 |
| Temas de FC |
J. Zubicaray Salegui, J. Sevilla Navarro
Hospital Infantil Universitario Niño Jesús
| Resumen
La anemia hemolítica se define por el acortamiento de la vida media del hematíe que puede ser aguda o crónica; esta deberá formar parte del diagnóstico diferencial de cualquier anemia normocítica o macrocítica. La hemólisis puede ocurrir por vía intravascular o extravascular, y se debe: a la pérdida de la deformabilidad que conduce a fagocitosis, a la destrucción mediada por anticuerpos o activación directa del complemento, o a la fragmentación debida a microtrombos o trauma mecánico directo, oxidación o destrucción celular directa. Clínicamente, los pacientes pueden presentar signos y síntomas como: anemia aguda, ictericia, hematuria, disnea, fatiga, taquicardia o hipotensión. Los resultados de las pruebas de laboratorio que confirman la hemólisis incluyen: reticulocitosis, así como aumento de lactato deshidrogenasa, aumento de bilirrubina no conjugada y disminución de los niveles de haptoglobina. La prueba de antiglobulina directa diferenciará, por lo general, las causas inmunes de las causas no inmunes. Asimismo, se debe realizar un frotis de sangre periférica, para identificar morfologías anormales de los glóbulos rojos. Todo ello, clasificará a las anemias hemolíticas en: hemoglobinopatías, membranopatías, enzimopatías, anemias inmunomediadas y anemias de causas extrínsecas no inmunes. |
| Abstract
Hemolytic anemia is defined as the reduction of the half-life of red blood cells, which can be an acute or chronic process, and it should be part of the differential diagnosis of any normocytic or macrocytic anemia. Hemolysis can occur intravascularly or extravascularly, and it is due to: loss of deformability that leads to phagocytosis, antibody-mediated destruction or direct complement activation, fragmentation due to microthrombi or direct mechanical trauma, oxidation, or direct cell destruction. Clinically, patients may present with signs and symptoms such as: acute anemia, jaundice, hematuria, dyspnea, fatigue, tachycardia, or hypotension. Laboratory tests that confirm hemolysis include reticulocytosis, increased lactate dehydrogenase and unconjugated bilirubin, and decreased haptoglobin levels. |
Palabras clave: Anemia hemolítica; Congénita; Pediatría.
Key words: Hemolytic anemia, Congenital, Pediatrics.
Pediatr Integral 2021; XXV (5): 233 – 240
Anemias hemolíticas: clasificación. Membranopatías. Enzimopatías. Anemia hemolítica autoinmune
Introducción
Las anemias hemolíticas son un conjunto de trastornos que se caracterizan por un acortamiento de la vida media del hematíe(1). Constituyen un grupo importante de anemias, con manifestaciones clínicas y analíticas comunes, pero con un origen y una fisiopatología distinta.
En función de la base fisiopatológica de la alteración, los podemos clasificar en dos grupos: intrínsecos (o corpusculares) y extrínsecos (extracorpusculares)(2) (Tabla I).

Los intrínsecos son mayoritariamente de origen congénito y se deben a alteraciones estructurales o funcionales de sus componentes fundamentales: hemoglobina (hemoglobinopatía), membrana (membranopatía) o enzimas (enzimopatías). Los defectos extrínsecos, en cambio, son adquiridos y se deben a alteraciones del entorno, una vez el hematíe ha abandonado la médula ósea. De una forma u otra, alteran su estructura y/o morfología, disminuyendo su capacidad de deformación, y por consiguiente, disminuyendo la vida media del hematíe. En cualquier caso, el resultado final es siempre un síndrome hemolítico de intensidad variable con aumento compensador de la eritropoyesis y aumento del número de reticulocitos. La hemólisis puede ser extravascular (en el sistema retículo-endotelial del bazo o del hígado) o intravascular, produciéndose directamente dentro de los vasos sanguíneos. Clínicamente, se caracterizan por tres signos: reticulocitosis, esplenomegalia e ictericia. La bilirrubina no conjugada o indirecta aumenta por incremento del catabolismo del hemo. Además, se produce disminución de la haptoglobina, debido a la unión de esta a la hemoglobina libre en sangre y su destrucción a nivel hepático.
En cuanto al diagnóstico general, requiere tres exploraciones básicas: 1) hemograma o examen hematológico básico; 2) recuento de reticulocitos o hematíes jóvenes circulantes; y 3) examen morfológico de la sangre a partir de un frotis(3-5).
En este capítulo, desarrollaremos las características básicas de las anemias hemolíticas más frecuentes.
Membranopatías: alteraciones de la membrana eritrocitaria
Introducción
La patología de la membrana eritrocitaria reside en la existencia de defectos congénitos de las proteínas que la constituyen.
Obedecen a defectos estructurales o funcionales de las proteínas de la membrana eritrocitaria. La membrana del hematíe es un sistema complejo construido por múltiples proteínas integrales y superficiales, que proporcionan al hematíe su característica forma de disco bicóncavo, así como la capacidad de deformarse o deformabilidad. Está constituida por una bicapa lipídica en la que flotan unas proteínas llamadas integrales. Recubriendo su cara interna, y en mínimo contacto con la Hb, se encuentra una red proteica bidimensional, fijada a la bicapa lipídica por proteínas integrales; esta red se conoce como el citoesqueleto de la membrana eritrocitaria(6). Las proteínas pueden formar parte de la bicapa lipídica (proteínas integrales) o formar parte del esqueleto (proteínas estructurales). De acuerdo a su movilidad electroforética en un gel de poliacrilamida, las proteínas de la membrana eritrocitaria se pueden clasificar en bandas o fracciones, lo que permite identificar alteraciones de estas en caso de patología.
Un defecto estructural o funcional en alguna de estas proteínas, puede alterar la integridad de todo el esqueleto de la membrana y dar lugar a una hemólisis, por lo que la patología de la membrana eritrocitaria reside en la existencia de defectos congénitos de las proteínas que la constituyen. En general, se trata de mutaciones en alguno de los genes que codifican estas proteínas y que se heredan mayoritariamente con carácter autosómico dominante, con hemólisis extravascular y con alteraciones morfológicas características en los hematíes que dan nombre y orientan al diagnóstico de cada entidad, como veremos a continuación (Fig. 1)(7).
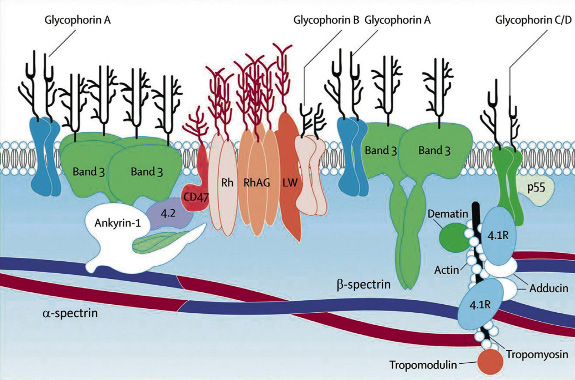
Figura 1. Representación esquemática de la membrana eritrocitaria. Las proteínas integrales de la membrana (banda 3, glucoforinas A, B, C, D y E, estomatina y proteínas de antígenos del Rh y grupos sanguíneos) atraviesan la capa lipídica. Las proteínas estructurales del citoesqueleto recubren la superficie interna (espectrina, actina, proteína 4.1, ankirina, proteína 4.2 y p55, adducina, dematina, actina, tropomiosina y tropomodulina). Las interacciones horizontales y verticales entre las proteínas son imprescindibles para el mantenimiento de la estructura de la membrana. (Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008; 372: 1411-26).
Esferocitosis hereditaria (EH)
La esferocitosis hereditaria cursa con un síndrome hemolítico crónico sobre el que pueden aparecer crisis aplásicas y crisis hemolíticas.
Es la membranopatía más común en la raza blanca, con una prevalencia estimada de 1/2.000(7). Obedece a un defecto de proteínas con interacción vertical del citoesqueleto que produce pérdida parcial de la misma (Fig. 1), con la consiguiente disminución de la relación superficie/volumen y aparición de esferocitos.
Manifestaciones clínicas
Tiene una expresividad clínica variable, aunque existe predominio de formas leves y moderadas. En general, sobre una base de hemólisis crónica se producen crisis hemolíticas, sobre todo en la infancia, en relación con infecciones virales y, menos frecuentes, episodios de eritroblastopenia por parvovirus B19. Los hallazgos clínicos son: anemia, ictericia y esplenomegalia. Como complicaciones en relación con la hemólisis, pueden observarse: colelitiasis, masas de eritropoyesis extramedular y sobrecarga de hierro. Las formas sintomáticas pueden debutar en el periodo neonatal, como enfermedad hemolítica no inmune. Antiguamente, debido a que a menudo, las pruebas de fragilidad osmótica y el examen morfológico no eran concluyentes en el recién nacido, se producía un retraso en el diagnóstico definitivo de estos casos; hecho que la implantación de la prueba de la fijación de eosina-5-maleimida (test de EMA) ha simplificado y mejorado.
Mecanismo molecular
El patrón de herencia es autosómico dominante en el 75% de los casos, y autosómico recesivo en el resto. En cuanto a la frecuencia de los distintos defectos o mutaciones, se estima que los defectos en el gen de la ankirina (40-60% de casos en el norte de Europa), beta-espectrina (20%), y banda 3 (15-25%), son las más frecuentes.
Diagnóstico
Para apoyar el diagnóstico clínico mencionado con anterioridad, el diagnóstico en el laboratorio se basa fundamentalmente en las siguientes pruebas.
Morfología eritrocitaria
Es característica la presencia de esferocitos o hematíes con forma redondeada, aspecto pequeño o intensamente coloreado, careciendo de aclaramiento central en el frotis de sangre periférica. Además, los índices eritrocitarios muestran un incremento de la concentración de la hemoglobina corpuscular media (CHCM), un incremento de la amplitud de distribución eritrocitaria (ADE) y un volumen corpuscular medio (VCN) normal o ligeramente disminuido.
Estudio de fragilidad osmótica eritrocitaria (ROE)
Durante muchos años, ha sido la prueba más utilizada para el diagnóstico de la EH. Mide la capacidad o habilidad del hematíe de incrementar su volumen cuando son sometidos a soluciones hipotónicas de cloruro de sodio. Debido a que los esferocitos tienen una relación de superficie/volumen disminuida, su capacidad de aumentar el volumen es también limitada, lisándose antes ante concentraciones crecientes de ClNa. Se debe de tener en cuenta que hasta un 20% de las esferocitosis hereditarias presentan un test de fragilidad normal en relación a la escasa presencia de esferocitos o por la reticulocitosis intensa.
Prueba de la fijación de eosina-5-maleimida (EMA binding test)
Esta técnica de citometría de flujo se basa en la medida de intensidad de fluorescencia en hematíes, que han sido incubados con el fluorocromo eosina-5-maleimida. Así, la fluorescencia estará disminuida en los casos de EH. Es especialmente sensible en la detección de la EH, por ello se está implementando actualmente, como técnica de referencia para el diagnóstico.
Existen otras técnicas como la ectacitometría de gradiente osmótico o la electroforesis de proteínas mediante gel de poliacrilamida, este último como paso previo al estudio molecular del gen afecto. Finalmente, puede completarse el diagnóstico mediante el diagnóstico molecular de las mutaciones genéticas. Permite el diagnóstico de certeza, siendo esencial para los pacientes en los que los test diagnósticos básicos son dudosos, en pacientes que requieren transfusiones o en situaciones complejas, como anemias hemolíticas en neonatos.
Tratamiento
El tratamiento es básicamente preventivo, a base de administración de ácido fólico ante incremento de hemólisis. Las transfusiones por su parte, se realizan a demanda. Cuando la anemia es muy intensa, puede realizarse una esplenectomía que, en la mayoría de los casos, normaliza la concentración de hemoglobina, desapareciendo la anemia (aunque no los esferocitos circulantes). La esplenectomía elimina o reduce la hemólisis, sin embargo, implica riesgo de problemas infecciosos por gérmenes encapsulados, y problemas trombóticos. Por ello, se considera indicación solo en membranopatías estructurales con fenotipo clínico grave. Según las guías británicas, la indicación se establece para esferocitosis graves (Hb mantenidas entre 6-8 g/dl), no antes de los 6 años y si es posible por laparoscopia(8). Antes de la cirugía, se exige vacunación reglada previa frente a gérmenes encapsulados (Neisseria meningitidis, Streptococcus pneumoniae y Haemophilus influenzae) y después de la misma, profilaxis con penicilina hasta la vida adulta(9-10).
Otras membranopatías
La eliptocitosis congénita y la estomatocitosis hereditaria son formas menos frecuentes de membranopatía.
Eliptocitosis congénita (EC)
Trastorno algo más leve que la EH que no siempre condiciona anemia hemolítica. En la EC, es característica la presencia de numerosos eliptocitos circulantes en sangre periférica. Obedece a un defecto de proteínas con interacción horizontal del citoesqueleto (principalmente Banda 4,1 y α-espectrina) (Fig. 1) que alteran la elasticidad de la membrana, impidiendo su recuperación después de un alargamiento. No existe pérdida de membrana y, por ello, la ROE y la CHCM son normales. La mayoría de los casos (80%) son asintomáticos o con anemia leve que, en ocasiones, suele pasar desapercibida. Esto implica que el diagnóstico de una eliptocitosis se puede hacer a cualquier edad y que su verdadera incidencia en una población pueda ser subestimada. En el periodo neonatal existe un cuadro clínico conocido como “poiquilocitosis neonatal”, caracterizado por anemia asociada a ascitis y edema, que suele resolverse espontáneamente durante las primeras semanas de vida. Existe poca evidencia sobre la eficacia de la esplenectomía, ya que por la gran inestabilidad de la membrana, los eritrocitos también se rompen fuera del bazo.
Estomatocitosis hereditaria
Finalmente, existe una forma muy rara de membranopatía cuya manifestación morfológica fundamental es la presencia de hematíes con una palidez central alargada en lugar de redonda. Existe un trastorno de la permeabilidad a los iones de sodio o potasio, por el cual el hematíe puede hidratarse (hidrocitosis congénita) o deshidratarse (xerocitosis congénita). La xerocitosis es el desorden más frecuente dentro de la estomatocitosis hereditarias (XC), con una incidencia de 1 en 50.000 nacimientos. Esta incidencia puede estar infravalorada, porque muchos casos con Hb en límites normales solo se van a manifestar, por un aumento de reticulocitos o larvados datos de hemólisis y van a pasar desapercibidos. La mayor parte de los casos descritos están causados por mutaciones del gen PIEZO1 (16q24.3), que se hereda de forma autosómica dominante. El dato más característico de los hematíes deshidratados es el aumento de la CHCM. Al contrario de la EH, la observación morfológica de sangre periférica es anodina y, a excepción de aquellos casos en los que se aprecian eritrocitos de forma irregular (xerocitos). En cuanto al tratamiento, a diferencia a lo que sucede en otras membranopatías, la XC no responde bien a la esplenectomía, y no se aconseja por el alto riesgo de problemas de trombosis arteriales y venosas. Por eso, se deberá descartar una estomatocitosis antes de programar una esplenectomía.
Eritroenzimopatías: las enzimas y sus alteraciones
Introducción
Las eritroenzimopatías son enfermedades hereditarias, debidas al déficit de alguna de las enzimas del hematíe que pueden localizarse en alguna de sus 4 vías metabólicas principales: 1) glucólisis anaerobia; 2) metabolismo antioxidante; 3) sistema diaforásico; y 4) metabolismo nucleotídico. Se engloban también dentro de las anemias raras o minoritarias, ya que su prevalencia en nuestra población es siempre inferior a 5 casos por 10.000 habitantes, siendo las más frecuentes: el déficit de glucosa-6-fosfato deshidrogenasa y el déficit de piruvato cinasa. El perfil o patrón de hemólisis puede variar en cada una de ellas: el déficit de piruvato cinasa cursa con hemólisis crónica, mientras que el déficit de glucosa-6-fosfato deshidrogenasa cursa con hemólisis aguda.
A diferencia de la EH, las eritroenzimopatías carecen de alteraciones morfológicas características y para su diagnóstico se requiere la cuantificación de la actividad enzimática en el hemolizado del paciente.
Déficit de piruvato-cinasa (PK)
Supone la primera causa de anemia hemolítica crónica no esferocítica.
Es una anemia hemolítica corpuscular por defecto enzimático en la vía anaerobia de producción de pirúvico y láctico del hematíe. Debido a la deficiencia de dicha enzima, se altera la generación de ATP y los eritrocitos no pueden mantener su contenido de agua y de potasio, volviéndose rígidos y, por consiguiente, disminuyéndose su vida media. El patrón de herencia es autosómico recesivo, los pacientes homocigotos o los dobles heterocigotos padecen un síndrome hemolítico con anemia de intensidad variable.
Manifestaciones clínicas
Cursa con un síndrome hemolítico crónico de intensidad variable y, habitualmente, de inicio neonatal. La anemia puede variar, desde formas moderadas o leves, incluso asintomáticas en caso de que la hemólisis sea compensada, hasta formas graves con intensa anemia, que se acompañan de retraso de crecimiento óseo y gonadal. Estos pacientes presentan sobrecarga de hierro incluso en pacientes no trasfundidos por el aumento de la absorción intestinal debido a la hemolisis crónica y la eritropoyesis ineficaz.
Mecanismo molecular
La PK funcional es un tetrámero formado por 4 subunidades idénticas y sintetizado por dos genes diferentes: PKM2 y PKLR. Hasta la actualidad, se han descrito mas de 300 mutaciones del gen PKLR responsables del déficit.
Diagnóstico
El diagnóstico de déficit de PK requiere la cuantificación de la actividad enzimática en el suero hemolizado. Si los leucocitos no se eliminan bien del preparado hemolizado, pueden enmascarar el déficit de PK eritrocitario, ya que los leucocitos tienen una actividad PK normal. Igualmente, la presencia de un elevado número de reticulocitos circulantes puede dar también falsos negativos, debido también a que la actividad PK de los reticulocitos es muy superior a la de los hematíes maduros. Por ello, si en presencia de intensa reticulocitosis se obtiene una actividad PK normal, debe confirmarse el resultado mediante la determinación del cociente entre la actividad PK y hexocinasa (HK)(11). No obstante, en la actualidad se recomienda realizar confirmación del diagnóstico con estudio molecular ya que la actividad enzimática no es fácil de estandarizar.
Tratamiento
El tratamiento del déficit PK es esencialmente sintomático, consistiendo en el soporte con concentrados de hematíes, quelación de hierro y, eventualmente, esplenectomía, aunque esta última no consigue normalizar las cifras de Hb. En los pacientes con síndrome hemolítico severo o intenso, puede optarse por un trasplante de progenitores hematopoyéticos, aunque se debe considerar su elevada morbimortalidad. Asimismo, en los últimos años, esta enzimopatía constituye una enfermedad candidata a ser tratada en aproximaciones de terapia génica. Del mismo modo, existen ensayos clínicos en marcha con nuevas moléculas con capacidad de estabilizar la proteína PK-R (NCT02476916, NCT03548220, NCT03559699)(12-13) con algunos resultados ya publicados.
Déficit de glucosa-6-fosfato deshidrogenasa (G6PD)
Las crisis hemolíticas desencadenadas por agentes oxidantes son características del déficit de glucosa-6-fosfato deshidrogenasa.
Es la eritroenzimopatía más frecuente, afectando a más de 400 millones de personas en todo el mundo. Está implicada en la ruta metabólica antioxidante de las pentosas, donde gracias a dicha enzima, se produce la nicotinamida adenindinucleótido fosfato reducido (NADPH), necesaria para mantener el glutatión reducido, que protege a la hemoglobina de la acción oxidativa del peróxido de hidrógeno y radicales oxidativos(14). Así, el déficit de G6PD reduce la concentración de glutatión reducido y disminuye la capacidad del hematíe para defenderse frente a agentes oxidantes, constituyendo una causa de hemólisis. Además, el déficit protege frente a la parasitación por el Plasmodium falciparum, explicando la mayor supervivencia de los portadores del déficit sobre los no portadores, así como su elevada prevalencia.
Manifestaciones clínicas
La manifestación clínica habitual es una crisis de hemólisis intravascular aguda, después de 24-72 horas de la ingesta o contacto con algún medicamento oxidante o habas (favismo), así como con algún agente infeccioso. Se presenta con intensa anemia y hemoglobinuria. Es típico que la anemia empeore hasta el día 7-8 de la crisis; sin embargo, una vez suprimido el agente oxidante, la concentración de Hb se recupera espontáneamente a los 15 días.
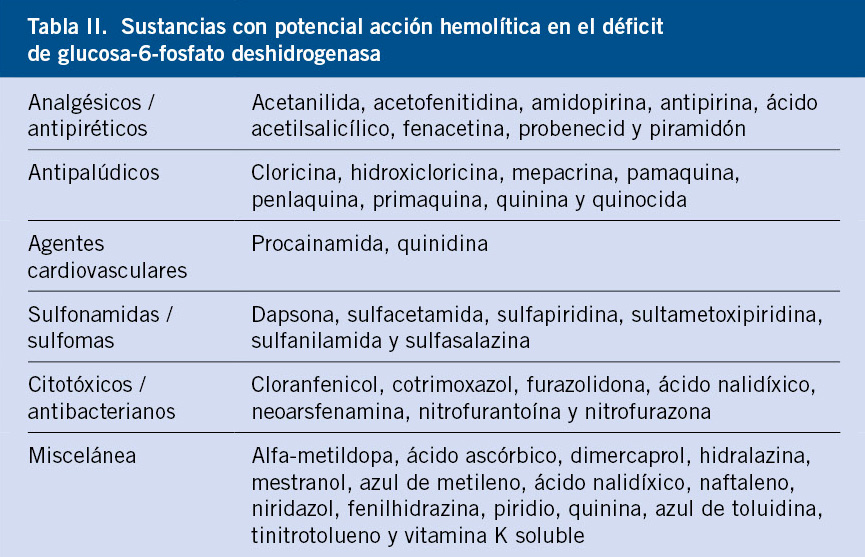
Debido a ello, los portadores de un déficit de G6PD pueden permanecer asintomáticos durante muchos años, hasta que se produce este contacto con las substancias que desencadenan la hemólisis. Entre los medicamentos que pueden ser causa de hemólisis en el déficit de G6PD destacan: ciertos analgésicos, sulfamidas, antipalúdicos y antibióticos (Tabla II).
Mecanismo molecular
La enzima G6PD es codificada por un gen situado en el cromosoma X, por lo que su herencia va ligada al sexo. Por lo tanto, los pacientes varones (hemicigotos o heterocigotos) son los que padecen la enfermedad, ya que la actividad enzimática es siempre <1%. La mayoría de las mujeres portadoras son asintomáticas, pero debido a la variabilidad aleatoria de la expresión del cromosoma X, durante el desarrollo pueden observarse portadoras heterocigotas con actividad G6PD prácticamente normal o significativamente disminuida, que puede dar lugar a crisis hemolíticas agudas de características superponibles a las de los varones heterocigotos.
Gracias a la secuenciación de cADN G6PD, se han identificado más de 300 mutaciones diferentes o combinaciones de mutaciones del gen G6PD. Las variantes se clasifican en cuatro grupos, según su comportamiento clínico: grupo I (variantes esporádicas), grupo II (variantes mediterráneas), grupo III (variantes africanas) y grupo IV (variantes asintomáticas).
Diagnóstico
El déficit de G6PD requiere la medida de la actividad enzimática en el hemolizado. Se debe tener en cuenta que la demostración de la disminución de actividad del enzima en los eritrocitos es más evidente después de varias semanas del episodio hemolítico, cuando remite la intensa reticulocitosis, ya que las células jóvenes poseen una actividad enzimática mayor. Normalmente, en los individuos varones portadores de la enzimopatía esta actividad es prácticamente nula, por lo que un individuo con déficit de G6PD puede resistir sin hemólisis, en condiciones normales, con una actividad de G6PD eritrocitaria alrededor del 1% de la normal. Como consecuencia de la agresión oxidativa, en estos pacientes, los hematíes suelen mostrar, además de anemia, una alteración morfológica característica conocida como excentrocitosis. Los cuerpos de Heinz aparecen durante las crisis y como las células que los contienen son eliminadas con rapidez de la circulación, pueden dejar de verse después de 2 o 3 días.
Tratamiento
La mejor medida terapéutica es la prevención de los episodios hemolíticos en los pacientes predispuestos, evitando la exposición a agentes oxidantes.
Anemia hemolítica autoinmune
La anemia hemolítica autoinmune presenta la prueba de Coombs directa positiva.
La anemia hemolítica autoinmune (AHAI) es un trastorno inmune caracterizado por la presencia de autoanticuerpos dirigidos contra antígenos de la membrana eritrocitaria, que producen acortamiento de la vida media de los hematíes. Estarían incluidas dentro de las anemias hemolíticas extrínsecas o extracorpusculares.
Aparece cuando existen anticuerpos dirigidos contra un componente de la membrana eritrocitaria con o sin participación del complemento, o cuando ciertos fármacos interaccionan con la membrana del hematíe y facilitan su fagocitosis por los macrófagos del bazo, el hígado y la médula ósea. El patrón en cada caso es característico y se determina por la naturaleza y la cantidad de autoanticuerpo producido. Los anticuerpos de naturaleza IgG preferentemente actúan a una temperatura ≥ 37ºC, mientras que los de naturaleza IgM son más activos a temperaturas frías (Tabla III).

Es una entidad rara en la infancia, con una incidencia estimada de 0,8-1,25 casos por 100.000 niños, aunque constituye la causa más frecuente de anemia hemolítica extracorpuscular(15-16). Puede aparecer a cualquier edad, con una mayor incidencia en torno a los tres o cuatro años. Etiológicamente, se pueden clasificar en AHAI secundarias (infección, hemopatías, enfermedades autoinmunes, tumores…) y primarias o idiopáticas. En población pediátrica, más de la mitad de los casos se presentan asociadas a otra patología, siendo un 40-50% AHAI idiopáticas(17). A diferencia de los adultos, con mayor frecuencia, suele presentarse como un cuadro autolimitado asociado a una infección viral. Sin embargo, los menores de dos años y adolescentes presentan más frecuentemente formas crónicas asociadas o no a enfermedades sistémicas, principalmente inmunodeficiencias o trastornos autoinmunes(18).
Diagnóstico
En todos los casos, el diagnóstico se basa en la demostración de la antiglobulina mediante positividad de la prueba de Coombs directa para IgG o IgM y complemento, que detecta el revestimiento de inmunoglobulinas o componentes del complemento en la superficie del hematíe. No obstante, se debe tener en cuenta que, de forma excepcional, existen casos de AHAI con Coombs directo negativo(19).
Tratamiento
La mayoría de las formas idiopáticas responden al tratamiento de primera línea, constituido por la corticoterapia (1-2 mg/kg/día hasta que se alcancen niveles de Hb mayores a 10 g/dL y descenso lento posterior en 4-6 meses). En general son de buen pronóstico, con tasas de respuesta de hasta 70-85%, aunque un 50% puede recaer y ser córtico-dependientes. La transfusión debe reservarse para casos graves con afectación hemodinámica. Las recaídas frecuentes y crónicas se asocian a formas más secundarias, donde el pronóstico depende de la enfermedad primaria. Como opciones de tratamientos para esas formas refractarias se pueden plantear, tales como: la esplenectomía, el rituximab u otros inmunomoduladores o inmunosupresores(20).
AHAI por anticuerpos fríos y hemolisina bifásica
En las anemias hemolíticas por anticuerpos fríos, causadas por IgM, los anticuerpos reaccionan contra el sistema antigénico eritrocitario I/i. Hay formas primarias, excepcionales en Pediatría, y formas secundarias a infecciones por Mycoplasma y Epstein-Barr, fundamentalmente. Se manifiesta por un síndrome hemolítico agudo menos sensible a los corticoides, pero generalmente autolimitado y prevenible, evitando la exposición al frío. En la hemoglobinuria paroxística a frigore, la hemolisina bifásica Donath-Landsteiner (IgG que reacciona a bajas temperaturas contra el antígeno P) fija grandes cantidades de complemento y los eritrocitos se hemolizan cuando la temperatura aumenta. Ocurre en el 30-40% de las AHAI en niños y, habitualmente, asocia historia de infección viral, varias semanas antes.
Función del pediatra de Atención Primaria
El pediatra de Atención Primaria podrá realizar la orientación diagnóstica de una anemia hemolítica a partir de la historia familiar y los signos y síntomas acompañantes. Se apoyará en las pruebas de diagnóstico o laboratorio básico para apoyar su sospecha diagnóstica (ver algoritmo al final del artículo). Tras el diagnóstico etiológico o definitivo por el especialista de área, el pediatra de Atención Primaria será fundamental para promover las medidas de prevención de las crisis hemolíticas, así como los cuidados posteriores a tratamientos específicos, como la esplenectomía.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1. Phillips J, Henderson AC. Hemolytic Anemia: Evaluation and Differential Diagnosis. Am Fam Physician. 2018; 98: 354-61.
2. González García H, Garrote Molpeceres R, Urbaneja Rodríguez E. Anemias hemolíticas en la infancia. Pediatr Integral. 2016; XX(5): 308-17.
3. Haley K. Congenital Hemolytic Anemia. Med. Clin. North Am. 2017; 101: 361-74.
4. Vives Corrons JL. Mañú Pereira MM, Trujillo JP, Surralles J, Sevilla J. Anemias raras y fallos medulares hereditarios. Arbor. 2018; 194: a463.
5. Arrizabalaga B. González F. Remacha A. Eritropatología. Ambos Marketing Services, S.L.; Nº 1 edición. 2017.
6.** Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013; 27: 167-78.
7.** Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008; 372: 1411-26.
8. Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis-2011 update. Br J Haematol. 2012; 156: 37-49.
9. Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015; 135: 1107-14.
10.** Iolascon A, Andolfo I, Barcellini W, Corcione F, Garçon L, De Franceschi L, et al. Working Study Group on Red Cells and Iron of the EHA. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017; 102: 1304-13.
11. Bianchi P, Fermo E, Glader B, Kanno H, Agarwal A, Barcellini W, et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: Consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am J Hematol. 2019; 94: 149-61.
12. Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019; 184: 721-34.
13.** Grace RF, Barcellini W. Management of pyruvate kinase deficiency in children and adults. Blood. 2020; 136: 1241-9.
14.** Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008; 371: 64-74.
15. Aladjidi N, Jutand MA, Beaubois C, Fernandes H, Jeanpetit J, Coureau G, et al. Reliable assessment of the incidence of childhood autoimmune hemolytic anemia. Pediatr Blood Cancer. 2017; 64: 1-19.
16. Chou ST, Schreiber AD. Autoimmune hemolytic anemia. In: Nathan and Oski’s Hematology and Oncology of Infancy and Childhood. 8th ed. Philadelphia: Saunders; 2015. p. 411.
17. Robert A, Brodsky MD. Warm Autoimmune Hemolytic Anemia. N Engl J Med. 2019; 381: 647-54.
18. Vagace JM, Bajo R, Gervasini G. Diagnostic and therapeutic challenges of primary autoimmune haemolytic anaemia in children. Arch Dis Child. 2014; 99: 668-73.
19.** Hill QA, Hill A, Berentsen S. Defining autoimmune hemolytic anemia: a systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019; 3: 1897-906.
20. Brodsky RA. Warm Autoimmune Hemolytic Anemia. N Engl J Med. 2019; 381: 647-54.
Bibliografía recomendada
- Hill QA, Hill A, Berentsen S. Defining autoimmune hemolytic anemia: a systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019; 3: 1897-906.
Actualización reciente de criterios de diagnóstico, criterios de severidad o fase de enfermedad, y criterios de respuesta a tratamientos.
- Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013; 27: 167-78.
Revisión ilustrativa de las membranopatías más importantes.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008; 372: 1411-26.
Exhaustiva revisión sobre las bases de la esferoticosis hereditaria.
- Iolascon A, Andolfo I, Barcellini W, Corcione F, Garçon L, De Franceschi L, et al. Working Study Group on Red Cells and Iron of the EHA. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017; 102: 1304-13.
Indicaciones actualizadas recientemente, para la esplenectomía en distintas anemias hemolíticas.
- Grace RF, Barcellini W. Management of pyruvate kinase deficiency in children and adults. Blood. 2020; 136: 1241-9.
Revisión y actualización muy reciente del manejo del déficit de piruvato cinasa y nuevos tratamientos.
- Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008; 371: 64-74.
Exhaustiva revisión sobre las bases del déficit de glucosa-6-fosfato deshidrogenasa.
| Caso clínico |
|
Paciente de 7 años que acude a Urgencias por ictericia y coluria de 24 horas de evolución. Sin fiebre, sin ingesta de medicamentos ni otros antecedentes relevantes. A la exploración, estable hemodinámicamente, no adenopatías ni megalias. Auscultación cardiaca taquirrítmica, soplo sistólico en foco aórtico III/IV y auscultación pulmonar sin hallazgos. Pruebas complementarias: Hemograma: leucocitos: 9,52×1.000/μL (4,5-14); neutrófilos: 7,17×1.000/μL; linfocitos: 1,91×1.000/μL; monocitos: 0,35×1.000/μL; Eosinófilos: *0,07×1.000/μL; Basófilos: 0,01×1.000/μL; hematíes: **2,28 mill./μL (3,9-5,3); hemoglobina: **6,6 g/dl (10,7-14,4); hematocrito: **20,7% (34,3-44,3); volumen corpuscular medio: 90,7 fl (75-95); hemoglobina corpuscular media: 29 pg (23,2-30,6); Concentración hemoglobina corp. media: 32,7 g/dl (29-35); índice de distribución eritrocitaria: *15,4% (11,6-15); plaquetas: 352×1.000/μL (150-400); VPM: 7,8 fL (5-10); reticulocitos: **11,53% (0,2-2); reticulocitos absolutos: **265,9×1.000/μL (24-84). Bioquímica sérica: glucosa *108 mg/dL (60-100); urea: 23,6 mg/dL (15,0-39,0); creatinina: 0,35 mg/dL (0,33- 0,73); ácido úrico: 2,47 mg/dL (2-5,1); proteínas totales: 6,3 g/dl (5,7-8); albumina: 4,2 g/dL (3,3-5,2); bilirrubina total: **6,85 mg/dL (0,2-1,3); bilirrubina conjugada: *0,65 mg/dL (0-0,3); lactatodeshidrogenasa (LDH): *624 U/L (110-295); transaminasa GOT: 43 U/L (20-57); transaminasa GPT: 22 U/L (17-43); gamma GT: 10 U/L (8-30); calcio: 9,53 mg/dL (8,8-10,8); calcio corregido por albúmina: 9,34 mg/dL; calcio corregido por proteínas: 10,12 mg/dL; fósforo: 4,3 mg/dL (2,7-5,3); fosfatasa alcalina: 225 U/L (140-351); sodio: 140 mEq/L (135-145); potasio: 3,8 mEq/L (3,4-5,5); cloro: 109 mEq/L (95-111). Proteínas: proteína C reactiva: 0,3 mg/dl (0,01-1); haptoglobinas: **<6 mg/dl (26-185). Test de Coombs directo: positivo (poliespecífico positivo; monoespecífico IgG positivo, C3d negativo). Otras pruebas: - Ecografía abdominal: hígado que impresiona de discretamente aumentado de tamaño. Leve esplenomegalia. - Rx tórax: sin alteraciones radiológicas aparentes. - Ferritina: 382 ng/ml. - B12 y folatos: 214 pg/mL (180-914); 12.53 ng/mL (3.9-23.9). - Hormonas tiroideas: normales. - Estudio de poblaciones linfocitarias e inmunoglobulinas: normal. - Sedimento de orina: hematuria macroscópica. - Otros estudios de autoinmunidad (ANA, FR, lúpico…): negativos.
Con los hallazgos descritos a su ingreso (hemograma, bioquímica y test de Coombs directo), diagnóstico de presunción de AHAI por anticuerpos calientes. Se inicia prednisona 1 mg/kg/día. Durante los primeros días de tratamiento, presenta anemización progresiva, con datos de hemólisis asociadas. Por dicho motivo y ante cuadro clínico de taquicardia y algún mareo ostortático asociado, precisa soporte con hemoderivados. Se transfunde lo mínimo para recuperar estabilidad hemodinámica y de forma lenta. Se completan estudios complementarios analíticos, así como pruebas de imagen para descartar etiologías secundarias como desencadenantes de la AHAI, siendo los resultados de momento negativos, confirmándose la sospecha inicial de anemia hemolítica idiopática por anticuerpos calientes. Alcanza respuesta a la corticoterapia a los 13 días de inicio, se inicia pauta descendente lenta, alcanzando y manteniendo la remisión completa.
|


 Anemia. Classification and diagnosis
Anemia. Classification and diagnosis