 |
| Temas de FC |
C. Jiménez Cobo, E. Sebastián Pérez, J. Sevilla Navarro
Unidad de Hemato-Oncología Pediátrica del Hospital Infantil Universitario Niño Jesús. Madrid
| Resumen
Las hemoglobinopatías constituyen un grupo de enfermedades hereditarias autosómicas recesivas ocasionadas por alteraciones en la cadena de globina. Se dividen en dos grandes grupos: alteraciones cuantitativas o síndromes talasémicos, caracterizados por la disminución de síntesis de globina, y alteraciones cualitativas o hemoglobinopatías estructurales (Hb S, Hb C y Hb E), en las que se produce un cambio de aminoácido en la estructura de la globina. Dentro de este último grupo destaca, por su elevada frecuencia, la drepanocitosis o enfermedad de células falciformes. La presentación clínica es muy variada dependiendo del mecanismo fisiopatológico de la enfermedad, siendo la anemia hemolítica crónica la manifestación más característica. El diagnóstico se establecerá por medio de técnicas de electroforesis o mediante el estudio de alteraciones moleculares. Aunque la transfusión tiene un papel central en el tratamiento de los casos más graves, no está exenta de complicaciones. En la actualidad, el trasplante de médula ósea es la única terapia potencialmente curativa. En los últimos años, el conocimiento de los mecanismos fisiopatológicos ha permitido la aparición de nuevas alternativas terapéuticas como la terapia génica. La posibilidad de un diagnóstico precoz facilita un control estrecho de los pacientes desde el inicio y la intervención precoz ante la aparición de complicaciones. |
| Abstract
Hemoglobinopathies are a group of autosomal recessive disorders involving the globin chains. They can be divided into two main groups: quantitative disorders or thalassemic syndromes, which result in defective synthesis of the alpha or beta globin chain, and qualitative Hb variants or structural hemoglobinopathies (Hb S, Hb C, and Hb E), in which there is an amino acid change in the globin structure. Within this last group, sickle cell disease stands out due to its high frequency. There is a wide range of clinical sings depending on the pathophysiological mechanism of the disease, being chronic hemolytic anemia the most relevant. To establish a correct diagnosis, electrophoresis techniques or molecular studies may be necessary. Although blood transfusions play a central role in the treatment of the most severe cases, they are not exempt from complications. Currently, bone marrow transplantation is the only potentially curative therapy. In the last few years, understanding of the pathophysiological mechanisms has allowed the discovery of new therapeutic alternatives such as gene therapy. Early diagnosis contributes to adequate patient follow-up and early intervention in case of complications. |
Palabras clave: Hemoglobinopatías; Anemia de células falciformes; Talasemia.
Key words: Hemoglobinopathies; Sickle cell anemia; Thalassemia.
Pediatr Integral 2021; XXV (5): 241.e1 – 241.e13
Hemoglobinopatías: talasemia y drepanocitosis
Introducción
Las hemoglobinopatías constituyen un grupo de enfermedades hereditarias autosómicas recesivas ocasionadas por alteraciones cuantitativas o cualitativas de la cadena de globina(1).
La hemoglobina es una hemoproteína tetramérica transportadora de oxígeno, que consta de cuatro subunidades o cadenas de globina iguales dos a dos (Fig. 1).
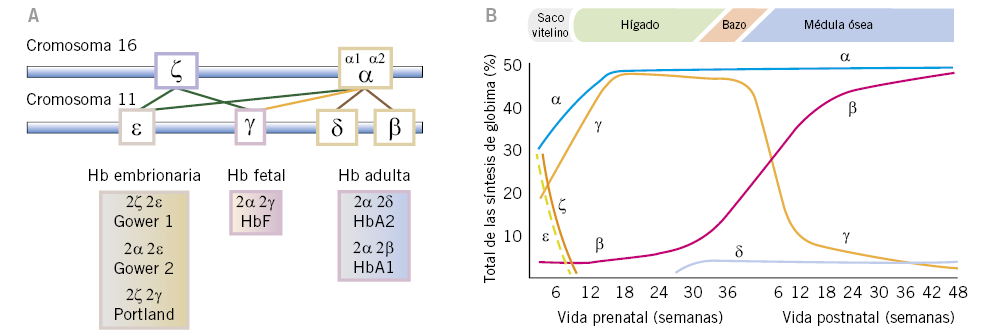
Figura 1. A. Representación de los genes que codifican las cadenas de globina. Tomada de: González García H. Anemias hemolíticas en la infancia. Pediatr Integral. 2012; XVI(5): 378-86. B. Evolución de la expresión de las distintas cadenas de hemoglobina normales a lo largo de la vida prenatal y postnatal. Tomada de: González Fernández FA, Ropero Gradilla P. Síndromes talasémicos. En: Oncología y Hematología Pediátricas, 3ª ed.
Cada cadena posee un grupo hemo con un átomo de hierro central en forma ferrosa, a través del cual cada subunidad es capaz de fijar una molécula de oxígeno. Existen seis tipos de cadenas de globina humana: alfa (α), beta (β), gamma (γ), delta (δ), épsilon (ε) y zeta (ζ). Las cadenas α y ζ están formadas por 141 aminoácidos y se denominan cadenas tipo α. Las cadenas β, γ, δ y ε son ligeramente más largas, están constituidas por 146 aminoácidos y se denominan tipo β o no α. De las combinaciones dos a dos de las diferentes cadenas de globina tipo α y tipo β se van a formar las diferentes hemoglobinas, en los períodos embrionario, fetal, neonatal y adulto. En el cromosoma 16, se encuentran los genes que codifican las cadenas α (α1 y α2) y las cadenas ζ. Las cadenas δ y β y sus variantes embrionarias ε y γ, se codifican en el cromosoma 11. Dichos genes se irán expresando durante el desarrollo, produciendo los diferentes tetrámeros de hemoglobina(2). Durante el primer mes de gestación, se formarán las hemoglobinas embrionarias (ζ2ε2, α2ε2 y ζ2γ2), las cuáles se irán sustituyendo a lo largo del embarazo por la hemoglobina fetal HbF (α2γ2), siendo esta la predominante desde las 8 semanas de vida embrionaria hasta el segundo mes de vida postnatal. El cambio de expresión de γ-globina a β-globina comienza durante el embarazo y se completa entre los seis meses y el año de vida; por lo que los neonatos con β talasemia no presentarán clínica al nacimiento, estableciéndose el diagnóstico a partir de los 6 meses. En el hematíe adulto, el 97% de la hemoglobina adulta será HbA (α2β2), siendo una pequeña proporción HbA2 (α2 δ2) (2%) y HbF (1%)(1,2).
Las hemoglobinopatías pueden clasificarse en dos grandes grupos: alteraciones cuantitativas o síndromes talasémicos, caracterizados por la disminución de síntesis de globina, y las alteraciones cualitativas o hemoglobinopatías estructurales, en las que se producen cadenas de globina anormales (hemoglobinas anómalas), por sustitución de uno o más aminoácidos. Dentro de este grupo destaca, por su elevada frecuencia, la drepanocitosis o enfermedad de células falciformes(3).
Las hemoglobinopatías son trastornos hereditarios frecuentes. Cerca del 7% de la población mundial es portadora de una hemoglobinopatía, estimándose entre 300.000 y 500.000 nacimientos al año de recién nacidos con un trastorno hereditario de la hemoglobina, siendo los más frecuentes la talasemia y la anemia de células falciformes. Estas alteraciones presentan mayor prevalencia en regiones tropicales y subtropicales como Oriente próximo y el área mediterránea, zonas que han sido o son áreas endémicas de paludismo, lo cual puede estar asociado a la probabilidad de producir cierta protección frente a dicha enfermedad. Por otro lado, no sólo la distribución de la malaria determina la prevalencia de las hemoglobinopatías en un área geográfica, existen además otros factores como la etnia, la consanguinidad y aislamiento geográfico. En la actualidad, se ha visto una generalización mundial de las hemoglobinopatías, debido al aumento de los movimientos migratorios(1), convirtiéndose en un problema de salud global emergente con un número cada vez mayor de casos en regiones originariamente no endémicas.
Talasemias(3,4)
Las talasemias (ver algoritmo al final del artículo) son un grupo heterogéneo de anemias hereditarias, ocasionadas por la disminución de la síntesis de una o varias cadenas de globina, debido a mutaciones en los genes que codifican dichas cadenas. Hablamos, por tanto, de una alteración cuantitativa(3).
Se han descrito una gran variedad de fenotipos que reciben su nombre en función de la cadena afecta, destacando por su frecuencia la α y β talasemia. Según el grado de síntesis de dichas cadenas podremos hablar α0 y β0 cuando no hay síntesis de la cadena afecta y de α+ y β+ cuando existe, pero en menor cantidad(4).
Fisiopatología(2,3,5)
La repercusión fisiopatológica vendrá determinada principalmente por la gravedad de la anemia y el grado de desequilibrio entre las cadenas de α y β-globina (Fig. 2).

Figura 2. Fisiopatología de la talasemia. Adaptado de: Taher AT, et al. Lancet 2018. MO: médula ósea.
La cadena producida en cantidad normal, al no poder unirse con la cadena deficitaria formará tetrámeros de cadenas libres que precipitarán en el interior celular. La interacción de estos con la membrana producirá su desestructuración, ocasionando una muerte celular precoz en la médula ósea (eritropoyesis ineficaz, dato característico de la talasemia) o en sangre periférica, ya que favorece el aclaramiento del eritrocito maduro en el sistema mononuclear fagocítico (hemólisis periférica). En el caso de la β-talasemia, las cadenas α en exceso son muy inestables, por lo que precipitarán en los precursores eritroides de la médula ósea, condicionando un importante componente de eritropoyesis ineficaz. En cambio, en la α-talasemia, las cadenas β libres formarán tetrámeros más estables que precipitarán más tardíamente en el hematíe maduro, predominando el componente de hemólisis en sangre periférica.
Al disminuir la síntesis de las cadenas de globina, se produce una hemoglobinación defectuosa, lo que condiciona la existencia de microcitosis (disminución del volumen corpuscular medio, VCM) e hipocromía (disminución de la concentración de hemoglobina corpuscular media, CHCM).
Todo ello llevará a una anemia hemolítica crónica que estimulará el aumento de progenitores eritroides en médula ósea, provocando alteraciones esqueléticas y osteoporosis. Por otro lado, la expansión de la función medular a otros tejidos (hemopoyesis extramedular) será responsable de la hepatoesplenomegalia y formación de masas paravertebrales de eritropoyesis. Además, la anemia, la hipoxia y la expansión medular llevan a una disminución de la hepcidina, que ocasiona una sobrecarga férrica por aumento de la absorción intestinal de hierro, que se verá agravada por la necesidad de terapia transfusional en estos pacientes.
Clínica(3,4,6,7)
α-talasemia
Dado que las cadenas alfa son necesarias para la eritropoyesis fetal y la producción de HbF, las alfa talasemias pueden ser sintomáticas intraútero. La presencia de cuatro genes α, dos de cada progenitor, condicionan las combinaciones genotípicas que se correlacionan con fenotipos clínicos específicos, dependiendo de la capacidad de síntesis de cadenas(1) (Tabla I).
La alteración en un solo gen α (α+ talasemia heterocigótica) se expresará en forma de talasemia silente, sin repercusión clínica, con valores de hemoglobina corpuscular media (HCM) y volumen corpuscular medio (VCM) dentro del límite bajo de la normalidad. La importancia de su diagnóstico radica en el consejo genético.
La pérdida de dos genes α cada uno en un alelo (α+ talasemia homocigótica) o en el mismo (α0 talasemia heterocigótica) dará lugar al rasgo talasémico, con niveles de hemoglobina en el límite inferior de la normalidad o anemia microcítica e hipocrómica leve.
La pérdida de tres genes α corresponderá a la Hemoglobina H (HbH) o talasemia intermedia. El exceso de cadenas β de globina precipita y forma una hemoglobina anormal característica, la Hb H, un tetrámero de β globina (β4). Se caracteriza por anemia hemolítica crónica, con gran variabilidad clínica y genética, pudiendo presentar desde una anemia leve a moderada, a precisar de transfusiones esporádicas en situaciones de estrés agudo o con caída en la concentración de hemoglobina (crisis hemolíticas por infecciones, embarazo o cirugía). En las formas graves, la clínica es superponible a la de la talasemia mayor (TM).
Por último, la pérdida de los cuatro genes α implicará la formación de Hb Bart o hydrops fetalis. Esta última constituye la forma más severa de α-talasemia, el cuadro clínico será similar a la hidropesía fetal, ocasionando en la mayoría de casos muertes intraútero (23-28 semanas) o poco después del nacimiento.
β-talasemia
Desde un punto de vista genético, en las β-talasemias se recibirá un gen β de cada progenitor, que se expresarán fenotípicamente en diferentes cuadros clínicos (Tabla I). La talasemia mayor es la forma más severa de presentación. Se caracteriza por anemia grave transfusión-dependiente. Los primeros síntomas serán secundarios a la hemólisis crónica, presentando: retraso en el crecimiento, palidez cutánea, ictericia y colelitiasis. Debido a la proliferación de progenitores eritroides, en médula ósea irán apareciendo alteraciones esqueléticas, sobre todo, en los casos más avanzados que no han recibido tratamiento, como osteopenia y osteoporosis, y deformidades óseas. Los puntos de hematopoyesis extramedular serán principalmente el bazo (esplenomegalia) y el hígado (hepatomegalia), pero se pueden activar en otros tejidos, generando masas paravertebrales de eritropoyesis. En aquellos pacientes en los que se inicia un programa de transfusión periódica precoz, el riesgo de complicaciones vendrá determinado por la sobrecarga de hierro postransfusional, pudiendo generar un daño orgánico a nivel cardiaco, hepático y endocrinológico.
Diagnóstico(5,8-11)
El diagnóstico de sospecha de la talasemia se basará en la clínica, la exploración física, los datos hematimétricos y la morfología del frotis de sangre periférica. Es importante realizar una anamnesis completa.
La microcitosis y la hipocromía constituyen un marcador diagnóstico de la enfermedad y se correlacionan con el grado de disminución de síntesis de la cadena afecta. La anemia microcítica es la alteración hematológica más común, por lo que ante la presencia de ésta en un hemograma, se deberá realizar un diagnóstico diferencial amplio que incluya: la anemia ferropénica, la anemia de trastornos crónicos y la anemia sideroblástica (Tabla II).
Puede resultar de utilidad la aplicación de índices o fórmulas para diferenciar entre talasemias y anemia ferropénica, como el índice de Mentzer (VCM/recuento de hematíes), en el que valores de menos de 13 apoyarán el diagnóstico de talasemia.
La presunción o alta probabilidad diagnóstica de talasemia la obtendremos mediante la electroforesis de hemoglobinas o cromatografía en el caso de la β-talasemia. Así, en la β-talasemia, el déficit de síntesis de cadena se verá reflejado por una disminución de HbA con elevación de niveles de HbA2 y/o HbF. Aunque, en los casos donde la mutación determina una disminución muy leve de la síntesis de cadena β, puede ser normal. De forma similar, en otras situaciones como la asociación de delta talasemias o variantes de la cadena delta o ferropenia severa entre otras, la HbA2 puede no estar aumentada. Por tanto, una HbA2 normal no excluye el diagnóstico de β talasemia(3). En estos supuestos, habrá que recurrir a otras técnicas moleculares para su identificación. En la α-talasemia, el resultado obtenido mediante electroforesis será normal, ya que el déficit de cadena se verá reflejado en una disminución proporcional de síntesis, tanto de HbA como de HbA2 y HbF. Por tanto, habrá que recurrir a estudios moleculares para su diagnóstico. Los pacientes con HbH sí se diagnostican en la electroforesis, ya que este tipo de Hb se demuestra en ella(9).
La confirmación diagnóstica la obtendremos mediante la identificación de la alteración genética a nivel molecular. Estas alteraciones pueden ser mutaciones puntuales (inserciones o sustituciones), que son las predominantes en las β talasemias; o mutaciones extensas (duplicaciones, deleciones y reagrupamientos), responsables de la mayoría de las β talasemias; (siendo la deleción de uno o varios genes β las más frecuentes). El estudio molecular no solo nos confirmará la presencia de una forma severa (talasemia mayor o talasemia intermedia), sino que nos va a permitir detectar a los pacientes en fase presintomática, planificar el manejo de las futuras complicaciones y realizar un consejo genético adecuado. De esta forma, se han desarrollado programas de cribado neonatal y diagnóstico prenatal en las primeras semanas de la gestación. Las técnicas más comunes para obtener material fetal son la biopsia de vellosidad corial y la amniocentesis(3).
Tratamiento(5,12-15)
El tratamiento de las formas sintomáticas incluye la corrección de la anemia, así como el de las diferentes complicaciones.
Las formas asintomáticas no necesitarán ningún tipo de tratamiento. Se ha recomendado un suplemento diario con 1 mg de ácido fólico en talasemia no dependiente de transfusión, aunque el aporte diario de la dieta suele ser suficiente como prevención de la anemia megaloblástica(3).
La transfusión tiene un papel central en el tratamiento de la talasemia. Sin un soporte transfusional continuo, muchos pacientes con talasemia mayor morirían por causa de la anemia.
El soporte transfusional mejora el desarrollo y crecimiento de los pacientes e inhibe la absorción intestinal de hierro. Además, la transfusión revierte las complicaciones derivadas de la hematopoyesis ineficaz (osteopenia, deformidades esqueléticas o focos de hematopoyesis extramedular) y de la hemolisis crónica (esplenomegalia, hipercoagulabilidad, hipertensión pulmonar e insuficiencia cardiaca), controlando así los mecanismos fisiopatológicos responsables del desarrollo y complicaciones(5).
Se recomienda un programa de transfusión regular que permita alcanzar concentraciones de hemoglobina, objetivo entre 9-10,5 g/dl con un intervalo de 2-4 semanas entre transfusiones. El nivel de hemoglobina no debe ser el único indicador para iniciar terapia transfusional (salvo Hb<5 g/dl), hay que basarse en los hallazgos clínicos y analíticos. Una anemia con una Hb <7 g/dl durante más de dos semanas, con exclusión de otras causas de anemia añadidas, se considera criterio para iniciar un programa transfusional. Mientras que en pacientes con Hb >7 g/dl, se deben considerar la presencia de síntomas y signos de anemia severa (retraso en el crecimiento, deformidad ósea, esplenomegalia, pseudotumores, etc.)(3,5).
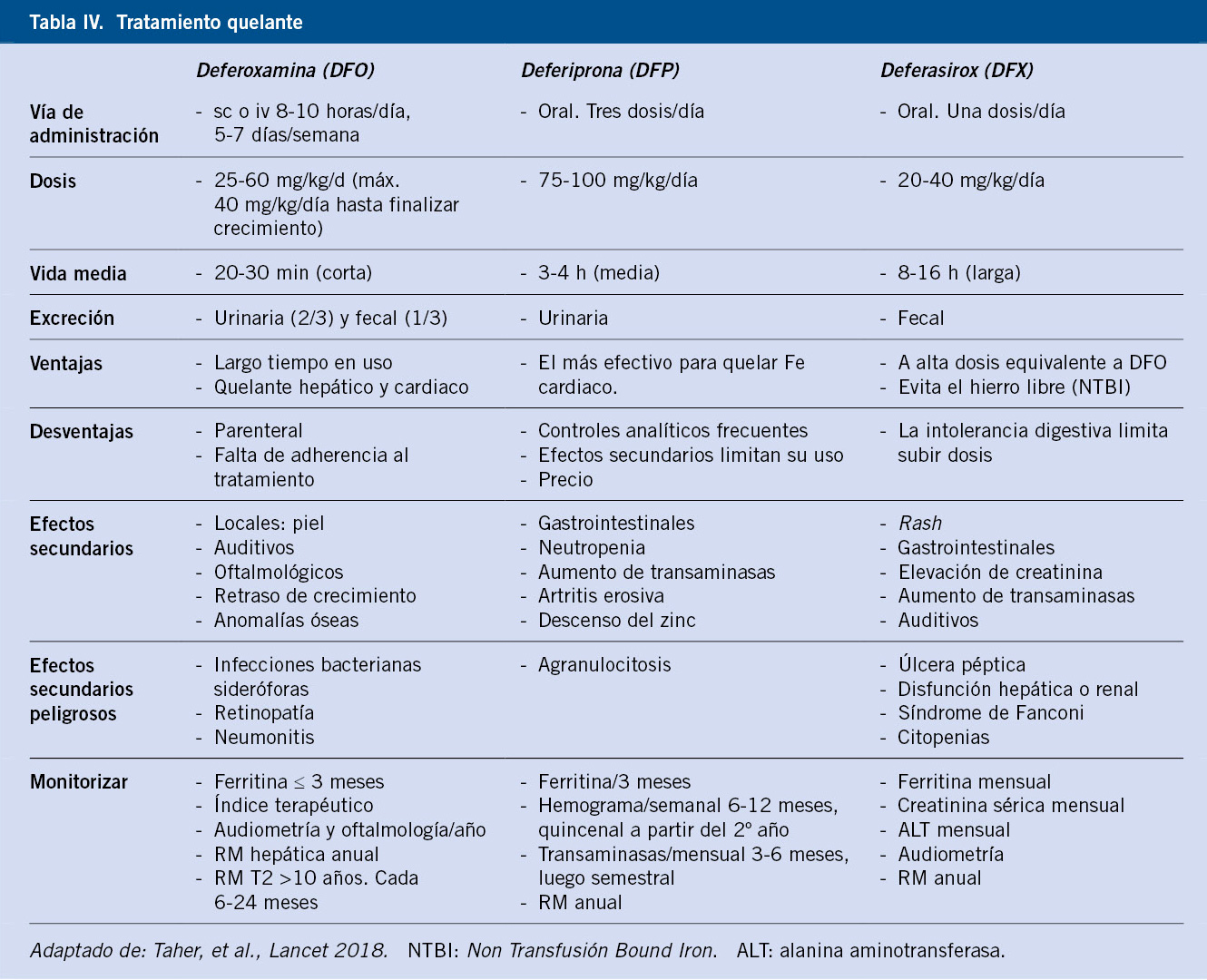
Sin embargo, la terapia transfusional asocia muchas de las complicaciones que sufren estos pacientes, como son, entre otras, el riesgo de aloinmunización y la sobrecarga férrica, la cual se ve favorecida por el aumento de la absorción intestinal de hierro. El hierro sérico no unido a transferrina se deposita en los órganos, produciendo lesiones por hemosiderosis a nivel endocrinológico, cardiaco y hepático, siendo la miocardiopatía la principal causa de muerte. La monitorización del exceso de hierro se realizará mediante los niveles de ferritina, la valoración de la concentración hepática de hierro (CHH) y la determinación del tiempo de relajación transversal T2 cardiaco en milisegundos (ms) (relacionado con la fracción de eyección) (Tabla III).
Estos umbrales, además de determinar el grado de sobrecarga férrica de un paciente (leve, moderado y grave), van a evaluar la respuesta al tratamiento quelante, permitiendo adaptar la dosis.
La ferritina es el método de valoración más utilizado, sin embargo sus niveles pueden fluctuar en respuesta a factores, como la infección o la inflamación, por lo que también se emplea el índice de saturación de transferrina. La resonancia magnética se ha establecido como gold estándar para la medición de CHH y el T2* cardiaco.
Las pautas de quelación se deben iniciar cuando la ferritina es > 1.000 μg/L o la CHH superior a 3,2 mg/g. El objetivo es mantener una CHH < 7 mg/g y una ferritina <1.500 μg/L. Actualmente, disponemos de tres quelantes de hierro para el tratamiento de la sobrecarga de hierro: deferoxamina (DFO), deferiprona (DFP) y deferasirox (DFX)(12) (Tabla IV). En los pacientes con grado de sobrecarga de hierro grave, se puede realizar una quelación intensiva basada en la combinación de estos fármacos.
Aunque la esplenectomía se ha aplicado como alternativa o complemento de la terapia transfusional, este procedimiento ha quedado relegado en la actualidad, ya que conlleva un aumento del riesgo de sepsis grave e infecciones por gérmenes encapsulados, y conduce a un estado de hipercoagulabilidad y, por tanto, a mayor riesgo de fenómenos tromboembólicos. Las recomendaciones actuales para la esplenectomía están restringidas a pacientes con(5):
• Incapacidad para recibir transfusión y terapia de quelación de hierro.
• Requerimientos de sangre de >225-250 ml/kg/año en ausencia de otros motivos como aloinmunización.
• Esplenomegalia clínicamente sintomática o hiperesplenismo.
Debido al riesgo de infección, se debe vacunar a los pacientes esplenectomizados, frente a bacterias encapsuladas (neumococo, meningococo y Haemophilus), al menos, dos semanas antes de la intervención. La esplenectomía se deber retrasar, si es posible, hasta los 5 años o más y, si se realiza antes, se recomienda profilaxis con penicilina V oral diaria (125 mg en <2 años y 250 mg en >2 años dos veces al día). En pacientes mayores de 5 años, se mantendrá, al menos, hasta dos años después de la cirugía.
El trasplante alogénico de progenitores hematopoyéticos es el único tratamiento curativo de la enfermedad(5). La fuente de progenitores hematopoyéticos de elección es la médula ósea de hermano HLA-idéntico. El resultado dependerá de la situación clínica previa del paciente. En pacientes con mayor número de transfusiones, aumenta el riesgo de aloinmunización, así como la probabilidad de rechazo y recurrencia de la enfermedad.
En los últimos años, gracias al conocimiento de los mecanismos fisiopatológicos de la talasemia han ido surgiendo nuevas líneas de tratamiento(13-15) centradas en mejorar:
• El desequilibrio de alfa/beta globina (inducción farmacológica de la Hb fetal, terapia génica y edición genómica).
• La eritropoyesis ineficaz (ruxolitinib, sotatercept y luspatercept).
• La disrregulación del hierro (análogos de la hepcidina, inhibidores de Tmprss6 e inhibidores de la ferroportina).
La terapia génica se basa en la corrección del defecto específico del gen de la talasemia añadiendo, mediante un vector, el gen corrector a la célula madre hematopoyética. De esa forma, se produce la sustitución de un gen enfermo por uno sano.
El aumento de la expresión de HbF en la β -talasemia reduce el desequilibrio α/β, disminuyendo la eritropoyesis ineficaz y la hemólisis, y aumentando la concentración total de hemoglobina. En las tres últimas décadas, se ha investigado el uso de fármacos, como inductores de HbF, sin embargo, estos estudios son limitados. Ente los fármacos más ampliamente estudiados, encontramos los agentes citotóxicos, como la hidroxiurea.
Complicaciones(3,5)
La mayoría de las complicaciones de los pacientes con talasemias serán consecuencia del alto grado de sobrecarga férrica.
Los pacientes con talasemia presentan mayor riesgo tromboembólico, sobre todo, en talasemia intermedia y pacientes esplenectomizados. Ante situaciones de riesgo de trombosis, hay que realizar una evaluación adecuada y valorar el inicio de anticoagulación.
Las complicaciones endocrinas son muy frecuentes en las talasemias y, entre sus causas principales, se encuentra la sobrecarga de hierro, por lo que es de relevancia asegurar un tratamiento quelante adecuado. Las principales endocrinopatías por orden de frecuencia son: hipogonadismo, talla baja, hipoparatiroidismo, hipotiroidismo, intolerancia a la glucosa/diabetes mellitus y la insuficiencia adrenal. El manejo de estas patologías se basa en intensificar la quelación, así como valorar el tratamiento hormonal sustitutivo en los casos necesarios.
La osteopenia es una de las complicaciones más frecuentes y de mayor morbimortalidad en el paciente adulto con talasemia. El diagnóstico debe basarse en la historia clínica, controles analíticos y la densitometría. Las pautas de prevención son las medidas terapéuticas más importantes. El tratamiento médico se basa en suplementos de calcio y vitamina D. En los casos de alto riesgo de fractura, se pueden emplear bifosfonatos.
La mayor causa de mortalidad en estos pacientes es la miocardiopatía ocasionada principalmente por el depósito férrico. La RMN es la prueba de elección diagnóstica. El tratamiento se basará en intensificar la terapia quelante para extraer el exceso de hierro depositado. A nivel hepático, esta sobrecarga conduce a fibrosis y cirrosis hepática, siendo la elastografía la técnica diagnóstica de elección, al ser un método indirecto. La quelación crónica revierte o estabiliza la fibrosis hepática.
Tras el fallo cardiaco, las infecciones son la segunda causa de mortalidad en pacientes con talasemia. La infección por parvovirus en estos pacientes puede ocasionar una “crisis aplásica transitoria” que precisará de una intensificación del soporte transfusional, al presentar una anemización variable y ausencia de progenitores en médula ósea. En el caso de pacientes esplenectomizados, hay que tener en cuenta el riesgo de infección por microorganismos encapsulados (S. pneumoniae, H. influenzae y N. meningitidis).
Finalmente, señalar la importancia del consejo genético (también en los portadores), así como del apoyo psicosocial de estos pacientes y sus familiares, ayudando a entender la enfermedad y aceptar las posibles complicaciones y dificultades asociadas.
Drepanocitosis
La drepanocitosis o enfermedad de células falciformes (ECF) es la hemoglobinopatía estructural más frecuente.
Las hemoglobinopatías estructurales son un gran grupo de enfermedades ocasionadas por mutaciones puntuales que conducen a la síntesis de cadenas de globina anómalas, dando lugar a variaciones de la hemoglobina (Hb S, Hb C, Hb E, Hb D). Entre ellas destaca por su frecuencia, la enfermedad de células falciformes.
La ECF constituye un trastorno hereditario de carácter autosómico recesivo, caracterizado por la presencia de hemoglobina falciforme (HbS) en los eritrocitos. Dicha hemoglobina surge por la sustitución del ácido glutámico en la posición 6 del gen β-globina por una molécula de valina(1). Este cambio permite a la HbS, polimerizarse en condiciones de baja oxigenación, generando hematíes en forma de hoz.
Distinguimos dos grandes grupos en función del número de alelos afectos. Los individuos heterocigóticos serán portadores asintomáticos con rasgo falciforme (HbAS). Por otro lado, los enfermos sintomáticos serán individuos homocigotos o doble heterocigotos, que presentan en un alelo el gen anormal de la Hb S y en el otro alelo, otro gen β– anormal por alteración estructural o talasémica (Hb SS, Hb SC, Hb S/β0 talasemia, Hb S/β+ talasemia) (Tabla V). Generalmente, los enfermos con HbSS y HbSβ0-talasemia presentan las formas más graves. La enfermedad falciforme HbSC puede tener las mismas complicaciones, pero generalmente más leves y tardías(16,17).

Fisiopatología
La hemoglobina S se caracteriza por polimerizarse en situaciones de baja oxigenación, alterando su solubilidad y, por tanto, distorsionando el glóbulo rojo, el cual adopta forma de hoz (falciformación). Esta deformidad ocasiona las denominadas crisis vasooclusivas, al impedir el paso de los hematíes por la microcirculación, ocasionando episodios intermitentes de oclusión vascular que causan isquemia tisular y disfunción orgánica aguda o crónica(16). La reoxigenación de los eritrocitos rompe el polímero de HbS, restaurando su forma habitual. Este proceso de falciformación y reoxigenación continúa hasta que la membrana del eritrocito se hace rígida generando células falciformes irreversibles, lo que favorece su destrucción generando una anemia hemolítica intravascular o eliminación extravascular por el sistema reticuloendotelial(18). Por otro lado, la saturación de la capacidad de filtro esplénico por el exceso de hematíes falciformes y los infartos esplénicos, acaban generando asplenia funcional, lo que incrementa el riesgo a infecciones por microorganismos encapsulados(1).
La polimerización de HbS, la vasooclusión y la anemia hemolítica generan una cascada de eventos patológicos que conducen a una amplia gama de complicaciones(17).
Diagnóstico
El momento diagnóstico de la enfermedad puede variar(19):
• En el caso de la existencia de antecedentes familiares, se han desarrollado programas de diagnóstico prenatal, en los que se puede llegar al diagnóstico de la enfermedad por medio de técnicas, como la biopsia de vellosidad corial y la amniocentesis(16).
• En el momento del nacimiento, el pilar diagnóstico radicará en el cribado neonatal de hemoglobinopatías, siendo la ECF la alteración más prevalente identificada(16).
• La sintomatología suele aparecer a partir de los 4-6 meses de vida, cuando disminuyen los niveles de HbF; de ahí, la importancia del cribado neonatal, ya que permite el diagnóstico precoz de la enfermedad y el inicio del tratamiento en fase presintomática, disminuyendo la morbimortalidad. Además, posibilita la educación sanitaria para detección de situaciones de alarma y complicaciones graves, ofrece consejo genético y detecta a portadores sanos, así como otras hemoglobinopatías(17). En general, las muestras para el cribado neonatal son muestras capilares de sangre seca del talón en papel de filtro. Hay que señalar que las pruebas de cribado no son pruebas diagnósticas. Será necesario realizar una prueba de confirmación mediante cromatografía o electroforesis de hemoglobina al año de vida aproximadamente, ya que en los primeros meses de vida del recién nacido, el resultado puede estar artefactado por la presencia elevada de HbF.
• Los casos de diagnóstico más tardío tienen lugar en aquellos individuos en los que no se ha realizado un cribado neonatal y que presentan un cuadro clínico sugestivo (eventos vasooclusivos, anemia hemolítica…), un hallazgo analítico casual o antecedentes familiares. El diagnóstico de sospecha vendrá dado por la presencia en el hemograma de una anemia hemolítica crónica normocítica y normocrómica(19), con valores de hemoglobina entre 6 y 9 g/dl que, aunque no es dependiente de transfusiones, puede precisarlas con una frecuencia variable en diversos tipos de crisis o complicaciones. Un VCM y HCM bajos nos pueden orientar al diagnóstico de genotipos de S/β-talasemia.
• En la bioquímica obtendremos datos que apoyen el diagnóstico de anemia hemolítica, como elevación de la bilirrubina, LDH y disminución de la haptoglobina. En el frotis de sangre periférica se evidenciará la morfología falciforme característica. El diagnóstico de confirmación se obtendrá mediante la cuantificación posterior del pico de HbS, mediante cromatografía líquida de alta resolución o electroforesis de hemoglobinas. La hemoglobina fetal en estos pacientes tiende a estar algo elevada como efecto protector frente a la hemoglobina S y tiene significado pronóstico.
• Los estudios moleculares se reservarán para aquellos casos dudosos en los que no se llegue a un diagnóstico, siendo raramente empleados como pruebas de primer escalón(16).
Tratamiento
La posibilidad de realizar un diagnóstico precoz permite la intervención prematura mediante la instauración del tratamiento basal consistente en(16):
Penicilina V
La profilaxis infecciosa se realiza mediante la administración de penicilina V. Se debe de iniciar a partir de los 2 meses de vida y mantenerse, al menos, hasta los 5 años. En el caso de esplenectomía, TPH (trasplante de progenitores hematopoyéticos) o enfermedad neumocócica no se debe suspender.
Vitamina D y ácido fólico
Se aconseja la administración de vitamina D (800 UI/día), realizando controles de 25-hidroxivitamina D cada 6-12 meses, ajustando tratamiento según niveles. La administración sistemática de ácido fólico no es necesaria si se consiguen los aportes suficientes mediante una dieta rica y variada.
Hidroxiurea
La hidroxiurea (HU), aunque su mecanismo de acción no se conoce totalmente, es un citostático inhibidor de la ribonucleótido reductasa, que actúa como agente estimulador de la producción de HbF, debido a que promueve una eritropoyesis más inmadura y disminuye la polimerización de la HbS. Esto se refleja en el hemograma con un aumento de la hemoglobina y el VCM, junto con una disminución de los reticulocitos(20). Además, disminuye los neutrófilos y las plaquetas, disminuyendo su interacción al endotelio y la adhesión de los hematíes al mismo y aumenta el óxido nítrico que es un potente vasodilatador. Su uso está recomendado en cualquier paciente con ECF SS o Sβ0-talasemia desde los 9 meses de edad, ya que se ha visto un aumento de los efectos clínicos beneficiosos durante el tratamiento, como son la reducción de las crisis vasooclusivas, el síndrome torácico agudo y las transfusiones. Asimismo, podría ser una alternativa a la transfusión crónica en la profilaxis primaria del ACVA en pacientes con eco-doppler transcraneal (EcoDTC) patológica, si esta se ha normalizado, y manteniendo controles trimestrales(19). Con respecto a los efectos secundarios, la trombocitopenia y leucopenia reversibles suelen ser los más frecuentes, por lo que es necesario un control clínico analítico estrecho durante el tratamiento. Se debe de informar sobre la posibilidad de esterilidad secundaria a la administración prolongada de HU en varones(16), así como ofrecer la posibilidad de criopreservación de semen en los pacientes con edad adecuada. Debido al riesgo de teratogenicidad, es necesario el empleo de métodos anticonceptivos durante el tratamiento y se debe interrumpir, al menos, 3 meses antes de la concepción. La dosis inicial recomendada es de 20 mg/kg/día vía oral, incrementando 5 mg/kg/d cada 8 semanas en >3-5 años, hasta dosis máxima (30-35 mg/kg/día) o toxicidad(16). La respuesta clínica puede aparecer a los 3-6 meses y se debe mantener el tratamiento de forma indefinida.
Terapia transfusional
Las indicaciones de transfusiones están asociadas a corregir el grado de anemia, así como a tratar o prevenir las complicaciones agudas o crónicas de la enfermedad, disminuyendo el porcentaje de HbS(16). Antes de la primera transfusión, se debe realizar fenotipo eritrocitario ampliado, junto con determinación del grupo sanguíneo. Se seleccionarán unidades que sean, al menos, compatibles para ABO, Rh (D, C, c, e) y Kell, para disminuir riesgo de aloinmunización. Además, se recomienda respectar la compatibilidad para los sistemas Kidd, Duffy y MNS.
Como ya hemos visto con anterioridad, las transfusiones no están exentas de complicaciones. En estos pacientes debemos tener en cuenta el riesgo de(21):
• Hiperviscosidad sanguínea: las transfusiones aumentan de forma exponencial la viscosidad sanguínea, aumentando el riesgo de desencadenar crisis vasooclusivas. No se debe sobrepasar el límite de 10-11 g/dL de Hb (o Htc 30-33%) salvo HbS <50%. Hay que tener en cuenta que la anemia crónica en estos pacientes es muy bien tolerada, por lo que la transfusión en pacientes con anemia asintomática está contraindicada salvo compromiso.
• Aloinmunización: más frecuente debido a la disparidad antigénica racial, algunas de las cuales no son detectadas con estudios serológicos, pudiendo ocasionar reacciones transfusionales hemolíticas tardías. En estos casos, está indicado el estudio del genotipo eritrocitario, para detectarlas y poder administrar sangre adecuada.
• Síndrome hiperhemolítico (hiperhemólisis postransfusional).
• Infecciones: la transmisión de agentes infecciosos ha disminuido en la actualidad, gracias a la selección de donantes y los estudios serológicos.
• Sobrecarga férrica.
Se distinguen varios tipos de pautas transfusionales(16,19):
• Transfusión simple: indicada en el tratamiento de anemia sintomática (secuestro esplénico, crisis eritroblastopénicas, cirugías). Los inconvenientes son la hiperviscosidad y la hipervolemia.
• Exanguinotransfusión: consiste en el recambio de los hematíes del paciente por hematíes normales, se puede realizar de forma manual o automatizada. Permite alcanzar la reducción de Hb S de forma más rápida y eficaz, sin aumentar el volumen ni el hematocrito. Indicada en crisis graves, como: accidentes cerebrovasculares, síndrome torácico agudo con mala evolución, crisis de dolor refractarias o priapismo. La exanguinotransfusión manual suele requerir más tiempo y se recambia la sangre total del paciente (hematíes y plasma); la automática es más rápida y suele conseguir menores HbS finales, por lo que se podrán espaciar visitas en los tratamientos crónicos. En la automática, se realiza solo recambio de hematíes (eritrocitaféresis). La indicación entre una u otra dependerá de la disponibilidad de personal y máquina. Hay que tener en cuenta que si la cifra de Hb de partida es menor de 7 g/dl, se debe realizar antes una transfusión simple.
• Régimen hipertransfusional: administrar hematíes de forma crónica, generalmente mensuales, con el objetivo de mantener una HbS baja (≤30%). Indicadas en accidentes cerebrovasculares, úlceras crónica y síndrome torácico agudo de repetición. Si se instaura un régimen hipertransfusional, se requerirá tratamiento quelante del hierro. En un programa de transfusión crónica, la exanguinotransfusión previene la sobrecarga férrica y mejora la calidad de vida por el número de visitas. Los inconvenientes de la exanguinotransfusión en este caso, serían un mayor coste y mayor número de unidades a las que se expone el paciente.
No está indicada la transfusión en determinadas situaciones como son: crisis vasooclusiva no complicada, anemia asintomática y la insuficiencia renal aguda que no se acompaña de fallo multiorgánico(16).
Trasplante de progenitores hematopoyéticos y terapia génica
Al igual que en las talasemias, el trasplante de progenitores hematopoyéticos es el único tratamiento potencialmente curativo. La fuente del donante preferiblemente será un hermano HLA idéntico. El trasplante estará indicado como recomendación general en pacientes menores de 17 años (preferiblemente antes de los 6 años) con ECF (HbSS, Sβ0 talasemia), sintomáticos y que dispongan de hermano HLA idéntico.
Con respecto a la aparición de nuevos tratamientos, la terapia génica sigue siendo la más prometedora. Hay algunos fármacos en estudio con diferentes mecanismos de acción, que se están utilizando en ensayos clínicos.
Complicaciones
La clínica de estos pacientes vendrá derivada de la anemia hemolítica crónica, la isquemia tisular y la susceptibilidad a las infecciones. Según los síntomas predominantes se pueden dividir en dos fenotipos clásicos:
1. Fenotipo hemólisis-disfunción endotelial: presentan niveles más bajos de hemoglobina y las manifestaciones están relacionadas con la vasculopatía (úlceras, priapismo, enfermedad cerebrovascular, hipertensión pulmonar y retinopatía).
2. Fenotipo vasooclusivo: presentan niveles más elevados de hemoglobina y eso produce más falciformación y, por tanto, mayor adhesión de los hematíes, causando daño órgano isquémico progresivo (crisis dolorosas vasooclusivas, síndrome torácico agudo, necrosis avascular, hipoesplenismo).
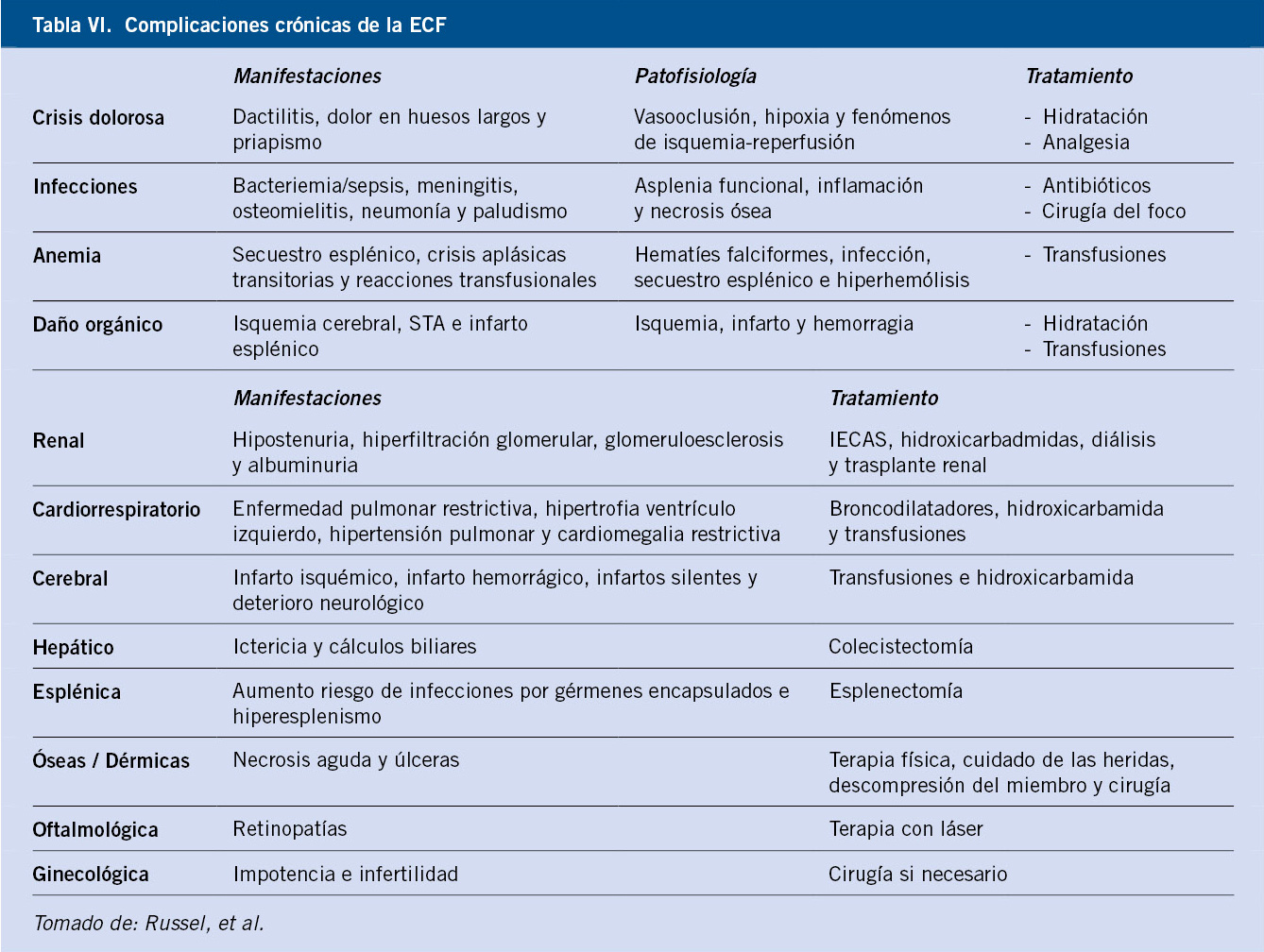
Todo ello, dará lugar a una serie de complicaciones que, según el momento de aparición, las podemos dividir en agudas o crónicas.
Complicaciones agudas
Crisis dolorosas o vasooclusivas(16,19): es la manifestación clínica más frecuente, producida por la oclusión de la microcirculación vascular en cualquier localización, provocando dolor e inflamación. Las más características son las crisis dolorosas óseas, ocasionadas por isquemia medular ósea. Pueden afectar a cualquier hueso, siendo más frecuentes en los huesos largos; aunque en lactantes, es típica la afectación de las falanges de manos y/o pies (dactilitis del lactante). Cursan con: dolor intenso, tumefacción, impotencia funcional y febrícula, pudiendo llegar a presentar repercusión analítica con leucocitosis y elevación de reactantes de fase aguda, lo cual dificulta el diagnóstico diferencial con la osetomielitis o la artritis séptica. El tratamiento se basa en analgesia, siendo posible el control de las crisis leves/moderadas en domicilio. En aquellos casos en los que se sospeche infección, será necesario iniciar antibioterapia, la radiografía suele ser normal en ambos procesos, y en la RM puede haber algunos signos específicos de osteomielitis, pero la confirmación se realizará con el cultivo. La necrosis avascular de la cabeza femoral o humeral suele aparecer en la segunda década de la vida, y ocurre debido a cambios degenerativos osteoarticulares secundarios a los fenómenos isquémicos a nivel local. El diagnóstico debe ser precoz, por medio de la resonancia magnética, de forma que permita instaurar la descarga del miembro afecto lo antes posible. En el caso de que la obstrucción microvascular se produzca a nivel de los vasos mesentéricos, hígado o bazo, los pacientes presentarán crisis de dolor abdominal que se tratarán con medidas conservadoras. Hay que tener en cuenta el riesgo aumentado en estos pacientes de litiasis biliar y colestasis intrahepática.
Reagudizaciones de la anemia basal o aplasia: en estos pacientes la anemia crónica es bien tolerada, sin embargo, la anemia puede agudizarse con valores de Hb < de 6 g/dl por distintas causas, como son el paludismo (en pacientes con antecedentes de viajes a zonas endémicas) o las crisis aplásicas o eritroblastopénicas por parvovirus B19. La infección por parvovirus B19 favorece la aparición de crisis vasooclusivas, el aumento de la respuesta inflamatoria y la eritroblastopenia. Se presenta de forma gradual, mostrando: fatiga, fiebre, aumento del tamaño del bazo, decaimiento y descenso brusco de la Hb y de reticulocitos, dato este último que nos tiene que hacer sospechar este diagnóstico(16). La cifra de hemoglobina desciende hasta muy por debajo del valor basal habitual, pudiendo persistir la ausencia de producción de eritrocitos de 7-14 días. El secuestro esplénico es una de las posibles etiologías de anemización aguda en estos pacientes. Supone una emergencia clínica y cursa con la aparición repentina de esplenomegalia por el cúmulo de sangre en el bazo. Se asocia a una elevada morbimortalidad, ya que puede generar una anemia grave, incluso shock hipovolémico. En el hemograma veremos un descenso de la hemoglobina y de las plaquetas, y un aumento de reticulocitos. El tratamiento consiste en reposición hidroelectrolítica y transfusión. La esplenectomía estará recomendada en caso de episodios de repetición. Finalmente, señalar la frecuencia de la acentuación de la hemólisis ante cualquier causa, ya sea infecciosa o vaso-oclusiva. En el caso de las crisis hiperhemolíticas postrasfusionales, la clínica se caracteriza por una crisis dolorosa con hemólisis intravascular, pobre respuesta reticulocitaria, caída de la hemoglobina por debajo del nivel pretransfusional y, en ocasiones, con estudio inmunohematológico negativo o que no explica la hemólisis, lo que dificulta el diagnóstico. El síndrome hiperhemolítico constituye una urgencia hematológica, el tratamiento se basará en interrumpir las transfusiones e inicio de corticoterapia y/o administración de inmunoglobulinas intravenosas(16).
Los episodios febriles sin foco pueden ser reflejo de infecciones graves, debido a la asplenia funcional, ya que aumenta el riesgo de infecciones por microorganismos encapsulados (neumococo, Haemophilus influenzae y Salmonella). Se aconseja prevenirlas mediante la administración de vacunas y penicilina oral en los primeros años de vida.
El síndrome torácico agudo (STA)(21) es una complicación grave que va a presentar el 50% de los pacientes, al menos, una vez en la vida, sobre todo, si la enfermedad no está controlada. Cursa con: fiebre, tos, disnea, dolor torácico y aparición de un infiltrado pulmonar nuevo en la radiografía de tórax. Puede coexistir con una infección (Mycoplasma, Chlamydia, S. pneumoniae y parvovirus) y/o embolismo graso, siendo frecuente en este último, el antecedente de episodio de dolor vasooclusivo. La imagen de la radiografía puede no aparecer hasta varios días después, por lo que se debe realizar una monitorización cardiorrespiratoria estrecha en los pacientes con clínica. El tratamiento incluye: antibioterapia empírica al diagnóstico, hidratación, oxigenoterapia, broncodilatadores si precisa, analgesia, transfusiones y, en casos graves, exanguinotransfusión.
La forma de presentación más frecuente del accidente cerebrovascular (ACV) en niños es el infarto cerebral. Es una de las complicaciones más devastadora que puede sufrir un niño con ECF (Tabla VI).
Solo un 8-10% de los casos presentarán síntomas compatibles con déficit neurológico, cefalea, alteración de la conciencia y/o crisis comiciales(22). La causa más frecuente de infarto cerebral es la obstrucción de las arterias carótida interna y de la cerebral media. El tratamiento requiere de la realización de exanguinotransfusión urgente hasta alcanzar niveles de HbS <20%. Estas lesiones pueden detectarse de forma precoz con ecodoppler transcraneal (Eco DTC), ya que la velocidad de la sangre es inversa al diámetro arterial. El riesgo de ACV aumenta en la primera década de vida, por ello se recomienda realizar a partir de los tres años, exploraciones seriadas con ecografía doppler transcraneal, evaluando el riesgo de ictus y realizando si hay alteración, prevención primaria mediante transfusiones que mantengan las tasas de HbS < 30%.
El priapismo es frecuente en niños mayores, a partir de los 12 años, y puede presentarse en forma de episodios prolongados de más de 24 horas de evolución o episodios repetidos de menor duración (2-3 h), generalmente nocturnos. El tratamiento consiste en medidas generales de hidratación y analgesia, siendo necesario, en ocasiones, maniobras de aspiración del cuerpo cavernoso.
Complicaciones crónicas
• Cardiovasculares: secundarias a la anemia crónica y al aumento del gasto cardiaco. Es habitual la cardiomegalia, la hipertrofia del ventrículo izquierdo y la hipertensión pulmonar tras síndromes torácicos repetidos.
• Respiratorio: se produce un daño pulmonar progresivo, que puede evolucionar a una fibrosis precoz, siendo importante el seguimiento mediante pruebas de función respiratoria.
• Vasculopatía cerebral: los pacientes pueden sufrir isquemia cerebral crónica, lo que se puede manifestar como infartos silentes y alteración de la función neurocognitiva. El diagnóstico se establece con la RM cerebral(22).
• Riñón: existe un aumento del flujo renal con incremento del filtrado glomerular, lo que puede generar hipostenuria, tubulopatías y aumento del riesgo de infecciones urinarias.
• Hígado y bazo: la función de filtración de hematíes alterados del bazo se ve sobrepasada, ocasionando una esplenomegalia compensadora, que evolucionará a una autoesplenia por fibrosis. La asplenia funcional es una causa importante de enfermedad y muerte en niños pequeños, debido al mayor riesgo de infección. También, se pueden ver en estos pacientes hepatomegalia y episodios de litiasis biliar.
• Huesos: los cambios esqueléticos se producen por expansión medular y los infartos óseos. Puede haber una afectación del crecimiento y el desarrollo.
• Órganos de los sentidos: pueden presentar retinopatía e hipoacusia sensorial.
• Piel: aparición de úlceras cutáneas en piernas por vasooclusión.
Seguimiento
El seguimiento de los pacientes con talasemia moderada-grave y ECF (Tabla VI) deberá de hacerse desde un punto de vista multidisciplinar, asegurando un manejo integral.
Antes de iniciar el tratamiento transfusional, se debe de hacer una valoración inicial que incluya(5,16):
• Historia clínica completa y minuciosa, incluyendo el momento del diagnóstico y la clínica acompañante.
• Exploración física completa, incluyendo: peso, talla y estadio de Tanner.
• Informar sobre la enfermedad, complicaciones agudas y graves, y realizando educación sanitaria. En el caso de los pacientes con drepanocitosis, se deberá insistir en las medidas higiénico-dietéticas: hidratación abundante, sobre todo, en situaciones de calor, diarrea o vómitos; realizar ejercicio físico evitando sobreesfuerzos en intensidad y/o duración; evitar inmersiones en apnea; la exposición al frío y estancias a grandes alturas o viajes en avioneta no presurizada; evitar prendas excesivamente ajustadas; evitar el consumo de alcohol y tabaco; y realizar control anual de la higiene dental.
• Hemograma, bioquímica completa, pruebas de coagulación, inmunoglobulinas, estudio del metabolismo del hierro y serologías, si han recibido transfusión.
• Fenotipado o genotipado eritrocitario completo.
• Comprobar el calendario vacunal.
• El estudio familiar (tanto de la enfermedad como de HLA) es recomendable.
• Descartar causas de acentuación de la anemia: niveles de Glucosa-6-fosfato deshidrogenasa (G-6PDH), ácido fólico y otros déficits nutricionales.
• Realizar consejo genético.
La frecuencia de las visitas dependerá de las necesidades de cada paciente, siendo más frecuentes durante el primer año. En cada visita habrá que preguntar por la aparición de nuevos síntomas y signos, y realizar una exploración física completa. Es fundamental comprobar el correcto cumplimiento del tratamiento, así como la actualización del calendario vacunal. A nivel analítico, se solicitará un hemograma con bioquímica que incluya: perfil hepático, renal, ionograma y ferritina, si transfusiones. Anualmente, habrá que realizar seguimiento a nivel hematológico, cardiológico, endocrinológico, digestivo, oftalmológico y auditivo. En los pacientes con formas HbSS/HbSβ0 de drepanocitosis, se solicitará control con ecografía doppler transcraneal desde los 2 hasta los 16 años. Es muy importante en estos pacientes, un apoyo psicosocial y escolar.
Función del pediatra de Atención Primaria:
• Conocer la enfermedad y aspectos esenciales del seguimiento y síntomas de alarma indicativos de derivación (Temperatura ≥ 38ºC, dolor agudo que no cede con analgesia, tos, dolor torácico y dificultad respiratoria, dolor abdominal o aumento del bazo, síntomas neurológicos, palidez, fatiga, irritabilidad, priapismo de dos o tres horas de persistencia y deshidratación).
• Asegurar el cumplimiento del calendario vacunal de estos pacientes, prestando especial atención a la vacunación frente a microorganismos encapsulados en pacientes con asplenia funcional.
• Garantizar un adecuado cumplimiento terapéutico y seguimiento de la enfermedad mediante revisiones.
• Realizar educación sanitaria y proporcionar al paciente información sobre su enfermedad y las complicaciones derivadas.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.** González García H. Anemias hemolíticas en la infancia. Pediatr Integral. 2012; XVI(5): 378-86.
2.*** Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018; 391: 155-67.
3.** González Fernández FA, Ropero Gradilla P. Síndromes Talasémicos. En: Madero L, Lassaletta A, Sevilla J. Hematología y Oncología Pediátrica. Ergon SA 2015.
4.** Vichinsky E. Non-transfusion-dependent thalassemia and thalassemia intermedia: epidemiology, complications, and management. Curr Med Res Opin. 2016; 32: 191-204.
5.*** Guía de Práctica Clínica de la Talasemia Mayor e Intermedia en Pediatría. Sociedad Española de Hematología y Oncología Pediátricas (SEHOP) 2015.
6.** Taher A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT). Nicosia (Cyprus): Thalassaemia International Federation. 2013.
7.** Farmakis D. Angastiniotis, M. Eleftheriou, A. A Short Guide for the Management of Transfusion Dependent Thalassaemia. 2017.
8.** Sebastián E, Sevilla J. Protocolo diagnóstico y tratamiento de la anemia microcítica en el adolescente. Medicine. 2018; 12: 3613-8.
9.** Sevilla Navarro J. Abordaje de la anemia microcítica, nuevas herramientas diagnósticas. En: AEPap ed. Curso de Actualización Pediatría 2010. Madrid: Exlibris Ediciones; 2010. p. 23-9.
10.** Brancaleoni V, Di Pierro E, Motta I, Cappellini MD. Laboratory diagnosis of thalassemia. Int J Lab Hematol. 2016; 38: 32-40.
11.** Viprakasit V, Ekwattanakit S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol Oncol Clin North Am. 2018; 32: 193-211.
12.** Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011; 118: 3479-88.
13.** Angulo Ureña G, Koss Hernández R, Monge Ortiz JM. Generalidades y tratamientos emergentes en la Beta-talasemia. Rev. méd. sinerg. 2020; 5: e549.
14.** Basack N. Nuevas estrategias para el tratamiento de la β talasemia. En: Hematología: vol. 23; Número Extraordinario XXIV Congreso Argentino de Hematología. 2019.
15.** Cappellini MD, Motta I. New therapeutic targets in transfusion-dependent and -independent thalassemia. Hematology Am Soc Hematol Educ Program; 2017. p. 278-83.
16.*** Guía de Práctica Clínica de la Enfermedad de células falciformes. Sociedad Española de Hematología y Oncología Pediátricas (SEHOP) 2019.
17.*** Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017; 376: 1561-73.
18.*** Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017; 390: 311-23.
19.** Cela de Julián E. Drepanocitosis y otras hemoglobinopatías estructurales. En: Madero L, Lassaletta A, Sevilla J. Hematología y Oncología Pediátricas. Ergon SA 2015.
20.** Hoppe C, Neumayr L. Sickle Cell Disease: Monitoring, Current Treatment, and Therapeutics Under Development. Hematol Oncol Clin North Am. 2019; 33: 355-71.
21.** Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers. 2018; 4: 18010.
22.** Hirtz D, Kirkham FJ. Sickle Cell Disease and Stroke. Pediatr Neurol. 2019; 95: 34-41.
Bibliografía recomendada
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018; 391: 155-67.
Completo artículo de revisión de la talasemia con gráficos ilustrativos, que recoge de forma adecuada, el tratamiento de las complicaciones en estos pacientes.
- Guía de Práctica Clínica de la Talasemia Mayor e Intermedia en Pediatría. Sociedad Española de Hematología y Oncología Pediátricas (SEHOP) 2015.
Imprescindible guía médica para la atención del paciente con talasemia. Incluye aspectos sobre clínica, tratamiento y complicaciones. Incluye tablas de seguimiento y hojas informativas para familias.
- Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017; 376: 1561-73.
Buena revisión de las drepanocitosis, con conceptos de patogénesis, diagnóstico, complicaciones y tratamiento.
- Guía de Práctica Clínica de la Enfermedad de células falciformes. Sociedad Española de Hematología y Oncología Pediátricas (SEHOP) 2019.
Guía de práctica clínica muy completa para el abordaje de pacientes con drepanocitosis. Contiene todos los aspectos asistenciales, información para familias y hoja informativa para urgencias.
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017; 390: 311-23.
Interesante actualización sobre enfermedad de células falciformes, fácil de leer gracias a su formato electrónico, con tablas muy ilustrativas.
| Caso clínico |
|
Varón de 4 años que ingresa por episodio de decaimiento, con vómitos y malestar general de 3-4 días de evolución, sin otra sintomatología previa asociada. Antecedentes personales: fractura de cúbito y radio en 2018. Sin otras enfermedades ni antecedentes de interés. Calendario vacunal completo, salvo antineumocócica y antimeningocócica. Antecedentes familiares: madre de 37 años, sana. Procedente de Senegal. Padre biológico desconocido. Dos hermanos de 18 y 12 años sanos. Exploración física: temperatura: 38,7ºC; FC: 130 lpm; FR: 30 rpm; TA: 108/79 mm Hg; SatO2: 96%. Palidez cutánea con tinte subictérico. No exantemas ni petequias. Destaca polipnea sin signos de dificultad respiratoria. Auscultación cardio-respiratoria: soplo sistólico grado II/VI. Precordio aumentado. Abdomen: marcada esplenomegalia hasta pala ilíaca. Pruebas complementarias: hemograma: hemoglobina: 5,2 g/dl; VCM: 80 fl; HCM: 22,4 pg; leucocitos: 20.200/ul (neutrófilos 13.360/ul); plaquetas: 135.000/ul. Bioquímica básica y renal normal. Bilirrubina: 1,36 mg/dl a expensas de indirecta. GOT: 320 U/L; LDH: 1.561 U/L. PCR: 4 mg/dl; PCT: 1,5 ng/ml. Coagulación: normal, salvo fibrinógeno: 550 mg/dl. Evolución: a su llegada se transfunden 2 concentrados de hematíes y se inicia antibioterapia empírica con cefotaxima, presentando mejoría neurológica y hemodinámica progresiva. Posteriormente, se mantiene con sueroterapia intravenosa. Durante el ingreso, permanece afebril con descenso progresivo de parámetros infecciosos y LDH. Mantiene parámetros de hemoglobina estables en torno a 8-9 mg/dl, sin necesidad de nuevas transfusiones. Disminución paulatina de la esplenomegalia hasta 1-2 cm del reborde costal. Al alta, se decide vacunación frente neumococo y meningococo B.
|


 Anemia. Classification and diagnosis
Anemia. Classification and diagnosis