 |
| Temas de FC |
M. Alós Díez*![]() , E. Frauca Remacha**
, E. Frauca Remacha**
*Facultativo Especialista de Pediatría. Servicio de Hepatología y Trasplante Hepático Infantil.
**Jefe de Sección de Hepatología y Trasplante Hepático Infantil
Hospital Universitario La Paz. Madrid
| Resumen
Las causas más frecuentes de hepatitis no infecciosa son la hepatitis autoinmune y la tóxica. Su presentación clínica es similar a una hepatitis aguda viral, por lo que es muy importante la sospecha diagnóstica. En la hepatitis autoinmune, el diagnóstico y tratamiento precoz evitan la evolución a cirrosis o la necesidad de trasplante hepático. La afectación hepática por tóxicos o medicamentos supone una de las principales causas dentro del diagnóstico diferencial ante un paciente con disfunción hepática con hipertransaminasemia o síndrome colestásico. En el caso de la hepatitis tóxica, su detección y suspensión del agente causal pueden evitar la progresión del daño hepático y, en casos como la hepatitis por acetaminofeno, permiten instaurar un tratamiento específico. Dada la idiosincrasia de algunos fármacos, requiere un alto índice de sospecha y, muchas veces, el diagnóstico se confirma tras la normalización de la función hepática tras la retirada del fármaco. |
| Abstract
The most common causes of non-infectious hepatitis are: autoimmune and toxic hepatitis. Its clinical presentation is similar to acute viral hepatitis, so diagnostic suspicion is very important. In autoimmune hepatitis, early diagnosis and treatment prevent the progression to cirrhosis or the need for liver transplantation. Liver involvement due to toxins or medications is one of the main causes in the differential diagnosis in a patient with liver dysfunction with hypertransaminasemia or cholestatic syndrome. In the case of toxic hepatitis, its detection and suspension of the causative agent can prevent the progression of liver damage, and in cases such as hepatitis due to acetaminophen, it allows specific treatment to be instituted. Given the idiosyncrasy of some drugs, a high index of suspicion is required and the diagnosis is often confirmed following normalization of liver function after drug withdrawal. |
Palabras clave: Hepatitis autoinmune; Hepatitis tóxica; Enfermedad de Wilson.
Key words: Autoimmune hepatitis; Toxic hepatitis; Wilson’s disease.
Pediatr Integral 2025; XXIX (1): 38 – 48
OBJETIVOS
• Comprender la patogenia de la hepatitis autoinmune, así como las alteraciones analíticas, para poder realizar el diagnóstico y establecer el tratamiento adecuado.
• Actualización del algoritmo diagnóstico de hepatitis autoinmune.
• Conocer las principales enfermedades metabólicas que presentan disfunción hepática.
• Actualización de la sospecha diagnóstica de enfermedad de Wilson, aproximación al diagnóstico y su tratamiento.
• Conocer la existencia de la hepatitis secundaria a fármacos.
Hepatitis autoinmunes, metabólicas y otras hepatitis no infecciosas
https://doi.org/10.63149/j.pedint.6
Introducción
La hepatitis es el cuadro clínico que asocia una elevación de aminotransferasas (AST/ALT) por encima de los rangos de normalidad. En muchas ocasiones, la hipertransaminasemia es el primer hallazgo de una hepatopatía subyacente. Las causas de hipertransaminasemia en la edad pediátrica son múltiples, por lo que el diagnóstico diferencial es amplio. Las causas más frecuentes de hepatitis no infecciosa son la hepatitis autoinmune y la tóxica. El despistaje de ambas entidades junto a otras, como la enfermedad de Wilson o la posibilidad de una enfermedad metabólica subyacente, es prioritario ante un niño con hipertransaminasemia sin proceso reactivo intercurrente asociado. La presentación clínica de estas entidades es similar a una hepatitis aguda viral, por lo que es muy importante la sospecha diagnóstica. El inicio precoz del tratamiento adecuado permite un buen control y una buena supervivencia a largo plazo en el caso de la hepatitis autoinmune y en la enfermedad de Wilson. En el caso de la hepatitis tóxica, la sospecha diagnóstica es esencial para la retirada del fármaco, si es posible(1-3).
Hepatitis autoinmune
La hepatitis autoinmune (HAI) es una enfermedad inflamatoria hepática de etiología autoinmune; sin tratamiento, avanza a cirrosis e insuficiencia hepática. El despistaje de HAI es prioritario ante un niño con hipertransaminasemia. El inicio precoz del tratamiento adecuado permite un buen control y una buena supervivencia a largo plazo.
La incidencia de las hepatopatías autoinmunes está aumentando en los últimos años, principalmente en las sociedades desarrolladas. Suelen tener un curso crónico, pudiendo asociar brotes de actividad inflamatoria que generan daño hepatocitario. Según el autoanticuerpo detectado, se clasifican en: HAI tipo I (ANA o AML) o HAI tipo II (LKM-1 o LC-1) (ANA: anticuerpos antinucleares; AML: anticuerpos antimúsculo liso; LKM: anticuerpos antimicrosomales tipo 1 de hígado y riñón; LC-1: anticuerpo anticitosol hepático tipo 1). Existe la hepatitis autoinmune seronegativa, también llamada HAI tipo III (presencia de disfunción hepática, hipergammaglobulinemia, datos en biopsia compatibles, pero autoanticuerpos negativos). Dada la posibilidad de solapamiento (“overlap”) con una colangitis esclerosante autoinmune, se aconseja realizar colangiografía a todos los pacientes(4,5).
Epidemiología
Su incidencia anual en Europa puede variar de 0,7-2/100.000 habitantes/año, mientras que su prevalencia es de 4-25/100.000.
La incidencia es mayor en el sexo femenino (4:1) y se presenta en todas las edades y grupos étnicos.
En la actualidad, su diagnóstico es más frecuente debido a un aumento real de su incidencia y al mayor índice de sospecha diagnóstica.
Fisiopatología
El mecanismo por el que se induce el daño hepático continúa siendo una incógnita. La hipótesis principal sugiere que la enfermedad se inicia tras un desencadenante ambiental, en un paciente genéticamente predispuesto, provocando una respuesta inmunitaria disregulada.
La lesión hepática se inicia y perpetúa por una respuesta inmune frente a autoantígenos hepáticos con fallo en los mecanismos inmunorreguladores. En la lesión hepática están implicados autoantígenos, el complejo mayor de histocompatibilidad y los receptores α/β de los linfocitos T. El reclutamiento masivo de células proinflamatorias, mayoritariamente linfocitos T CD4 helper, seguidos por CD8, células “natural killer” (NK), macrófagos, linfocitos B y células plasmáticas, reconoce dicho autoantígeno induciendo un efecto citotóxico en cascada.
El péptido del autoantígeno, abrazado por una molécula HLA clase II, es presentado por una célula presentadora de antígeno a un CD4 T helper naive (Th0), el cual se activa y desencadena una cascada de reacciones inmunes determinadas principalmente por las citoquinas: interleuquina 2 (IL-2) e interferón γ, las cuales provocarán el daño celular. Otras citoquinas implicadas en la citolisis de los hepatocitos son: la IL-1, el factor de necrosis tumoral α, la IL-4, IL-10, IL-13, IL-17, IL-22 y el ligando de quimoquina CCL-20. Se conoce que existe un defecto de la inmunorregulación que afecta a las células T reguladoras, particularmente en el debut y durante los brotes de la enfermedad. Entre los factores ambientales desencadenantes, se han propuesto fármacos, virus (VHA, SARS-CoV2, sarampión, VEB) o la vacunación.
La mayor comprensión de los mecanismos involucrados en la patogénesis contribuirá al desarrollo de nuevos tratamientos, como la transferencia de linfocitos T reguladores autólogos expandidos y específicos de antígeno, para restaurar la tolerancia a antígenos hepáticos(5).
Genética
La hepatitis autoinmune tiene una asociación muy sólida con el locus (HLA)-DRB1 del antígeno leucocitario humano. Se sugiere que los pacientes con HAI tipo 1 y 2 tienen diferente base genética. La HAI tipo I se asocia al HLA DRB1*03, y también el HLA DRB1*04 predispone a dicha enfermedad en la población adulta, pero no en la edad pediátrica. La HAI tipo 2 se asocia al DRB1*07.
La hepatitis autoinmune tipo 2 puede formar parte de un desorden autosómico recesivo monogénico llamado síndrome de poliendocrinopatía autoinmune tipo 1 (poliendocrinopatía autoinmune, candidiasis crónica y distrofia ectodérmica)(5).
Criterios diagnósticos de hepatitis autoinmune (Tablas I y II)


Requiere elevación de transaminasas y de IgG, autoanticuerpos positivos y criterios histológicos en la biopsia (hepatitis de interfase), junto con la exclusión de otras enfermedades que recuerdan a la hepatitis autoinmune, como la enfermedad de Wilson, déficit de alfa-1 antitripsina, infección viral (virus de la hepatitis A, B, C, VEB) y fármacos(6).
Clínica
Se debe sospechar en un niño con síntomas agudos o crónicos de hepatitis en ausencia de otras etiologías y, sobre todo, si en él o en sus familiares coexisten enfermedades autoinmunes.
La mayoría de los niños presentan al inicio una clínica similar a una hepatitis aguda ictérica prolongada y un 10 % (sobre todo, pacientes LKM1 y menores de 2 años) debutan como fallo hepático agudo. El resto inician una clínica insidiosa de enfermedad hepática o, un pequeño porcentaje, se diagnostican de forma casual estando asintomáticos o en el seguimiento de una hipertransaminasemia.
Los antecedentes familiares de enfermedades autoinmunes o la asociación en el mismo paciente de tiroiditis autoinmune, enfermedad inflamatoria intestinal, artritis reumatoide, celiaquía, vitíligo, déficit de IgA (40 % de las HAI tipo 2) o diabetes mellitus tipo 1 deben hacer sospechar HAI. Hasta un 40 % de los niños con HAI pueden tener enfermedad autoinmune extrahepática.
Excluidos los niños con debut como fallo hepático, la clínica es similar en la tipo 1 y en la tipo 2. La exploración física es normal en un 25 % de los pacientes. La hepatomegalia es el hallazgo más frecuente; la esplenomegalia puede indicar la existencia de una hepatopatía crónica. En la exploración física de los pacientes se puede apreciar ictericia. En los casos evolucionados se pueden apreciar estigmas de hepatopatía crónica(6,7).
Bioquímica
Puede existir cualquier grado de elevación de transaminasas (AST/ALT), con o sin elevación de otros marcadores, como la fosfatasa alcalina o la gamma glutamil-transferasa (GGT), y puede acompañarse de colestasis. La concentración de gammaglobulinas séricas o IgG total suele ser mayor de 1,5 veces su límite superior. Hasta un 20 % de niños no muestran elevación de gammaglobulinas.
El hallazgo más frecuente es la elevación de AST y ALT. La elevación de gamma glutamil-transferasa (GGT), bilirrubina y fosfatasa alcalina (FA) suele ser menos marcada que en otras hepatopatías. En caso de que los marcadores de colestasis estén muy elevados, considerar la posibilidad de síndrome de solapamiento. Es característica la elevación de gammaglobulinas séricas a expensas de IgG.
Los niveles de cobre, ceruloplasmina y alfa-1 antitripsina, así como las serologías de virus hepatotropos, deben ser normales.
Otras alteraciones bioquímicas, como el INR (índice internacional normalizado del tiempo de protrombina) alargado o la hipoalbuminemia, orientan a insuficiencia hepática(3,6,7).
Autoanticuerpos
Seropositividad de ANA, AML a título mayor de 1:20 y de LKM y anti-LC1 mayor de 1:10 en niños, mediante inmunofluorescencia indirecta en sustrato fresco de roedores, que incluye riñón, hígado y estómago.
Los autoanticuerpos más típicos son los ANA, antimúsculo liso (AML) y anti-LKM y anti-LC1.
Los autoanticuerpos no son patognomónicos ni específicos de la enfermedad y su expresión puede variar en el curso de la hepatitis autoinmune. Un título bajo no excluye el diagnóstico ni uno alto lo confirma en ausencia de otros datos.
En niños se ha demostrado, en la evolución de la hepatitis autoinmune, correlación entre el grado de actividad de la enfermedad y el nivel de IgG y autoanticuerpos(1,2,6,7).
Subclasificaciones
Hepatitis autoinmune tipo 1
Se asocia con ANA y anti-músculo liso (AML) positivo. Los ANA son poco específicos de HAI, porque también se elevan en otras entidades sistémicas de base autoinmune, en niños obesos y, en ocasiones, en hepatitis reactivas a procesos intercurrentes.
Un 30 % presentan AML, la mitad como único marcador serológico y la otra mitad junto a los ANA.
Hepatitis autoinmune tipo 2
La presencia de anti-LKM1 y/o de anti-LC1 define la HAI tipo 2. Los anti-LKM también se pueden encontrar en infección crónica por VHC.
Hepatitis autoinmune tipo 3 o seronegativa
La prevalencia de enfermedad celiaca en niños con HAI es del 19 % y, muchas veces, es seronegativa. Se trata de niños con alteración en la bioquímica hepática, niveles elevados de IgG y daños histológicos compatibles con HAI, habiendo excluido otras causas de disfunción hepática, pero sin positividad en los autoanticuerpos.
En todos los tipos de HAI se debe hacer cribado de enfermedad inflamatoria intestinal (EII) mediante calprotectina fecal y de posible síndrome de overlap mediante colangio-RMN(6).
Histología
La diana de la lesión es el hepatocito. Por tanto, existirá hepatitis con actividad moderada o grave con necrosis periportal, conocida como hepatitis de interfase, con o sin lesión lobular o necrosis en puentes centro-portal, siendo el infiltrado portal predominantemente linfoplasmocitario.
La hepatitis de interfase es el hallazgo típico de la hepatitis autoinmune, si bien no es patognomónico de la misma. Consiste en una infiltración densa de la placa limitante compuesta de linfocitos y células plasmáticas. En las formas agudas y durante los periodos de enfermedad activa, se puede encontrar una hepatitis panlobular con necrosis en puente. Otros datos sugestivos son las células de Kupffer y la formación de hepatocitos en rosetas. Si el daño en los conductos biliares es llamativo, es importante descartar un síndrome de solapamiento.
No deben existir lesiones biliares, granulomas, depósitos de cobre u otros cambios que sugieran otra etiología. Los hallazgos de ductopenia o colangitis destructiva añadida pueden apoyar el diagnóstico de colangitis esclerosante autoinmune descrito en niños (“overlap”). En niños con hepatitis autoinmune, principalmente HAI tipo 1, se recomienda realizar colangiorresonancia magnética para descartar síndrome de solapamiento. La presencia de sobrecarga de hierro o esteatosis sugiere diagnósticos alternativos, como hepatitis C, enfermedad de Wilson o toxicidad por fármacos. Existe algún grado de fibrosis en prácticamente todos los pacientes. La histología inicial es de cirrosis en la mitad de los niños (Tabla III)(6,8).

Tratamiento
El objetivo de tratamiento de las HAI es prevenir la progresión de la enfermedad y obtener la remisión completa (estimada en >80 %); esto es, ausencia de síntomas, normalidad de transaminasas y mejoría de la inflamación histológica. El 50 % de los casos consigue además normalizar niveles de IgG y negativización o títulos bajos de anticuerpos.
El tratamiento se debe iniciar en cuanto se establezca el diagnóstico. Una de las características de la hepatitis autoinmune es que la gran mayoría responde a la terapia inmunosupresora.
La pauta convencional se basa en metilprednisolona con o sin azatioprina (v. Algoritmo al final del artículo). Se inicia a 2 mg/kg/día (máx. 60 mg/día)
con descenso gradual durante 6-8 semanas hasta dosis de mantenimiento (0,1-0,2 mg/kg/día o 2-4 mg/día). La azatioprina (1-2 mg/kg/día, máx. 100 mg/día), como ahorrador de corticoide, se añade tras la mejoría bioquímica inicial. Se debe monitorizar la evolución bioquímica de la HAI estrechamente para ajustar el corticoide, así como vigilar los posibles efectos adversos de la azatioprina (citopenias o pancreatitis bioquímica) (Tabla IV)(1,2,6).
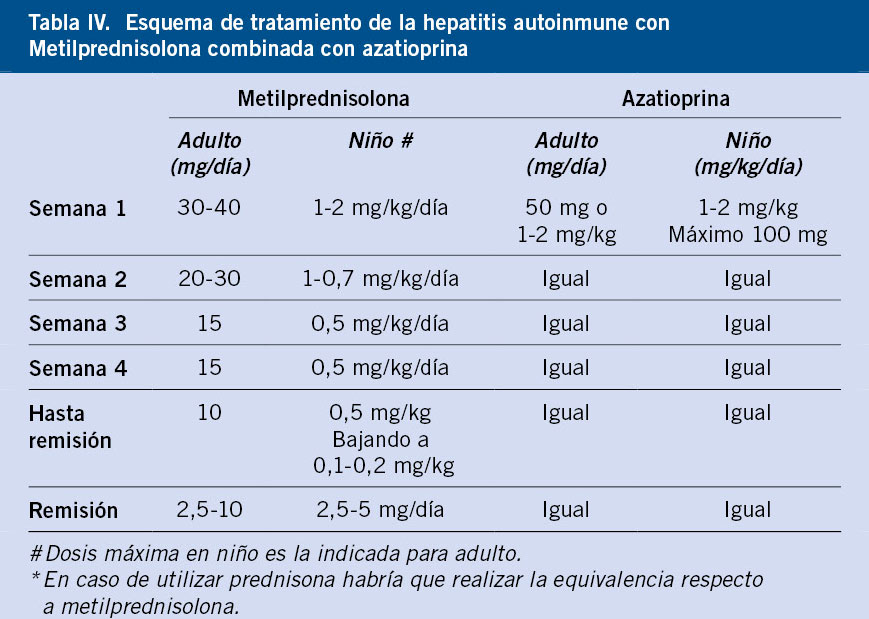
El tratamiento es indefinido dada la alta tasa de recaídas tras su retirada. Solo en una minoría de pacientes con rigurosa normalización clínica, bioquímica e histológica, se considera monoterapia con azatioprina, y un porcentaje bajo permite la retirada completa a largo plazo del tratamiento. Solo se puede plantear la retirada de tratamiento en casos seleccionados, nunca antes de los 2-3 años de haberlo iniciado y siempre que mantengan función hepática normal, IgG en rango y autoanticuerpos negativos o débilmente positivos, al menos, durante un año. Un 10 % de pacientes con hepatopatía muy evolucionada al diagnóstico requiere trasplante hepático evolutivamente.
Para el manejo de las recaídas, se aumentará de forma transitoria la dosis de metilprednisolona con posterior disminución progresiva, tras evidenciar mejoría del perfil hepático, hasta llegar a la dosis de mantenimiento.
Existen tratamientos de segunda línea para los fallos de inducción a la remisión como para los casos con recaídas frecuentes. Estos fármacos son, por orden de frecuencia: el micofenolato mofetil (MMF), seguido de los inhibidores calcineurínicos (tacrólimus)(9).
• Respuesta completa: supone la desaparición de los síntomas y la normalización de la función hepática con gammaglobulinas en rango y negativización de los anticuerpos (ANA y AML <1:20, LKM y LC1 <1:10) durante el primer año tras el diagnóstico. Se considera, además, que dicha respuesta es mantenida si persiste durante, al menos, 6 meses con terapia de mantenimiento.
• Recaída: es el restablecimiento de la actividad de la enfermedad después de la inducción de remisión, bien al suspender el tratamiento o bien al intentar reducirlo. Ocurre hasta en el 40 % de los casos y los pacientes con recaída muestran mayor riesgo de progresión a cirrosis.
• Fallo del tratamiento: es el empeoramiento clínico o de laboratorio a pesar del tratamiento. Se produce en menos del 10 % de los niños. La respuesta incompleta se caracteriza por mejoría de la clínica, la función hepática e histología, pero sin llegar a la remisión. Estos casos son indicación de fármacos de segunda línea(2,8).
Enfermedad de Wilson
La enfermedad de Wilson (EW) es un trastorno autosómico recesivo (gen ATP7B) que condiciona un acúmulo progresivo de cobre en el hígado con extensión secundaria a otros tejidos. En la infancia predomina la clínica hepática, más frecuentemente en forma de disfunción leve (hipertransaminasemia) asintomática. El tratamiento con quelantes de cobre o sales de zinc normaliza la función hepática en los pacientes sin cirrosis.
La enfermedad de Wilson (EW) es un trastorno autosómico recesivo (gen ATP7B) que condiciona un acúmulo progresivo de cobre en el hígado con extensión secundaria a otros tejidos. La prevalencia es de 1/30.000 personas. La instauración de un tratamiento precoz es vital porque mejora el pronóstico, por lo que siempre debe estar en el diagnóstico diferencial de una disfunción hepática(3,10,11).
Clínica
La forma de presentación es muy heterogénea. En la infancia predomina la clínica hepática en forma de disfunción leve (hipertransaminasemia en ausencia de síntomas), hepatopatía crónica (cirrosis, hepatomegalia, ascitis, varices esofágicas, ictericia) o fallo hepático agudo (asociado con frecuencia a hemólisis). En paralelo a la lesión hepática pueden aparecer síntomas neurológicos (disfunción extrapiramidal y cerebelosa), anillo de Kayser-Fleischer, anemia hemolítica (Coombs directo negativo), tubulopatía por cobre, etc.(10,11).
Diagnóstico
El diagnóstico requiere de un alto índice de sospecha. Se debe solicitar ceruloplasmina ante cualquier disfunción hepática. El estudio genético es una herramienta fundamental para confirmar el diagnóstico.
El diagnóstico requiere de un alto índice de sospecha. Se debe solicitar ceruloplasmina ante cualquier disfunción hepática en niños mayores de 1 año. La ceruloplasmina baja (<20 mg/dL) es el test de mayor sensibilidad para el diagnóstico de sospecha. Otros hallazgos diagnósticos dependen del estadio avanzado de la enfermedad, como el cobre en orina de 24 horas elevado (>40-100 µg/día) y el cobre en suero. Los niños presintomáticos no suelen tener cupruria elevada. La biopsia hepática presenta datos de inflamación, fibrosis y esteatosis no específicos. El cobre en tejido hepático es la base del diagnóstico de certeza y requiere que el paciente no tenga colestasis; ya que, independientemente de la etiología, la colestasis aumenta el cobre intrahepático. Se obtienen dos cilindros conservados en fresco, procesados inmediatamente o congelados a –20 °C. El valor normal es <50 µg/g de tejido seco y el valor característico de EW es >250 µg/g. De esta forma, la biopsia sirve como criterio diagnóstico y del grado de severidad de la enfermedad (fibrosis)(11).
El estudio genético es una herramienta fundamental para el diagnóstico: variantes patogénicas en ambos alelos del gen ATP7B confirman el diagnóstico. El hecho de que no todas las posibles variantes asociadas a la enfermedad sean conocidas actualmente supone que en algunos pacientes con EW no se identifiquen. Además, permite el despistaje de la enfermedad en los familiares de primer grado(10,11).
Tratamiento
El tratamiento normaliza la función hepática en los pacientes sin cirrosis. Se utilizan quelantes de cobre en los casos sintomáticos y quelantes de cobre o sales de zinc (disminuyen la absorción intestinal de cobre) en los asintomáticos.
El tratamiento en la enfermedad de Wilson (EW) es de por vida y su objetivo es normalizar la función hepática en los pacientes sin cirrosis. En pacientes cirróticos con insuficiencia hepática al diagnóstico (tiempo de protrombina <50 %), el tratamiento consigue revertir la insuficiencia en el 50 % de los casos.
Se utilizan quelantes de cobre en los casos sintomáticos y quelantes de cobre o sales de zinc en los asintomáticos:
• Quelantes de cobre: D-penicilamina (20 mg/kg/día en 2-3 dosis) o trientina (20 mg/kg/día en 2-3 dosis). Vigilar efectos secundarios de D-penicilamina (exantema, citopenias, lupus, síndrome nefrótico) que indicarían cambio de tratamiento a trientina. Control de cumplimiento: cupruria de 24 horas elevada (200-500 µg/día).
• Sales de zinc (<50 kg, 75 mg/día en 3 dosis; >50 kg, 150 mg/día de zinc elemento). Control de cumplimiento: cupruria normal con zinc elevado en sangre y orina.
Se aconseja moderar el consumo de alimentos que contienen cobre (chocolate, frutos secos, moluscos, setas). Los pacientes requieren controles clínicos y analíticos cada tres-seis meses.
Los pacientes con hepatopatía avanzada que no responden a tratamiento médico y aquellos que debutan con fallo hepático (“Wilson fulminante”) requieren trasplante hepático(10,11).
Hepatitis metabólica (Tabla V)(12)

Ante el proceso diagnóstico de un paciente con disfunción hepática por una enfermedad metabólica, resulta útil partir de la alteración analítica inicial o predominante para establecer el diagnóstico diferencial.
Parámetros a valorar ante una sospecha de hepatitis metabólica (12):
• Glucemia.
• Gasometría venosa o arterial.
• Lactato.
• Cetonemia.
• Amonio.
• Aminoácidos en plasma y ácidos orgánicos en orina.
Hepatitis tóxica
Introducción
Una de las funciones primarias del hígado es la biotransformación de fármacos, suplementos y productos de herbolario en compuestos que puedan ser metabolizados y excretados de forma segura por el organismo. Cuando esto no sucede así, se produce un daño hepático por tóxicos que puede ser predecible y dosis dependiente, como es el caso del acetaminofeno (la intoxicación por paracetamol) o idiosincrásico, no dosis dependiente. Estos últimos casos requieren de un alto índice de sospecha para realizar el diagnóstico y retirar el agente causal.
La afectación hepática por tóxicos o medicamentos supone una de las principales causas dentro del diagnóstico diferencial al evaluar a un paciente con hipertransaminasemia o síndrome colestásico. Su incidencia real en Pediatría se desconoce, ya que muchos casos son asintomáticos(1,2,13,14).
Fisiopatología
El concepto de hepatitis tóxica engloba cualquier grado de afectación hepática, desde la elevación asintomática de transaminasas hasta el fallo hepático agudo.
El hígado metaboliza y excreta los fármacos en tres pasos:
• Fase 1 activación: el citocromo P450 inserta un residuo de oxígeno en la medicación, haciéndola más hidrosoluble y a la vez más tóxica.
• Fase 2 detoxificación: mediante conjugación del metabolito, aumenta la hidrosolubilidad y se neutraliza la toxicidad.
• Fase 3: excreción del producto hidrosoluble, se transporta al canalículo y se segrega a la bilis.
La mayoría de las veces, la hepatotoxicidad se produce por acumulación de metabolitos de fase 1, siguiendo un patrón de toxicidad intrínseca o idiosincrásica.
Desde un punto de vista conceptual, se pueden establecer tres categorías en las hepatitis tóxicas.
Los fármacos con hepatotoxicidad intrínseca causan daño hepático previsible, dosis dependiente y con periodos de latencia cortos (horas, días). El paradigma es el paracetamol (acetaminofeno) que se convierte por medio de las monooxigenasas del CYP 450 en NAPQI (N-acetil-p benzoquinona imina), que es neutralizada por el glutatión. En grandes dosis, si se sobrepasa la reserva de glutatión, la NAPQI daña los hepatocitos. La toxicidad del acetaminofeno puede ser revertida por la N-acetilcisteína que restaura la producción de glutatión(15).
La mayoría del resto de fármacos causa hepatotoxicidad de forma idiosincrásica, no dosis dependiente, con un periodo de latencia variable (días, semanas). Suele deberse a variabilidad individual de las fases 1 y 2 como resultado de interacciones complejas por factores genéticos, edad, sexo, hormonas, estado nutricional y patología hepática previa (podría aumentar la toxicidad de fármacos debido a la disminución de su metabolismo y aclaramiento) con factores adquiridos (polimedicación, alcohol y tabaco) y respuesta de adaptación inmune asociado a HLA. Es menos frecuente que la intrínseca, pero son las que presentan la mayoría de fármacos(16).
Más del 50 % de los fármacos que producen hepatotoxicidad es por causar disfunción mitocondrial. Los niños con mutaciones de enzimas mitocondriales tienen mayor susceptibilidad a este tipo de daño hepático. Los polimorfismos del gen POLG (gen gamma de la DNA polimerasa mitocondrial) se han asociado a fallo hepático por valproato.
La toxicidad idiosincrásica puede ser causada también por un mecanismo inmunoalérgico que asemeja a un daño autoinmune. Es el caso de minociclina, metildopa, diclofenaco, azitromicina, amoxicilina, valproato, fenitoína, lamotrigina e isoniazida. En estos casos, el metabolito de fase 1 se ligaría a proteínas del hepatocito, formando neoantígenos que, en algunos individuos, desencadenarán una respuesta inmune. Suelen tener un periodo corto de latencia, pueden detectarse autoanticuerpos (64 % en Pediatría) y, en el caso de la reintroducción del fármaco, la sintomatología reaparece de forma precoz. Es difícil de diferenciar de la hepatitis autoinmune. La ausencia de reactivación tras la retirada de la medicación inmunosupresora descarta la hepatitis autoinmune clásica.
En Pediatría, el principal grupo de fármacos causante de hepatitis tóxica son los antibióticos (50 % de los casos), seguido de los fármacos para el sistema nervioso central (antiepilépticos, medicación para el TDAH). Como fármaco aislado, el más frecuente es la minociclina, empleada para el tratamiento del acné (13 % casos)(13,14).
Clínica
El tiempo de latencia desde que se tomó la medicación y los síntomas es difícil de precisar; la toxicidad puede ocurrir incluso semanas después de suspender el fármaco.
Establecer un diagnóstico de certeza es complicado. Los pacientes pueden tener un curso subclínico con solo alteraciones de laboratorio. En muchas ocasiones, se trata de un diagnóstico de exclusión. En la evaluación clínica de un paciente con hipertransaminasemia o colestasis, realizar una anamnesis detallada que recoja la ingesta de medicación, productos de herboristería y suplementos dietéticos. Se puede encontrar información actualizada de la toxicidad hepática de los diferentes fármacos en la web LiverTox: https://www.ncbi.nlm.nih.gov/books/NBK547852/.
Cuando el daño es fundamentalmente de los hepatocitos, la clínica es muy similar a una hepatitis aguda viral con náuseas, vómitos, anorexia y aumento de transaminasas. Lo más frecuente es la afectación mixta de hepatocitos y colangiocitos, con clínica de hepatitis y síntomas colestáticos (ictericia y prurito). La enfermedad puede progresar a fallo hepático agudo. Algunos fármacos ocasionan importante daño endotelial y producen enfermedad venoclusiva con ictericia, hepatomegalia y ascitis. Cuando los metabolitos inducen una reacción inmunoalérgica, puede haber síntomas acompañantes de fiebre, rash, artralgias y edema facial(13,14,16).
Diagnóstico
El diagnóstico de hepatotoxicidad es a menudo de exclusión, basado en la historia clínica, presentación, laboratorio y evolución.
Se debe descartar: hepatitis virales, esteatohepatitis no alcohólica, hepatitis autoinmune, enfermedad de Wilson, causa metabólica y enfermedad mitocondrial; y en los casos que predominan los síntomas colestáticos: colangitis esclerosante y enfermedad pancreático-biliar.
En la mayoría de ocasiones, el diagnóstico se confirma con la normalización de la función hepática tras la retirada del fármaco o tóxico.
El papel de la biopsia hepática para establecer el diagnóstico es discutido; se reserva para casos seleccionados, principalmente orientada a descartar otras causas de enfermedad hepática, ya que no hay hallazgos patognomónicos de hepatotoxicidad. Se recomienda realizar biopsia en aquellos en los que no haya disminuido un 50 % la cifra de transaminasas en 1-2 meses, o un 50 % la cifra de bilirrubina en los 6 meses siguientes a la retirada del tóxico. Otra excepción sería para establecer el diagnóstico diferencial con una hepatitis autoinmune clásica. Es común observar necrosis hepatocelular, muchas veces acompañada de esteatosis con infiltrado de eosinófilos, en ocasiones prominente(17).
Tratamiento
Lo más importante del tratamiento es la retirada causal y el tratamiento de soporte. Solo es eficaz el tratamiento con N-acetilcisteína en el caso del paracetamol y la L-carnitina para el valproato.
Se ha probado el tratamiento empírico con corticoides en pauta corta, habitualmente 6 meses, en casos de colestasis progresiva y en curso clínico similar a una hepatitis autoinmune, pero la eficacia no ha sido probada. No se dispone de suficientes datos que avalen el uso de ácido ursodeoxicólico en los pacientes con patrón colestático.
En algunos casos se produce descompensación aguda, siendo causante de fallo hepático agudo en niños, sobre todo causado por paracetamol (acetaminofeno). Si se producen datos de fallo hepático (coagulopatía, encefalopatía o hipoglucemia), los niños deben ser remitidos rápidamente a un centro donde se realice trasplante hepático(13,14).
En otros casos, la evolución es hacia una enfermedad hepática crónica, a pesar de la retirada del agente tóxico. En el resto de casos, se produce recuperación en un periodo de días a meses(18).
En todos los pacientes con hepatitis tóxica, tanto idiosincrásica como intrínseca, se debe monitorizar la función hepática y el seguimiento clínico tras su retirada hasta la normalización de los valores analíticos y la desaparición de la sintomatología clínica. Evitar la reintroducción del fármaco sospechoso salvo no disponer de terapia alternativa eficaz en caso de riesgo vital y a menor dosis. La reintroducción del fármaco puede generar hepatotoxicidad de forma más precoz y grave, con mayor riesgo de fallo hepático agudo (Tabla VI)(18).

Función del pediatra de Atención Primaria
Se debe tener en cuenta cualquier grado de elevación de transaminasas como posible indicador de enfermedad hepática. Es importante el seguimiento del paciente ante la detección de una elevación de transaminasas por cualquier contexto, hasta la resolución o el diagnóstico de la causa por parte de su pediatra de Atención Primaria. Es importante recordar que la hipertransaminasemia puede ser la manifestación clínica inicial de una hepatopatía crónica no diagnosticada, como una hepatitis autoinmune o una enfermedad de Wilson, si persiste la hipertransaminasemia en el control evolutivo. Se debe remitir al especialista para realizar un estudio etiológico de disfunción hepática con ceruloplasmina, inmunoglobulinas, y autoanticuerpos. En la mayoría de las ocasiones, el pediatra de Atención Primaria es el primer contacto del niño con el sistema sanitario para poder detectar enfermedades hepáticas subyacentes y signos de alarma, así como manejo multidisciplinar con el especialista para detectar posibles brotes de enfermedad de base y complicaciones y monitorizar la adherencia al tratamiento.
Conflicto de intereses
No hay conflicto de interés en la elaboración del presente manuscrito ni fuente de financiación.
Bibliografía
Los asteriscos muestran el interés del artículo a juicio de los autores.
1.** Marugán de Miguelsanz JM, Torres Hinojal MC, Alonso Vicente C. Aproximación diagnóstica al paciente con enfermedad hepática. Pediatr Integral. 2020; XXIV: 6-14. Disponible en: https://www.pediatriaintegral.es/publicacion-2020-01/aproximacion-diagnostica-al-paciente-con-enfermedad-hepatica/.
2.** Ros Arnal I, Reyes Andrade J, Mercadal Hally M, Blesa Baviera LC, García Tirado D, Campuzano Martín SH, et al. Actuación diagnóstica ante hipertransaminasemia en pediatría: documento de consenso de Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP), Asociación Española de Pediatría de Atención Primaria (AEPap) y Sociedad Española de Pediatría de Atención Primaria (SEPEAP). Anales de Pediatría. 2022; 96: 448.e1-e11. Disponible en: https://doi.org/10.1016/j.anpedi.2022.04.002.
3.** Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver disease in children. 4th ed. Cambridge University Press. 2014.
4. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol. 2015; 63: 971-1004.
5. Camarena Grande C. Hepatitis no infecciosas. Pediatr Integral. 2020; XXIV: 28-37. Disponible en: https://www.pediatriaintegral.es/publicacion-2020-01/hepatitis-no-infecciosas/.
6.** Mieli-Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Statement. J Pediatr Gastroenterol Nutr. 2018; 66: 345-60. Disponible en: https://doi.org/10.1097/mpg.0000000000001801.
7. Mack CL, Adams D, Assis DN, Kerkar N, Manns MP, Mayo MJ, et al. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines from the American Association for the Study of Liver Diseases. Hepatology. 2020; 72: 671-722. Disponible en: https://doi.org/10.1002/hep.31065.
8.** Samyn M, Indolfi G, Vergani D, Mieli-Vergani G; Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. Liver Biopsy is Indicated before Attemting Treatment Withdrawal in Children with AIH: Commentary by the ESPGHAN HepCom. J Pediatr Gastroenterol Nutr. 2023; 77: e63-e64. Disponible en: https://doi.org/10.1097/mpg.0000000000003858.
9. Lohse AW, Sebode M, Jorgensen MH, Ytting H, Karlsen TH, Kelly D, et al; European Reference Network on Hepatological Disease (ERN RARE-LIVER): International Autoimmune Hepatitis Group (IAIHG). Second-line and third-line for autoimmune hepatitis: A position statement from the European Reference Network on Hepatological Diseases and the International Autoimmune Hepatitis Group. Hepatol. 2020; 73: 1496-1506. Disponible en: https://doi.org/10.1016/j.jhep.2020.07.023.
10. Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology; 2008; 47: 2089-111.
11.** Socha P, Janczyk W, Dhawan A, Baumann U, D’Antiga L, Tanner S, et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018; 66: 334-44. Disponible en: https://doi.org/10.1097/mpg.0000000000001787.
12. Jara P, Díaz MC, Hierro L, Camarena C, de la Vega Á, Frauca E, eds. Enfermedad hepática en el niño. Madrid: Tile Von Spain. 2014.
13.** Amin MN, Harpavat S, Leung DH. Drug-induced liver injury in children. Curr Opin Pediatr. 2015; 27: 625-33.
14. European Association for the Study of the Liver; Clinical Practice Guideline Panel: Chair; Panel members; EASL Governing Board representative. EASL Clinical Practice Guidelines: Drug-induced liver injury. J Hepatol. 2019; 70: 1222-61.
15. Shi Q, Yang X, Greenshaw JJ, Salminen AT, Russotti GM, Salminen WF. Drug-Induced liver injury in children: Clinical observations, animal models, and regulatory status. Int J Toxicol. 2017; 36: 365-79.
16. Molleston JP, Fontana RJ, López MJ, Kleiner DE, Gu J, Chalasani N. Characteristics of idiosyncratic drug-induced liver injury in children: Results from the DILIN prospective study. J Ped Gastroenterol Nutr. 2011; 53: 182-9.
17. Amin MD, Harpavat S, Leung DH. Drug-induced liver injury in children. Curr Opin Pediatr. 2015; 27: 625-33.
18. Zhu Y, Li Y, Wang J, Liu S, Wang L, Zhao Y, et al. Causes, features, and outcomes of drug-induced liver injury in 69 children from China. Gut Liver. 2015; 9: 525-33.
Bibliografía recomendada
– Mieli-Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Statement. J Pediatr Gastroenterol Nutr. 2018; 66: 345-60. Disponible en: https://doi.org/10.1097/mpg.0000000000001801.
Artículo de posicionamiento realizado por un grupo de expertos y los miembros del Comité de Hepatología de la Sociedad Europea de Gastroenterología, Hepatología y Nutrición Pediátrica (ESPGHAN), centrado en el diagnóstico, tratamiento y seguimiento de la enfermedad hepática autoinmune en el niño. Se revisan los artículos publicados en los últimos 30 años.
– Mack CL, Adams D, Assis DN, Kerkar N, Manns MP, Mayo MJ, et al. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines from the American Association for the Study of Liver Diseases. Hepatology. 2020; 72: 671-722. Disponible en: https://doi.org/10.1002/hep.31065.
Actualización de las guías clínicas de algoritmo diagnóstico y tratamiento en hepatitis autoinmune de la Sociedad Americana de Estudio de Enfermedades Hepáticas (AASLD).
– Socha P, Janczyk W, Dhawan A, Baumann U, D’Antiga L, Tanner S, et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018; 66: 334-44. Disponible en: https://doi.org/10.1097/mpg.0000000000001787.
Artículo de posicionamiento realizado por un grupo de expertos y los miembros del Comité de Hepatología de la Sociedad Europea de Gastroenterología, Hepatología y Nutrición Pediátrica (ESPGHAN), centrado en el diagnóstico, tratamiento y seguimiento de la enfermedad de Wilson en el niño. Se revisan los artículos publicados en los últimos 25 años.
– Amin MD, Harpavat S, Leung DH. Drug-induced liver injury in children. Curr Opin Pediatr. 2015; 27: 625-33.
Revisión de la etiología, epidemiología, fisiopatología y tratamiento de la hepatitis tóxica en niños.
| Caso clínico |
|
Niña de 12 años que consulta por ictericia conjuntival de 24 horas de evolución junto a cuadro de astenia de 1 mes de evolución con náuseas y dolor abdominal asociado. Afebril, deposiciones con color. No ha tomado paracetamol ni otros medicamentos. Sin ingesta de productos de herbolario. Antecedentes familiares: madre con enfermedad de Graves y hermano con diabetes mellitus tipo 1. Sin ambiente epidémico familiar. Exploración física: buen estado general. Ictericia generalizada. Bien nutrida. Sin circulación colateral ni otros datos de hepatopatía. Auscultación cardiopulmonar: rítmica, sin soplos. Buena entrada de aire bilateral. Abdomen: blando, depresible, no doloroso, sin datos de irritación peritoneal. Hepatomegalia: 1-2 traveses. No se palpa bazo. Se extrae analítica sanguínea y ecografía abdominal (con 4 horas de ayuno): analítica de sangre: hemoglobina: 12,6 g/dL; Ecografía abdominal: hígado con aumento de ecogenicidad periportal, edema de pared vesicular y adenopatías en hilio hepático, todo ello en relación con cuadro de hepatitis. No se visualiza dilatación de vía biliar. Sin datos de hepatopatía. Se trata de una hepatitis ictérica sin datos de insuficiencia hepática, dado que el tiempo de protrombina (TP) mejora tras la administración de vitamina K. Las pruebas de laboratorio para estudio de disfunción hepática han resultado normales, salvo IgG 2.790 mg/dL con ANA + (1/160) y AML (1/320) y las serologías virales negativas. Se realiza biopsia hepática, cuyo resultado muestra lesiones histológicas características de autoinmunidad (hepatitis crónica activa con infiltrado portal de predominio linfocitario, presencia de células plasmáticas, rosetas y presencia de células inflamatorias [eosinófilos y neutrófilos]) y excluye otras patologías (ausencia de depósitos de cobre, no gránulos compatibles con alfa-1 antitripsina, no colestasis); además, no aporta datos compatibles con hepatopatía evolucionada (grado de fibrosis o cirrosis). Ante los resultados de la biopsia, se establece el diagnóstico de hepatitis autoinmune y se inicia tratamiento. En los controles evolutivos presenta transaminasas, GGT y bilirrubina normales, mantiene hipergammaglobulinemia, aunque en descenso, y negativiza los ANA.
|


 Diagnosis in liver disease
Diagnosis in liver disease