha desarrollado el emicizumab (HEMLIBRA®), de administración subcutánea. Es un anticuerpo monoclonal biespecífico humanizado que ha sido desarrollado mediante tecnología de ADN recombinante. Se une a los factores IXa y X, formando el complejo necesario en la cascada de la coagulación para la hemostasia eficaz, imitando en parte la función del factor VIII y no induce ni aumenta el desarrollo de inhibidores contra el factor. Ha sido autorizado para profilaxis de episodios hemorrágicos en pacientes con hemofilia A, que presentan inhibidores del factor VIII en todos los grupos de edad(21-23).
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.** Álvarez Guisasola FJ, González H. Exploración del funcionalismo hemostático. Cap. 360. En: Moro M, Málaga S, Madero L, editores. Tratado de Pediatría (M Cruz) 11ª Ed. Panamericana; 2014. p. 1866-70.
2. Mann KG, Brummel-Ziedins K. Blood Coagulation. Cap 26. En: Orkin SH, Nathan DG, et al, editors. Nathan and Oski´s Hematology of Infancy and Childhood. 7th ed. Philadelphia: Saunders Elsevier; 2009; p. 1399-424.
3.** Scott JP, Raffini LJ, Montgomery RR, Flood VH. Hemorrhagic and thrombotic diseases. En: Kliegman RM, Stanton BF, Gemme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics, 20 ed. Elsevier; 2016. p. 2379-408.
4.** Cervera Bravo A, Álvarez Román MT. Fisiopatología y trastornos de la coagulación hereditarios más frecuentes. Pediatr Integral. 2016; XX(5): 318-30.
5.*** Kalot MA, Al-Khatib M, Connell NT, Flood V, Brignardello-Petersen R, James P, et al. An international survey to inform priorities for new guidelines on von Willebrand disease. Haemophilia. 2020; 26: 106-16.
6. Batlle J, Pérez-Rodríguez A, Costa Pinto J, Lourés Fraga E, Rodríguez Trillo A, López-Fernández MF. Avances en el síndrome de von Willebrand o enfermedad de von Willebrand adquirida: aspectos más novedosos. Hematológica/edición española. 2011; 96: 35-42.
7. Bowman M, Hopman WM, Rapson D, Lillicrap D, James P. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost. 2010; 8: 213-6.
8.*** Baronciani L, Goodeve A, Peyvandi F. Diagnóstico molecular de la enfermedad de von Willebrand. Haemophilia. 2017; 23: 188-97.
9.*** Ng CJ, Di Paola J. von Willebrand Disease: Diagnostic Strategies and Treatment Options. Pediatr Clin North Am. 2018; 65: 527-41.
10. Sharma R, Flood VH. Advances in the diagnosis and treatment of von Willebrand disease. Blood. 2017; 130: 2386-91.
11.*** Leebeek FW, Eikenboom JC. Von Willebrand’s Disease. N Engl J Med. 2016; 375: 2067-80.
12. Favaloro EJ. Toward a new paradigm for the identification and functional characterization of von Willebrand disease. Semin Thromb Hemost. 2009; 35: 60-75.
13. Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008; 14: 171-232.
14. Bowman M, Riddel J, Rand ML, Tosetto A, Silva M, James PD. Evaluation of the diagnostic utility for von Willebrand disease of a pediatric bleeding questionnaire. J Thromb Haemost. 2009; 7: 1418-21.
15.** Mannucci PM. New therapies for von Willebrand disease. Hematology Am Soc Hematol Educ Program; 2019. p. 590-5.
16.** Heijdra JM, Cnossen MH, Leebeek FWG. Current and Emerging Options for the Management of Inherited von Willebrand Disease. Drugs. 2017; 77: 1531-47.
17. Franchini M, Mannucci PM. Von Willebrand factor (Vonvendi®): the first recombinant product licensed for the treatment of von Willebrand disease. Expert Rev Hematol. 2016; 9: 825-30.
18. Iorio A, Stonebraker JS, Chambost H, Makris M, Coffin D, Herr C, et al. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males: A Meta-analytic Approach Using National Registries. Ann Intern Med. 2019; 171: 540-6.
19.** Keith Hoots W, Shapiro AD, Heiman M. Genetics of hemophilia A and B. UpToDate. Versión: marzo de 2021. Actualizado el 16 de octubre de 2019. Consultado el 22 de marzo de 2021. Disponible en: www.uptodate.com.
20. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014; 12: 1935-9.
21. Hoots WK, Shapiro AD. Treatment of bleeding and perioperative management in hemophilia A and B. UpToDate. Versión: marzo de 2021. Actualizado el 10 de marzo de 2021. Consultado el 22 de marzo de 2021. Disponible en: www.uptodate.com.
22.** Hoots WK, Shapiro AD. Hemophilia A and B: Routine management including prophylaxis. UpToDate. Versión: marzo de 2021. Actualizado el 23 de septiembre de 2020. Consultado el 22 de marzo de 2021. Disponible en: www.uptodate.com.
23. Muto A, Yoshihashi K, Takeda M, Kitazawa T, Soeda T, Igawa T, et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014; 12: 206.
Bibliografía recomendada
– Baronciani L, Goodeve A, Peyvandi F. Diagnóstico molecular de la enfermedad de von Willebrand. Haemophilia. 2017; 23: 188-97.
Este artículo aporta una descripción muy detallada de la estructura molecular y funciones fisiológicas del factor von Willebrand. Además, aborda cómo llegar al diagnóstico de los subtipos de la enfermedad, bien a través de análisis especiales o mediante técnicas moleculares.
– Ng CJ, Di Paola J. von Willebrand Disease: Diagnostic Strategies and Treatment Options. Pediatr Clin North Am. 2018; 65: 527-41.
Artículo importante y didáctico para entender los distintos subtipos de enfermedad de von Willebrand y su fisiopatología, así como para su abordaje terapéutico.
– Leebeek FW, Eikenboom JC. Von Willebrand’s Disease. N Engl J Med. 2016; 375: 2067-80.
Magnífica revisión de la enfermedad de von Willebrand, con excelentes ilustraciones que ayudan a comprender la fisiopatología de los diferentes subtipos de la enfermedad.
| Caso clínico |
|
Niña de 8 años que acude derivada por su pediatra por epistaxis de repetición bilaterales desde hace un año, sin predominio estacional y alargamiento del APTT (tiempo de tromboplastina parcial activada). No refieren otros sangrados en mucosas, pero sí equimosis en zonas expuestas a traumatismos (pretibiales). Antecedentes personales Embarazo refieren normal y controlado. Parto vaginal eutócico a término. No cirugías ni seguimientos por otros especialistas. Antecedentes familiares Madre: 38 años, refieren embolia pulmonar a los 35 años que recibió tratamiento con acenocumarol durante un año, con estudio de trombofilia negativo. Padre: 37 años. Refieren antecedentes de epistaxis de repetición desde la infancia en padre, tía y en abuelo de la rama paterna, sin estudios adicionales. Exploración física Equimosis pretibiales bilaterales, menores de 1 cm, no a otros niveles. Rinoscopia: zona de narina derecha con ingurgitación de vasos, sin sangrado activo. Resto de exploración por aparatos normal. Pruebas complementarias Hemograma: Hb: 12,9 g/dL; VCM: 83,1 fl; HCM: 27,5 pg; Leucocitos: 8.200/mm3 (56% neutrófilos); Plaquetas: 368.000/mm3. Coagulación: TP, INR y TT: normales. TTPA: prolongado (42,2 segundos). Se solicita determinación de factores VIII, IX, XI y XII que resultaron normales. Se determina nuevamente TTPA con resultado de 34,6 segundos, normal para su edad.
|

 Anemia. Classification and diagnosis
Anemia. Classification and diagnosis